
Postbiotics as Mitochondrial Modulators in Inflammatory Bowel Disease: Mechanistic Insights and Therapeutic Potential

 ,
,  ,
, 
Abstract
1. Introduction
2. Mitochondrial Dysfunction in IBD
2.1. Oxidative Stress in IBD
2.2. Mitochondrial Energy Metabolism and Barrier Integrity in IBD
2.3. Apoptosis and Cell Death Pathways in IBD Pathogenesis
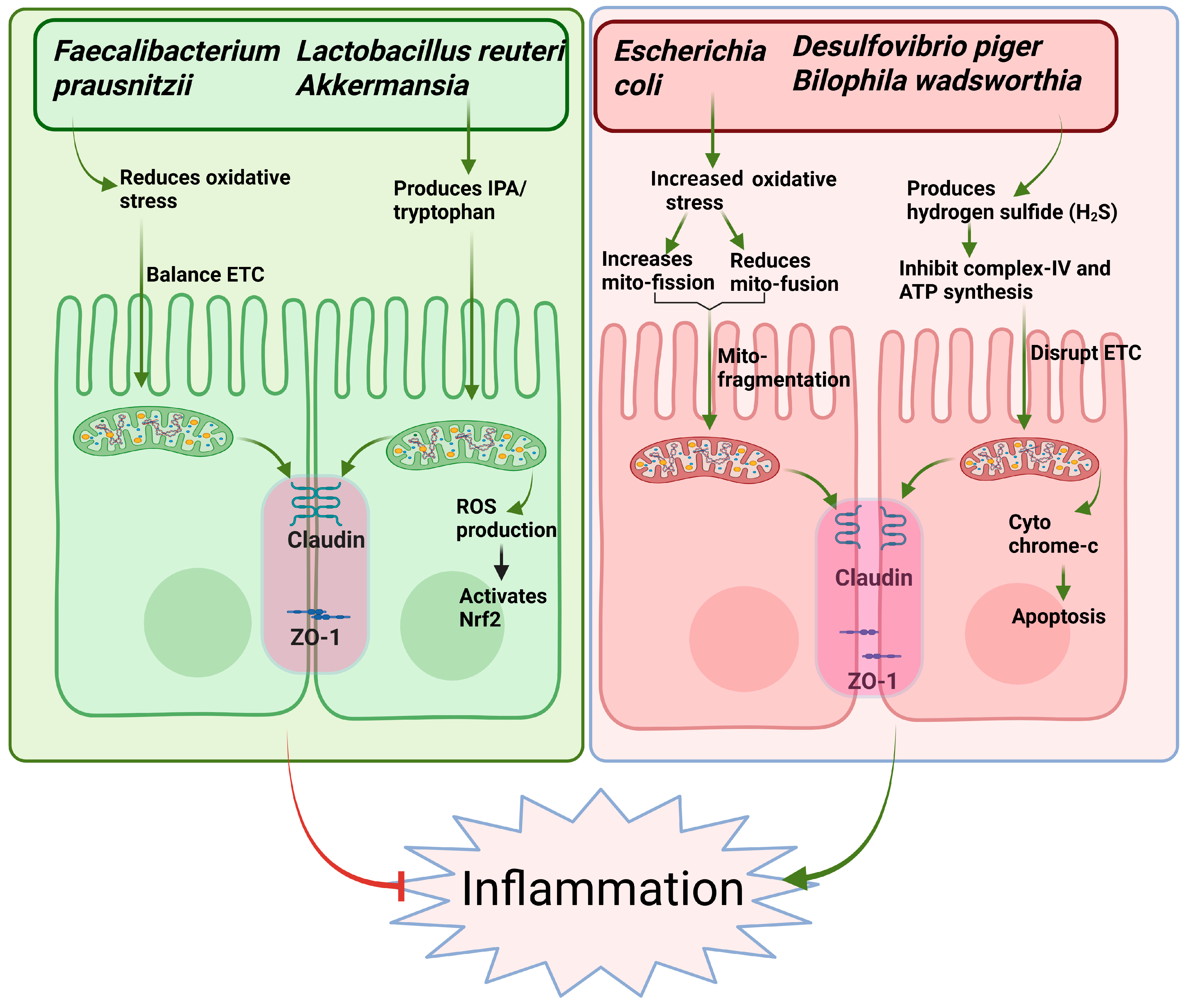
3. Microbiota–Mitochondria Crosstalk in IBD Pathogenesis
4. Postbiotics in Inflammatory Bowel Disease: Insights from Preclinical and Clinical Studies
Clinical Studies on Postbiotics in IBD Patients: Evidence of Translational Impact
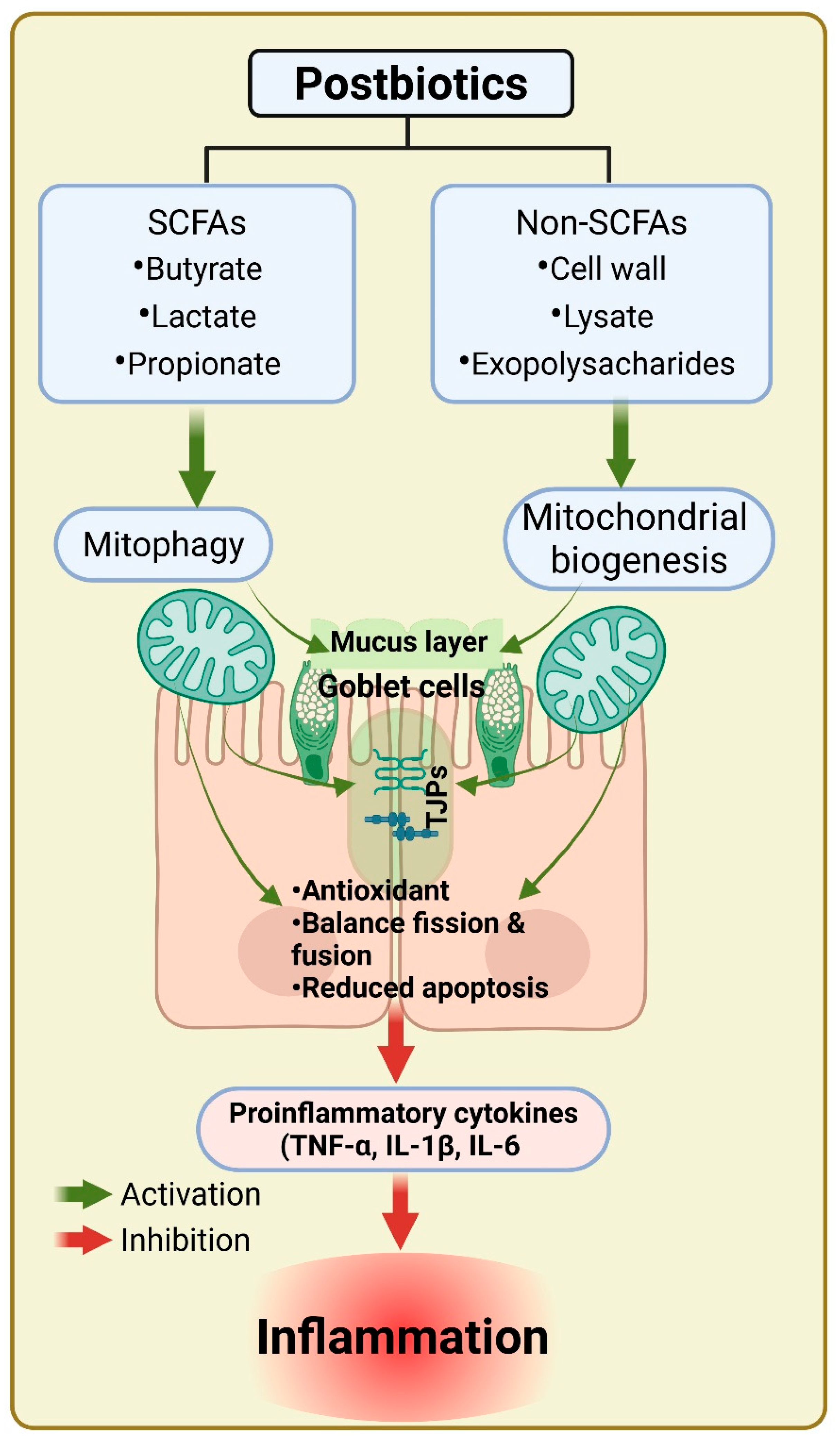
5. Postbiotics in Mitochondrial Dysfunction and IBD Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Asefa, Z.; Belay, A.; Welelaw, E.; Haile, M. Postbiotics and their biotherapeutic potential for chronic disease and their feature perspective: A review. Front. Microbiomes 2025, 4, 1489339. [Google Scholar] [CrossRef]
- Prajapati, S.K.; Lekkala, L.; Yadav, D.; Jain, S.; Yadav, H. Microbiome and Postbiotics in Skin Health. Biomedicines 2025, 13, 791. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar]
- Pedersen, S.S.; Ingerslev, L.R.; Olsen, M.; Prause, M.; Billestrup, N. Butyrate functions as a histone deacetylase inhibitor to protect pancreatic beta cells from IL-1β-induced dysfunction. FEBS J. 2024, 291, 566–583. [Google Scholar] [CrossRef]
- Sabahi, S.; Homayouni Rad, A.; Aghebati-Maleki, L.; Sangtarash, N.; Ozma, M.A.; Karimi, A.; Hosseini, H.; Abbasi, A. Postbiotics as the new frontier in food and pharmaceutical research. Crit. Rev. Food Sci. Nutr. 2023, 63, 8375–8402. [Google Scholar] [CrossRef]
- Jarmakiewicz-Czaja, S.; Zielińska, M.; Sokal, A.; Filip, R. Genetic and epigenetic etiology of inflammatory bowel disease: An update. Genes 2022, 13, 2388. [Google Scholar] [CrossRef]
- Vuyyuru, S.K.; Kedia, S.; Sahu, P.; Ahuja, V. Immune-mediated inflammatory diseases of the gastrointestinal tract: Beyond Crohn’s disease and ulcerative colitis. JGH Open 2022, 6, 100–111. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Mirsepasi-Lauridsen, H.C.; Vallance, B.A.; Krogfelt, K.A.; Petersen, A.M. Escherichia coli pathobionts associated with inflammatory bowel disease. Clin. Microbiol. Rev. 2019, 32, e00060-18. [Google Scholar] [CrossRef]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of inflammatory bowel disease: Innate immune system. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef]
- Schippa, S.; Conte, M.P. Dysbiotic events in gut microbiota: Impact on human health. Nutrients 2014, 6, 5786–5805. [Google Scholar] [CrossRef]
- Pastorelli, L.; De Salvo, C.; Mercado, J.R.; Vecchi, M.; Pizarro, T.T. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: Lessons learned from animal models and human genetics. Front. Immunol. 2013, 4, 280. [Google Scholar] [CrossRef]
- Taurog, J.D.; Richardson, J.A.; Croft, J.; Simmons, W.A.; Zhou, M.; Fernández-Sueiro, J.L.; Balish, E.; Hammer, R.E. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 1994, 180, 2359–2364. [Google Scholar] [CrossRef]
- Sartor, B.R. Targeting enteric bacteria in treatment of inflammatory bowel diseases: Why, how, and when. Curr. Opin. Gastroenterol. 2003, 19, 358–365. [Google Scholar] [CrossRef]
- Martyniak, A.; Medyńska-Przęczek, A.; Wędrychowicz, A.; Skoczeń, S.; Tomasik, P.J. Prebiotics, probiotics, synbiotics, paraprobiotics and postbiotic compounds in IBD. Biomolecules 2021, 11, 1903. [Google Scholar] [CrossRef]
- Mazumder, S.; Bindu, S.; De, R.; Debsharma, S.; Pramanik, S.; Bandyopadhyay, U. Emerging role of mitochondrial DAMPs, aberrant mitochondrial dynamics and anomalous mitophagy in gut mucosal pathogenesis. Life Sci. 2022, 305, 120753. [Google Scholar] [CrossRef]
- Sánchez-Quintero, M.J.; Rodríguez-Díaz, C.; Rodríguez-González, F.J.; Fernández-Castañer, A.; García-Fuentes, E.; López-Gómez, C. Role of mitochondria in inflammatory bowel diseases: A systematic review. Int. J. Mol. Sci. 2023, 24, 17124. [Google Scholar] [CrossRef]
- Haque, P.S.; Kapur, N.; Barrett, T.A.; Theiss, A.L. Mitochondrial function and gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 537–555. [Google Scholar] [CrossRef]
- Goudie, L. Investigating the Consequences of DRP1 and Fis1 Mediated Mitochondrial Fission in Colitis: In Pursuit of a Novel Therapeutic Target for IBD. Master’s Thesis, University of Calgary, Calgary, AB, Canada, 2019. [Google Scholar]
- Mottawea, W.; Chiang, C.-K.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S. Altered intestinal microbiota–host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef]
- Clark, A.; Mach, N. The crosstalk between the gut microbiota and mitochondria during exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747. [Google Scholar] [CrossRef]
- Wang, S.; Wang, P.; Wang, D.; Shen, S.; Wang, S.; Li, Y.; Chen, H. Postbiotics in inflammatory bowel disease: Efficacy, mechanism, and therapeutic implications. J. Sci. Food Agric. 2025, 105, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Reductive stress in inflammation-associated diseases and the pro-oxidant effect of antioxidant agents. Int. J. Mol. Sci. 2017, 18, 2098. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Song, M.-H.; Oh, J.-W.; Keum, Y.-S.; Saini, R.K. Pro-oxidant actions of carotenoids in triggering apoptosis of cancer cells: A review of emerging evidence. Antioxidants 2020, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Alula, K.M.; Jackson, D.N.; Smith, A.D.; Kim, D.S.; Turner, K.; Odstrcil, E.; Kaipparettu, B.A.; Dassopoulos, T.; Venuprasad, K.; Feagins, L.A. Targeting mitochondrial damage as a therapeutic for ileal Crohn’s disease. Cells 2021, 10, 1349. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological activities of reactive oxygen and nitrogen species: Oxidative stress versus signal transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef]
- Panayotova, M.; Penkova, M. Measurement of oxidative stress-related markers in gastro-intestinal damages in Bulgarian pediatric patients. Bulg. Chem. Commun. 2024, 56, 142–147. [Google Scholar] [CrossRef]
- Banerjee, S.; Fu, Q.; Shah, S.K.; Melnyk, S.B.; Sterneck, E.; Hauer-Jensen, M.; Pawar, S.A. C/EBPδ protects from radiation-induced intestinal injury and sepsis by suppression of inflammatory and nitrosative stress. Sci. Rep. 2019, 9, 13953. [Google Scholar] [CrossRef]
- Rezaie, A.; Ghorbani, F.; Eshghtork, A.; Zamani, M.J.; Dehghan, G.; Taghavi, B.; Nikfar, S.; Mohammadirad, A.; Daryani, N.E.; Abdollahi, M. Alterations in Salivary Antioxidants, Nitric Oxide, and Transforming Growth Factor-β1 in Relation to Disease Activity in Crohn’s Disease Patients. Ann. N. Y. Acad. Sci. 2006, 1091, 110–122. [Google Scholar] [CrossRef]
- Aghdassi, E.; Wendland, B.E.; Steinhart, H.A.; Wolman, S.L.; Jeejeebhoy, K.; Allard, J.P. Antioxidant vitamin supplementation in Crohn’s disease decreases oxidative stress: A randomized controlled trial. Off. J. Am. Coll. Gastroenterol. | ACG 2003, 98, 348–353. [Google Scholar]
- Esworthy, R.S.; Aranda, R.; Martín, M.G.; Doroshow, J.H.; Binder, S.W.; Chu, F.-F. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 281, G848–G855. [Google Scholar] [CrossRef]
- Liu, G.; Yu, L.; Fang, J.; Hu, C.-A.A.; Yin, J.; Ni, H.; Ren, W.; Duraipandiyan, V.; Chen, S.; Al-Dhabi, N.A. Methionine restriction on oxidative stress and immune response in dss-induced colitis mice. Oncotarget 2017, 8, 44511. [Google Scholar] [CrossRef]
- D’Odorico, S.; Bortolan, R.; Cardin, R.; D’Inca’, D.; Martines, A.; Ferronato, G.C.; Sturniolo, A. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand. J. Gastroenterol. 2001, 36, 1289–1294. [Google Scholar] [CrossRef]
- Nair, J.; Gansauge, F.; Beger, H.; Dolara, P.; Winde, G.; Bartsch, H. Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn’s disease, ulcerative colitis, and chronic pancreatitis. Antioxid. Redox Signal. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- Bär, F.; Bochmann, W.; Widok, A.; Von Medem, K.; Pagel, R.; Hirose, M.; Yu, X.; Kalies, K.; König, P.; Böhm, R. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology 2013, 145, 1055–1063.e3. [Google Scholar] [CrossRef]
- Nishikawa, M.; Oshitani, N.; Matsumoto, T.; Nishigami, T.; Arakawa, T.; Inoue, M. Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br. J. Cancer 2005, 93, 331–337. [Google Scholar] [CrossRef]
- Theiss, A.L.; Laroui, H.; Obertone, T.S.; Chowdhury, I.; Thompson, W.E.; Merlin, D.; Sitaraman, S.V. Nanoparticle-based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm. Bowel Dis. 2011, 17, 1163–1176. [Google Scholar] [CrossRef]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020, 69, 1928–1938. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and oxidative stress: The role of aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Li, W.; Jiang, W.-S.; Su, Y.-R.; Tu, K.-W.; Zou, L.; Liao, C.-R.; Wu, Q.; Wang, Z.-H.; Zhong, Z.-M.; Chen, J.-T. PINK1/Parkin-mediated mitophagy inhibits osteoblast apoptosis induced by advanced oxidation protein products. Cell Death Dis. 2023, 14, 88. [Google Scholar] [CrossRef]
- Cunningham, K.E.; Vincent, G.; Sodhi, C.P.; Novak, E.A.; Ranganathan, S.; Egan, C.E.; Stolz, D.B.; Rogers, M.B.; Firek, B.; Morowitz, M.J. Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) protects against experimental murine colitis. J. Biol. Chem. 2016, 291, 10184–10200. [Google Scholar] [CrossRef] [PubMed]
- Mancini, N.L.; Goudie, L.; Xu, W.; Sabouny, R.; Rajeev, S.; Wang, A.; Esquerre, N.; Al Rajabi, A.; Jayme, T.S.; van Tilburg Bernandes, E. Perturbed mitochondrial dynamics is a novel feature of colitis that can be targeted to lessen disease. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 287–307. [Google Scholar] [CrossRef]
- Chernyavskij, D.; Galkin, I.; Pavlyuchenkova, A.; Fedorov, A.; Chelombitko, M. Role of Mitochondria in Intestinal Epithelial Barrier Dysfunction in Inflammatory Bowel Disease. Mol. Biol. 2023, 57, 1024–1037. [Google Scholar] [CrossRef]
- Goll, R.; van Beelen Granlund, A. Intestinal barrier homeostasis in inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Mucus and the goblet cell. Dig. Dis. 2013, 31, 305–309. [Google Scholar] [CrossRef]
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in intestinal mucosal defense and inflammation: Learning from clinical and experimental studies. Front. Immunol. 2020, 11, 2054. [Google Scholar] [CrossRef]
- Bergstrom, K.S.; Kissoon-Singh, V.; Gibson, D.L.; Ma, C.; Montero, M.; Sham, H.P.; Ryz, N.; Huang, T.; Velcich, A.; Finlay, B.B. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010, 6, e1000902. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Shen, J. The roles and functions of Paneth cells in Crohn’s disease: A critical review. Cell Prolif. 2021, 54, e12958. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Blikslager, A.T.; Moeser, A.J.; Gookin, J.L.; Jones, S.L.; Odle, J. Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 2007, 87, 545–564. [Google Scholar] [CrossRef]
- Martel, J.; Chang, S.-H.; Ko, Y.-F.; Hwang, T.-L.; Young, J.D.; Ojcius, D.M. Gut barrier disruption and chronic disease. Trends Endocrinol. Metab. 2022, 33, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Gelberg, H. Pathophysiological mechanisms of gastrointestinal toxicity. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2017; p. 139. [Google Scholar]
- Caruso, R.; Lo, B.C.; Núñez, G. Host–microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef]
- Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the fine-tuning of colonic homeostasis: Implication for inflammatory bowel diseases. Int. J. Mol. Sci. 2021, 22, 3061. [Google Scholar] [CrossRef]
- Salvi, P.S.; Cowles, R.A. Butyrate and the intestinal epithelium: Modulation of proliferation and inflammation in homeostasis and disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef]
- Santhanam, S.; Venkatraman, A.; Ramakrishna, B.S. Impairment of mitochondrial acetoacetyl CoA thiolase activity in the colonic mucosa of patients with ulcerative colitis. Gut 2007, 56, 1543–1549. [Google Scholar] [CrossRef]
- Girardin, M.; Dionne, S.; Goyette, P.; Rioux, J.; Bitton, A.; Elimrani, I.; Charlebois, P.; Qureshi, I.; Levy, E.; Seidman, E.G. Expression and functional analysis of intestinal organic cation/L-carnitine transporter (OCTN) in Crohn’s disease. J. Crohn’s Colitis 2012, 6, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Günther, C.; Neumann, H.; Neurath, M.F.; Becker, C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut 2013, 62, 1062–1071. [Google Scholar] [CrossRef]
- Goretsky, T.; Dirisina, R.; Sinh, P.; Mittal, N.; Managlia, E.; Williams, D.B.; Posca, D.; Ryu, H.; Katzman, R.B.; Barrett, T.A. p53 mediates TNF-induced epithelial cell apoptosis in IBD. Am. J. Pathol. 2012, 181, 1306–1315. [Google Scholar] [CrossRef]
- Piguet, P.F.; Vesin, C.; Donati, Y.; Barazzone, C. TNF-induced enterocyte apoptosis and detachment in mice: Induction of caspases and prevention by a caspase inhibitor, ZVAD-fmk. Lab. Investig. A J. Tech. Methods Pathol. 1999, 79, 495–500. [Google Scholar]
- Ramachandran, A.; Madesh, M.; Balasubramanian, K.A. Apoptosis in the intestinal epithelium: Its relevance in normal and pathophysiological conditions. J. Gastroenterol. Hepatol. 2000, 15, 109–120. [Google Scholar] [CrossRef]
- Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Autophagy and apoptosis in inflammatory bowel disease. Gastroenterol. Insights 2023, 14, 598–636. [Google Scholar]
- Dirisina, R.; Katzman, R.B.; Goretsky, T.; Managlia, E.; Mittal, N.; Williams, D.B.; Qiu, W.; Yu, J.; Chandel, N.S.; Zhang, L. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011, 141, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Garcia Carbonell, R. The Role of RIPK1 in TNF-Induced Cell Death in Intestinal Epithelial Cells. Ph.D. Thesis, Universitat de Barcelona, Barcelona, Spain, 2017. [Google Scholar]
- Vereecke, L.; Vieira-Silva, S.; Billiet, T.; Van Es, J.H.; Mc Guire, C.; Slowicka, K.; Sze, M.; Van Den Born, M.; De Hertogh, G.; Clevers, H. A20 controls intestinal homeostasis through cell-specific activities. Nat. Commun. 2014, 5, 5103. [Google Scholar] [CrossRef]
- Webster, J.D.; Vucic, D. The balance of TNF mediated pathways regulates inflammatory cell death signaling in healthy and diseased tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Qian, J.; Zhao, W.; Miao, X.; Li, L.; Zhang, D. Sam68 modulates apoptosis of intestinal epithelial cells via mediating NF-κB activation in ulcerative colitis. Mol. Immunol. 2016, 75, 48–59. [Google Scholar] [CrossRef]
- Prajapati, S.K.; Shah, R.; Alford, N.; Mishra, S.P.; Jain, S.; Hansen, B.; Sanberg, P.; Molina, A.J.; Yadav, H. The triple alliance: Microbiome, mitochondria, and metabolites in the context of age-related cognitive decline and Alzheimer’s disease. J. Gerontol. Ser. A 2023, 78, 2187–2202. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Duan, L. The role of microbiota-mitochondria crosstalk in pathogenesis and therapy of intestinal diseases. Pharmacol. Res. 2022, 186, 106530. [Google Scholar] [CrossRef]
- Mancini, N.L.; Rajeev, S.; Jayme, T.S.; Wang, A.; Keita, Å.V.; Workentine, M.L.; Hamed, S.; Söderholm, J.D.; Lopes, F.; Shutt, T.E. Crohn’s disease pathobiont adherent-invasive E coli disrupts epithelial mitochondrial networks with implications for gut permeability. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 551–571. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Han, B.; Sivaramakrishnan, P.; Lin, C.-C.J.; Neve, I.A.; He, J.; Tay, L.W.R.; Sowa, J.N.; Sizovs, A.; Du, G.; Wang, J. Microbial genetic composition tunes host longevity. Cell 2017, 169, 1249–1262.e13. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.J.; Wang, M.C. Microbial metabolites regulate host lipid metabolism through NR5A–Hedgehog signalling. Nat. Cell Biol. 2017, 19, 550–557. [Google Scholar] [CrossRef]
- Kushkevych, I.; Cejnar, J.; Treml, J.; Dordević, D.; Kollar, P.; Vítězová, M. Recent advances in metabolic pathways of sulfate reduction in intestinal bacteria. Cells 2020, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Stummer, N.; Feichtinger, R.G.; Weghuber, D.; Kofler, B.; Schneider, A.M. Role of hydrogen sulfide in inflammatory bowel disease. Antioxidants 2023, 12, 1570. [Google Scholar] [CrossRef]
- Kushkevych, I.; Dordević, D.; Vítězová, M. Toxicity of hydrogen sulfide toward sulfate-reducing bacteria Desulfovibrio piger Vib-7. Arch. Microbiol. 2019, 201, 389–397. [Google Scholar] [CrossRef]
- Davies, J.; Mayer, M.J.; Juge, N.; Narbad, A.; Sayavedra, L. Bacteroides thetaiotaomicron enhances H2S production in Bilophila wadsworthia. Gut Microbes 2024, 16, 2431644. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J. Microbiota-derived indole metabolites promote human and murine intestinal homeostasis through regulation of interleukin-10 receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef]
- Li, J.; Zou, P.; Xiao, R.; Wang, Y. Indole-3-propionic acid alleviates DSS-induced colitis in mice through macrophage glycolipid metabolism. Int. Immunopharmacol. 2025, 152, 114388. [Google Scholar] [CrossRef]
- Konopelski, P.; Mogilnicka, I. Biological effects of indole-3-propionic acid, a gut microbiota-derived metabolite, and its precursor tryptophan in mammals’ health and disease. Int. J. Mol. Sci. 2022, 23, 1222. [Google Scholar] [CrossRef]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Luo, Y.; An, Y.; Wu, X. The mechanism of action of indole-3-propionic acid on bone metabolism. Food Funct. 2025, 16, 406–421. [Google Scholar] [CrossRef]
- Shanmugham, M.; Bellanger, S.; Leo, C.H. Gut-derived metabolite, trimethylamine-N-oxide (TMAO) in cardio-metabolic diseases: Detection, mechanism, and potential therapeutics. Pharmaceuticals 2023, 16, 504. [Google Scholar] [CrossRef]
- Bordoni, L.; Petracci, I.; Feliziani, G.; de Simone, G.; Rucci, C.; Gabbianelli, R. Gut microbiota-derived trimethylamine promotes inflammation with a potential impact on epigenetic and mitochondrial homeostasis in Caco-2 cells. Antioxidants 2024, 13, 1061. [Google Scholar] [CrossRef]
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ahmadi, S.; Nagpal, R.; Jain, S.; Mishra, S.P.; Kavanagh, K.; Zhu, X.; Wang, Z.; McClain, D.A.; Kritchevsky, S.B.; et al. Lipoteichoic acid from the cell wall of a heat killed Lactobacillus paracasei D3-5 ameliorates aging-related leaky gut, inflammation and improves physical and cognitive functions: From C. elegans to mice. Geroscience 2020, 42, 333–352. [Google Scholar] [CrossRef]
- Chen, G.; Ran, X.; Li, B.; Li, Y.; He, D.; Huang, B.; Fu, S.; Liu, J.; Wang, W. Sodium butyrate inhibits inflammation and maintains epithelium barrier integrity in a TNBS-induced inflammatory bowel disease mice model. EBioMedicine 2018, 30, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.-C.; Wang, Y.; Wang, Z.-B.; Liu, W.-Y.; Sun, S.; Li, L.; Su, D.-F.; Zhang, L.-C. Propionate ameliorates dextran sodium sulfate-induced colitis by improving intestinal barrier function and reducing inflammation and oxidative stress. Front. Pharmacol. 2016, 7, 253. [Google Scholar] [CrossRef]
- Luceri, C.; Femia, A.P.; Fazi, M.; Di Martino, C.; Zolfanelli, F.; Dolara, P.; Tonelli, F. Effect of butyrate enemas on gene expression profiles and endoscopic/histopathological scores of diverted colorectal mucosa: A randomized trial. Dig. Liver Dis. 2016, 48, 27–33. [Google Scholar] [CrossRef]
- Dou, X.; Gao, N.; Yan, D.; Shan, A. Sodium butyrate alleviates mouse colitis by regulating gut microbiota dysbiosis. Animals 2020, 10, 1154. [Google Scholar] [CrossRef]
- Jourova, L.; Satka, S.; Frybortova, V.; Zapletalova, I.; Anzenbacher, P.; Anzenbacherova, E.; Hermanova, P.P.; Drabonova, B.; Srutkova, D.; Kozakova, H. Butyrate treatment of DSS-induced ulcerative colitis affects the hepatic drug metabolism in mice. Front. Pharmacol. 2022, 13, 936013. [Google Scholar] [CrossRef]
- Chorawala, M.R.; Chauhan, S.; Patel, R.; Shah, G. Cell wall contents of probiotics (Lactobacillus species) protect against lipopolysaccharide (LPS)-induced murine colitis by limiting immuno-inflammation and oxidative stress. Probiotics Antimicrob. Proteins 2021, 13, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.-S.; Park, M.Y.; Shin, J.-H.; Kim, J.Y.; Kwon, O. Lysate of probiotic lactobacillus plantarum K8 modulate the mucosal inflammatory system in dextran sulfate sodium-induced colitic rats. Korean J. Food Sci. Anim. Resour. 2014, 34, 829. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Peng, C.; Sun, Y.; Zhang, T.; Feng, C.; Zhang, W.; Huang, T.; Yao, G.; Zhang, H.; He, Q. Both viable Bifidobacterium longum subsp. infantis B8762 and heat-killed cells alleviate the intestinal inflammation of DSS-induced IBD rats. Microbiol. Spectr. 2024, 12, e03509-23. [Google Scholar] [CrossRef]
- Ueno, N.; Fujiya, M.; Segawa, S.; Nata, T.; Moriichi, K.; Tanabe, H.; Mizukami, Y.; Kobayashi, N.; Ito, K.; Kohgo, Y. Heat-killed body of lactobacillus brevis SBC8803 ameliorates intestinal injury in a murine model of colitis by enhancing the intestinal barrier function. Inflamm. Bowel Dis. 2011, 17, 2235–2250. [Google Scholar] [CrossRef]
- Lührs, H.; Gerke, T.; Müller, J.; Melcher, R.; Schauber, J.; Boxberger, F.; Scheppach, W.; Menzel, T. Butyrate inhibits NF-κB activation in lamina propria macrophages of patients with ulcerative colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef]
- Segain, J.; De La Blétiere, D.R.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.; Galmiche, J. Butyrate inhibits inflammatory responses through NFκB inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef]
- Jin, Y.; Xu, X.; Huang, K.; Liang, Z. Pre-Administration of Saccharomyces boulardii-Derived Postbiotics Effectively Prevents Dextran Sulfate Sodium-Induced Colitis in Mice. Foods 2025, 14, 1109. [Google Scholar] [CrossRef]
- Enck, P.; Zimmermann, K.; Menke, G.; Müller-Lissner, S.; Martens, U.; Klosterhalfen, S. A mixture of Escherichia coli (DSM 17252) and Enterococcus faecalis (DSM 16440) for treatment of the irritable bowel syndrome–a randomized controlled trial with primary care physicians. Neurogastroenterol. Motil. 2008, 20, 1103–1109. [Google Scholar] [CrossRef]
- Andresen, V.; Gschossmann, J.; Layer, P. Heat-inactivated Bifidobacterium bifidum MIMBb75 (SYN-HI-001) in the treatment of irritable bowel syndrome: A multicentre, randomised, double-blind, placebo-controlled clinical trial. Lancet Gastroenterol. Hepatol. 2020, 5, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yoo, J.I.; Ma, H.W.; Park, I.S.; Son, M.; Shin, Y.; Kim, K.B.; Kim, S.W.; Park, S.J.; Park, J. Anti-inflammatory properties of butyrate-producing atypical Escherichia coli in a murine colitis model. Intest. Res. 2022, 21, 266–269. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, W.; Zhang, T.; He, Q.; Kwok, L.-Y.; Tan, Y.; Zhang, H. Heat-killed Bifidobacterium bifidum B1628 may alleviate dextran sulfate sodium-induced colitis in mice, and the anti-inflammatory effect is associated with gut microbiota modulation. Nutrients 2022, 14, 5233. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wu, S.; Xie, Y.; Liu, H.; Gao, X.; Zhang, H. Live and heat-killed cells of Lactobacillus plantarum Zhang-LL ease symptoms of chronic ulcerative colitis induced by dextran sulfate sodium in rats. J. Funct. Foods 2020, 71, 103994. [Google Scholar] [CrossRef]
- Liu, C.; Qi, X.; Li, D.; Zhao, L.; Li, Q.; Mao, K.; Shen, G.; Ma, Y.; Wang, R. Limosilactobacillus fermentum HF06-derived paraprobiotic and postbiotic alleviate intestinal barrier damage and gut microbiota disruption in mice with ulcerative colitis. J. Sci. Food Agric. 2024, 104, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, D.; Gulati, G.; Avadhani, R.; HM, R.; Soumya, K.; Kumari, A.; Gupta, A.; Dwivedi, D.; Kaushik, J.K.; Grover, S. Postbiotic Lipoteichoic acid of probiotic Lactobacillus origin ameliorates inflammation in HT-29 cells and colitis mice. Int. J. Biol. Macromol. 2023, 236, 123962. [Google Scholar] [CrossRef]
- Chen, K.; Luo, H.; Li, Y.; Han, X.; Gao, C.; Wang, N.; Lu, F.; Wang, H. Lactobacillus paracasei TK1501 fermented soybeans alleviate dextran sulfate sodium-induced colitis by regulating intestinal cell function. J. Sci. Food Agric. 2023, 103, 5422–5431. [Google Scholar] [CrossRef]
- Sawada, D.; Sugawara, T.; Ishida, Y.; Aihara, K.; Aoki, Y.; Takehara, I.; Takano, K.; Fujiwara, S. Effect of continuous ingestion of a beverage prepared with Lactobacillus gasseri CP2305 inactivated by heat treatment on the regulation of intestinal function. Food Res. Int. 2016, 79, 33–39. [Google Scholar] [CrossRef]
- Tarrerias, A.; Costil, V.; Vicari, F.; Letard, J.; Adenis-Lamarre, P.; Aisene, A.; Batistelli, D.; Bonnaud, G.; Carpentier, S.; Dalbies, P. The effect of inactivated Lactobacillus LB fermented culture medium on symptom severity: Observational investigation in 297 patients with diarrhea-predominant irritable bowel syndrome. Dig. Dis. 2011, 29, 588–591. [Google Scholar] [CrossRef]
- Huang, F.; Huang, S. The Combined Beneficial Effects of Postbiotic Butyrate on Active Vitamin D3-Orchestrated Innate Immunity to Salmonella Colitis. Biomedicines 2021, 9, 1296. [Google Scholar] [CrossRef]
- Gonçalves, P.; Martel, F. Butyrate and colorectal cancer: The role of butyrate transport. Curr. Drug Metab. 2013, 14, 994–1008. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.; Zhu, J.; Lin, Q.; Yu, M.; Wen, J.; Feng, J.; Hu, C. Sodium butyrate ameliorates oxidative stress-induced intestinal epithelium barrier injury and mitochondrial damage through AMPK-mitophagy pathway. Oxid. Med. Cell. Longev. 2022, 2022, 3745135. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef]
- Pereira, C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative stress and DNA damage: Implications in inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 2403–2417. [Google Scholar] [CrossRef]
- Izuddin, W.I.; Humam, A.M.; Loh, T.C.; Foo, H.L.; Samsudin, A.A. Dietary postbiotic Lactobacillus plantarum improves serum and ruminal antioxidant activity and upregulates hepatic antioxidant enzymes and ruminal barrier function in post-weaning lambs. Antioxidants 2020, 9, 250. [Google Scholar] [CrossRef]
- Dameshghian, M.; Tafvizi, F.; Tajabadi Ebrahimi, M.; Hosseini Doust, R. Anticancer potential of Postbiotic derived from Lactobacillus brevis and Lactobacillus casei: In vitro analysis of breast Cancer cell line. Probiotics Antimicrob. Proteins, 2024; Epub ahead of print. [Google Scholar]
- Wu, Y.; Hu, A.; Shu, X.; Huang, W.; Zhang, R.; Xu, Y.; Yang, C. Lactobacillus plantarum postbiotics trigger AMPK-dependent autophagy to suppress Salmonella intracellular infection and NLRP3 inflammasome activation. J. Cell. Physiol. 2023, 238, 1336–1353. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Liu, J.; Yu, P.; Qiu, T.; Jiang, S.; Yu, R. Unlocking the power of probiotics, postbiotics: Targeting apoptosis for the treatment and prevention of digestive diseases. Front. Nutr. 2025, 12, 1570268. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.-J.; Khan, F.; Tabassum, N.; Cho, K.-J.; Kim, Y.-M. Controlling biofilm and virulence properties of Gram-positive bacteria by targeting wall teichoic acid and lipoteichoic acid. Int. J. Antimicrob. Agents 2023, 62, 106941. [Google Scholar] [CrossRef]
- Wang, P.; Wang, S.; Wang, D.; Li, Y.; Yip, R.C.S.; Chen, H. Postbiotics-peptidoglycan, lipoteichoic acid, exopolysaccharides, surface layer protein and pili proteins—Structure, activity in wounds and their delivery systems. Int. J. Biol. Macromol. 2024, 274, 133195. [Google Scholar] [CrossRef]
- Yan, F.; Liu, L.; Dempsey, P.J.; Tsai, Y.-H.; Raines, E.W.; Wilson, C.L.; Cao, H.; Cao, Z.; Liu, L.; Polk, D.B. A Lactobacillus rhamnosus GG-derived soluble protein, p40, stimulates ligand release from intestinal epithelial cells to transactivate epidermal growth factor receptor. J. Biol. Chem. 2013, 288, 30742–30751. [Google Scholar] [CrossRef]
- Seth, A.; Yan, F.; Polk, D.B.; Rao, R. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC-and MAP kinase-dependent mechanism. Am. J. Physiol.-Gastrointest. Liver Physiol. 2008, 294, G1060–G1069. [Google Scholar] [CrossRef]
- Guglielmetti, S.; Tamagnini, I.; Minuzzo, M.; Arioli, S.; Parini, C.; Comelli, E.; Mora, D. Study of the adhesion of Bifidobacterium bifidum MIMBb75 to human intestinal cell lines. Curr. Microbiol. 2009, 59, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Martorell, P.; Alvarez, B.; Llopis, S.; Navarro, V.; Ortiz, P.; Gonzalez, N.; Balaguer, F.; Rojas, A.; Chenoll, E.; Ramon, D. Heat-treated Bifidobacterium longum CECT-7347: A whole-cell postbiotic with antioxidant, anti-inflammatory, and gut-barrier protection properties. Antioxidants 2021, 10, 536. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Strains | Postbiotics | Study Type | Therapeutic Outcomes | References |
|---|---|---|---|---|
| Atypical Escherichia coli | Butyrate | In vivo | Anti-inflammatory; improves gut integrity | [104] |
| Bifidobacterium bifidum B1628 | Heat killed cells | In-vivo DSS-induced colitis model | Anti-inflammatory; increase beneficial gut microbiome | [105] |
| Lactobacillus plantarum | Heat-killed cells | DSS-induced colitis mice | Immunomodulator; increase tight junction proteins | [23,106] |
| Limosilactobacillus fermentum HF06 | Heat-killed cells | DSS-induced colitis mice | Decrease intestinal barrier damage | [107] |
| Lactobacillus | Lipoteichoic acid | DSS-induced colitis model | Decrease proinflammatory and increase anti-inflammatory cytokines | [108] |
| Lactobacillus paracasei TK1501 | Lipoteichoic acid and peptidoglycan | DSS-induced colitis mice | Ameliorate the mucin-2 expression and boost the phagocytosis | [109] |
| Lactobacillus spp. | Cell wall contents | Lipopolysaccharide-induced colitis rats | Mitigate immune-mediated inflammation and oxidative stress | [95] |
| Lactobacillus gasseri CP2305 | Fermented milk-based beverage | Human | Increase the concentrations of short-chain fatty acids and the population of Clostridium in fecal matter | [110] |
| Lactobacillus LB | Inactivated fermented culture medium | Human | Reduce pain scores and improve quality of life in diarrhea-predominant irritable bowel syndrome | [111] |
| Bifidobacterium bifidum MIMBb75 | Heat-inactivated | Human | Alleviates IBS and symptoms | [103] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prajapati, S.K.; Yadav, D.; Katiyar, S.; Jain, S.; Yadav, H. Postbiotics as Mitochondrial Modulators in Inflammatory Bowel Disease: Mechanistic Insights and Therapeutic Potential. Biomolecules 2025, 15, 954. https://doi.org/10.3390/biom15070954
Prajapati SK, Yadav D, Katiyar S, Jain S, Yadav H. Postbiotics as Mitochondrial Modulators in Inflammatory Bowel Disease: Mechanistic Insights and Therapeutic Potential. Biomolecules. 2025; 15(7):954. https://doi.org/10.3390/biom15070954
Chicago/Turabian StylePrajapati, Santosh Kumar, Dhananjay Yadav, Shweta Katiyar, Shalini Jain, and Hariom Yadav. 2025. "Postbiotics as Mitochondrial Modulators in Inflammatory Bowel Disease: Mechanistic Insights and Therapeutic Potential" Biomolecules 15, no. 7: 954. https://doi.org/10.3390/biom15070954
APA StylePrajapati, S. K., Yadav, D., Katiyar, S., Jain, S., & Yadav, H. (2025). Postbiotics as Mitochondrial Modulators in Inflammatory Bowel Disease: Mechanistic Insights and Therapeutic Potential. Biomolecules, 15(7), 954. https://doi.org/10.3390/biom15070954