
Presenilin-1 Familial Alzheimer Mutations Impair γ-Secretase Cleavage of APP Through Stabilized Enzyme–Substrate Complex Formation



Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of FAD-Mutant γ-Secretase Expression Vectors
2.2. Expression and Purification of γ-Secretase Complexes
2.3. In Vitro γ-Secretase Activity Assays
2.4. LC-MS/MS Analysis of Tri- and Tetrapeptide Products
2.5. Quantification of AICD Products by MALDI-TOF MS
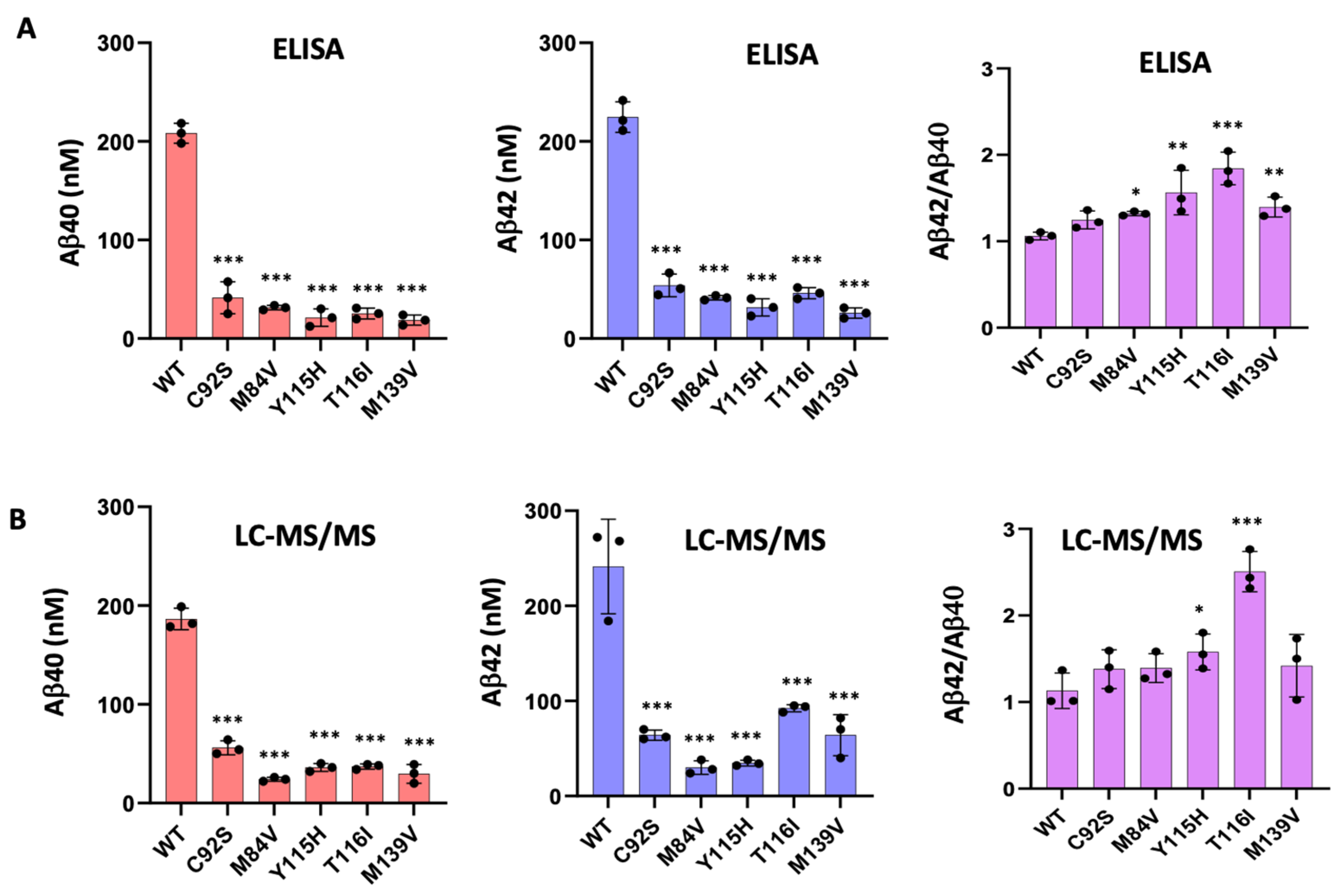
2.6. Quantification of Aβ40 and Aβ42 by ELISA
3. Results
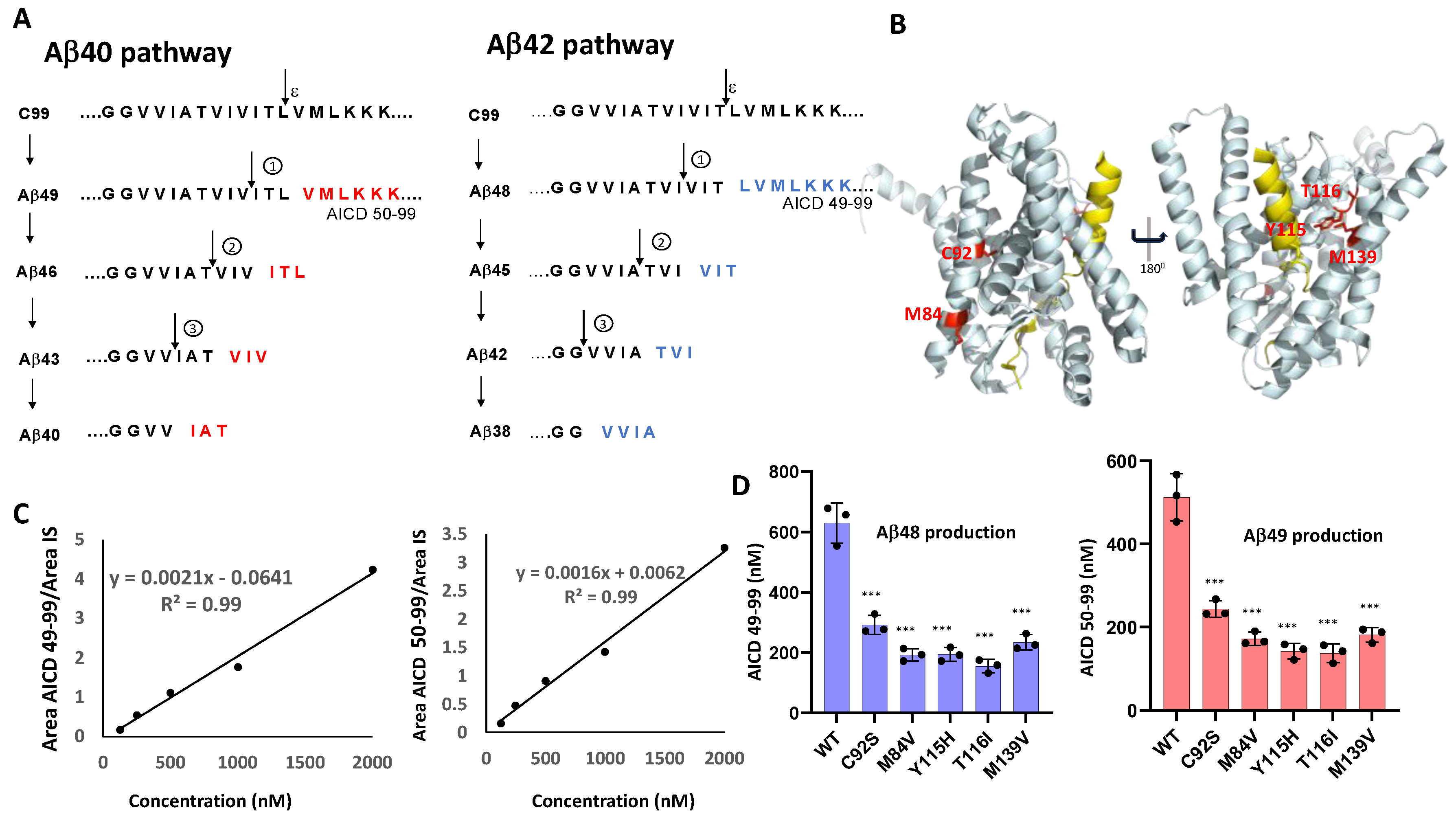
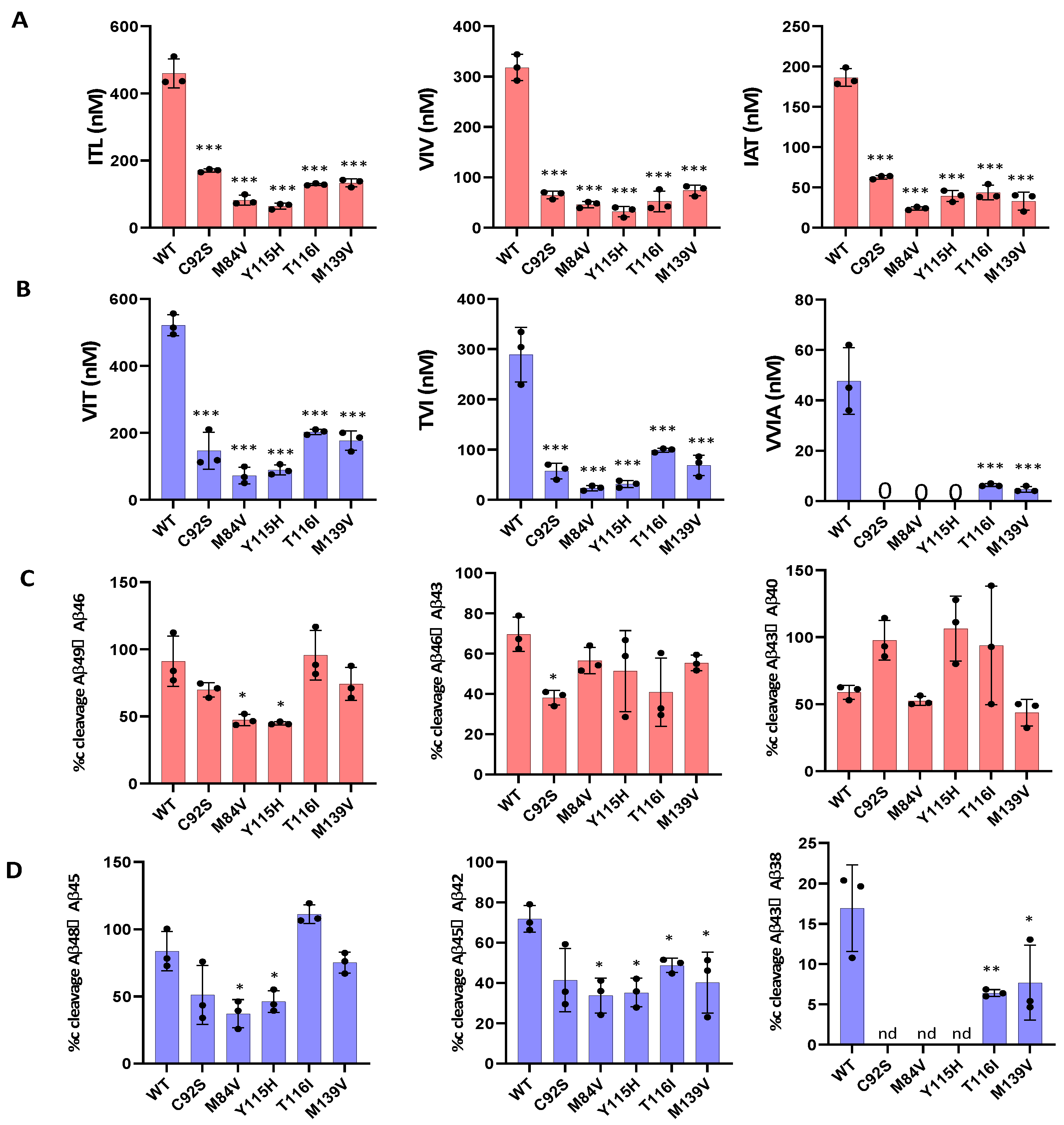
3.1. FAD PSEN1 Mutations Impair Proteolysis of C99 by γ-Secretase
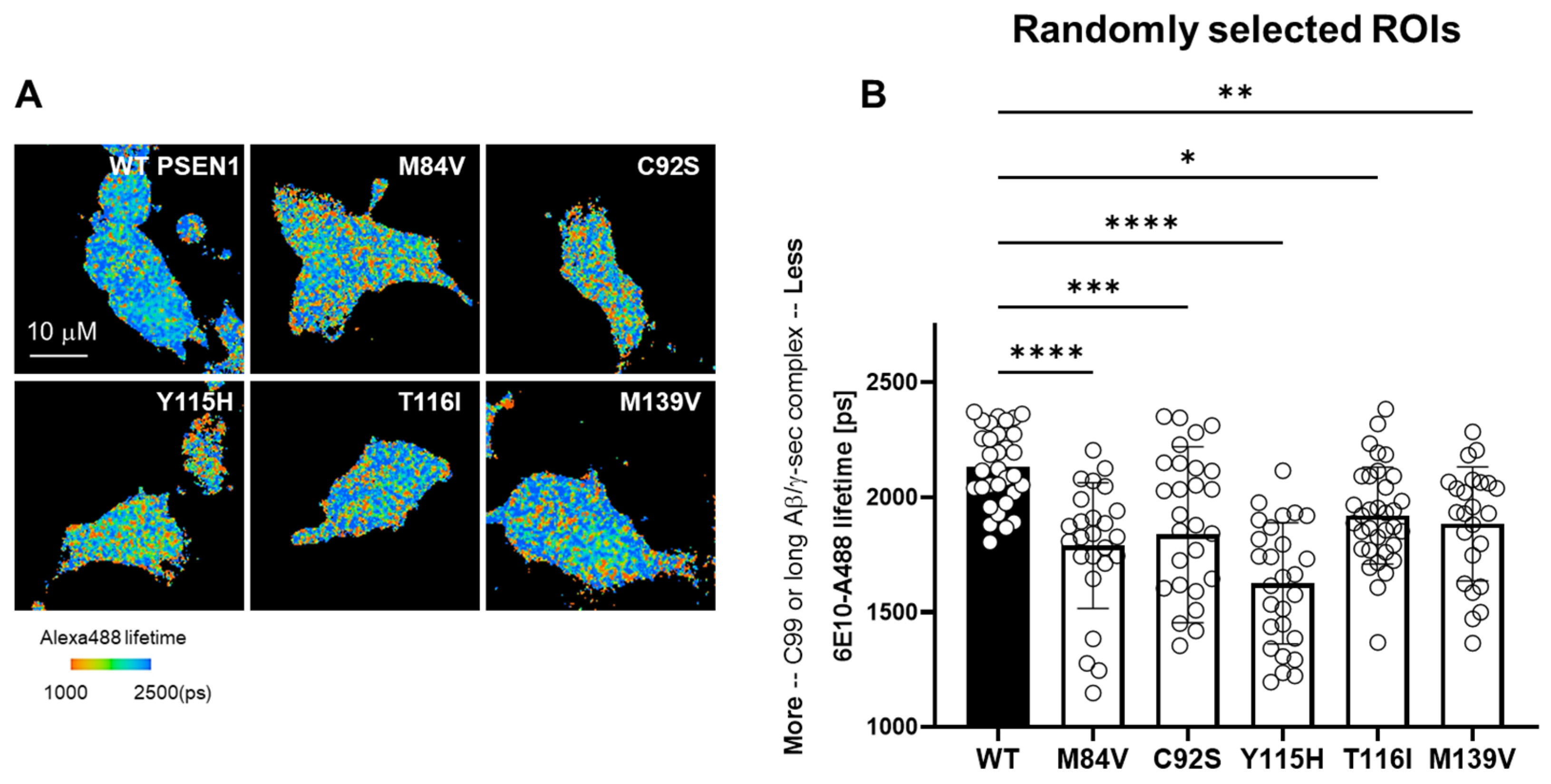
3.2. FAD Mutations Stabilize the γ-Secretase/Substrate Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β-protein |
| AD | Alzheimer’s disease |
| AICD | APP intracellular domain |
| APP | Amyloid β-protein precursor |
| FAD | Familial Alzheimer’s disease |
| HEK | Human embryonic kidney |
| LC-MS/MS | Liquid chromatography coupled to tandem mass spectrometry |
| MALDI-TOF | Matrix-assisted laser desorption/ionization time-of-flight |
| MS | Mass spectrometry |
| Q-TOF | Quadrupole time-of-flight |
| FILM | Fluorescence lifetime imaging microscopy |
References
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimers Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure and function of the γ-secretase complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Guner, G.; Lichtenthaler, S.F. The substrate repertoire of γ-secretase/presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of the β-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Presenilin, γ-Secretase, and the search for pathogenic triggers of Alzheimer’s disease. Biochemistry 2025, 64, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Zhao, R.; Nagarajan, V.; Maesako, M.; Do, H.; Noorani, A.; Overmeyer, C.; Bhattarai, S.; Douglas, J.T.; Saraf, A.; et al. Familial Alzheimer’s disease mutations stabilize synaptotoxic γ-secretase–substrate complexes. Cell Rep. 2024, 43, 101123. [Google Scholar] [CrossRef] [PubMed]
- Arafi, P.; Devkota, S.; Williams, E.; Maesako, M.; Wolfe, M.S. Alzheimer-mutant γ-secretase complexes stall amyloid β-peptide production. eLife 2025, 13, RP102274. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.A.; Liu, L.; Schultz, A.P.; Fitzpatrick, C.D.; Levin, R.; Bellier, J.-P.; Shirzadi, Z.; Joseph-Mathurin, N.; Chen, C.D.; Benzinger, T.L.S.; et al. γ-Secretase activity, clinical features, and biomarkers of autosomal dominant Alzheimer’s disease: Cross-sectional and longitudinal analysis of the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS). Lancet Neurol. 2024, 23, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Williams, T.D.; Wolfe, M.S. Familial Alzheimer’s disease mutations in amyloid protein precursor alter proteolysis by γ-secretase to increase amyloid β-peptides of ≥45 residues. J. Biol. Chem. 2021, 296, 100281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-M.; Lai, M.-T.; Xu, M.; Huang, Q.; DiMuzio-Mower, J.; Sardana, M.K.; Shi, X.-P.; Yin, K.-C.; Shafer, J.A.; Gardell, S.J. Presenilin-1 is linked with γ-secretase activity in the detergent-solubilized state. Proc. Natl. Acad. Sci. USA 2000, 97, 6138–6143. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lauro, B.M.; Wolfe, M.S.; Selkoe, D.J. Hydrophilic loop 1 of presenilin-1 and the APP GxxxG transmembrane motif regulate γ-secretase function in generating Alzheimer-causing Aβ peptides. J. Biol. Chem. 2021, 296, 100393. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aβ49 | Aβ46 | Aβ43 | Aβ40 | Aβ48 | Aβ45 | Aβ42 | Aβ38 | |
|---|---|---|---|---|---|---|---|---|
| WT | 52.5 | 142 | 131.5 | 186.4 | 108.2 | 132.3 | 241.3 | 47.6 |
| C92S | 74.3 | 105.1 | 8.8 | 56.1 | 145.5 | 82.6 | 64.5 | nd |
| M84V | 90.2 | 36.3 | 22.1 | 24.4 | 120.7 | 42.3 | 30.1 | nd |
| Y115H | 78.4 | 32.7 | −4.3 | 36.7 | 104.7 | 54.6 | 34.6 | nd |
| T116I | 8.7 | 76.3 | 15.3 | 37.4 | −46.8 | 104.1 | 92.3 | 6.3 |
| M139V | 48.1 | 59.3 | 44.3 | 29.6 | 57.6 | 108.2 | 64.6 | 4.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devkota, S.; Maesako, M.; Wolfe, M.S. Presenilin-1 Familial Alzheimer Mutations Impair γ-Secretase Cleavage of APP Through Stabilized Enzyme–Substrate Complex Formation. Biomolecules 2025, 15, 955. https://doi.org/10.3390/biom15070955
Devkota S, Maesako M, Wolfe MS. Presenilin-1 Familial Alzheimer Mutations Impair γ-Secretase Cleavage of APP Through Stabilized Enzyme–Substrate Complex Formation. Biomolecules. 2025; 15(7):955. https://doi.org/10.3390/biom15070955
Chicago/Turabian StyleDevkota, Sujan, Masato Maesako, and Michael S. Wolfe. 2025. "Presenilin-1 Familial Alzheimer Mutations Impair γ-Secretase Cleavage of APP Through Stabilized Enzyme–Substrate Complex Formation" Biomolecules 15, no. 7: 955. https://doi.org/10.3390/biom15070955
APA StyleDevkota, S., Maesako, M., & Wolfe, M. S. (2025). Presenilin-1 Familial Alzheimer Mutations Impair γ-Secretase Cleavage of APP Through Stabilized Enzyme–Substrate Complex Formation. Biomolecules, 15(7), 955. https://doi.org/10.3390/biom15070955