
Synthesis and Cytotoxicity Evaluation of Denitroaristolochic Acids: Structural Insights and Mechanistic Implications in Nephrotoxicity

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis
2.1.1. Synthesis of 3-Bromo-4,5-dihydroxybenzaldehyde (2)
2.1.2. Synthesis of 3-Bromo-4,5-methylenedioxybenzaldehyde (3)
2.1.3. Synthesis of Methyl 3-Bromo-4,5-methylenedioxybenzoate (4)
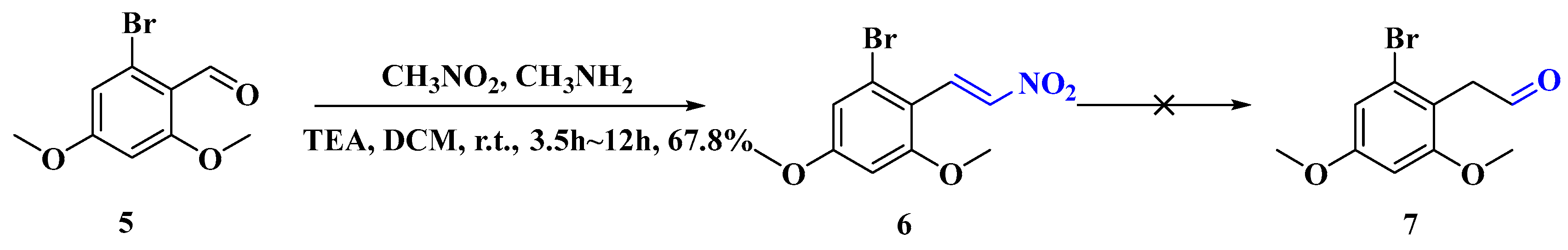
2.1.4. Synthesis of (E)-1-Bromo-3,5-dimethoxy-2-(2-nitrovinyl)benzene (6)
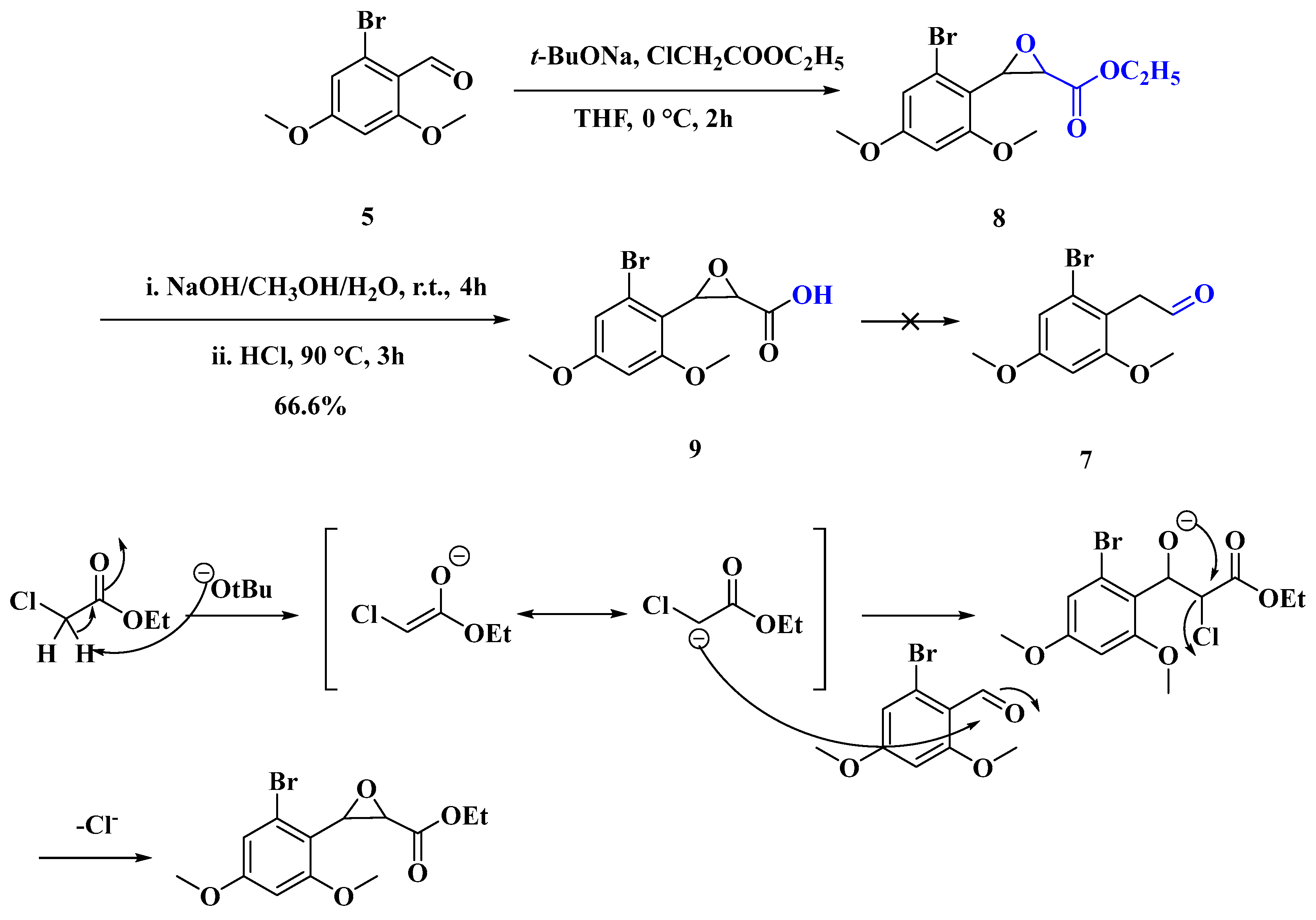
2.1.5. Synthesis of 3-(2-Bromo-4,6-dimethoxyphenyl)oxirane-2-carboxylic Acid (9)
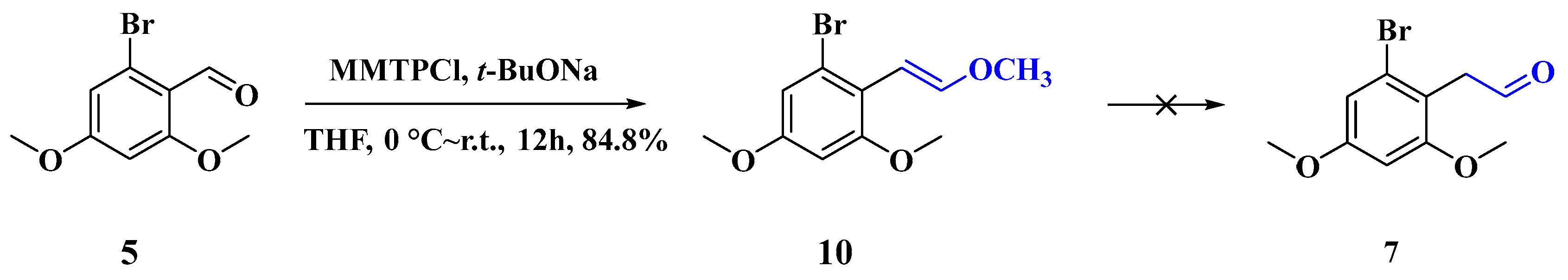
2.1.6. Synthesis of (E)-1-Bromo-3,5-dimethoxy-2-(2-methoxyvinyl)benzene (10)
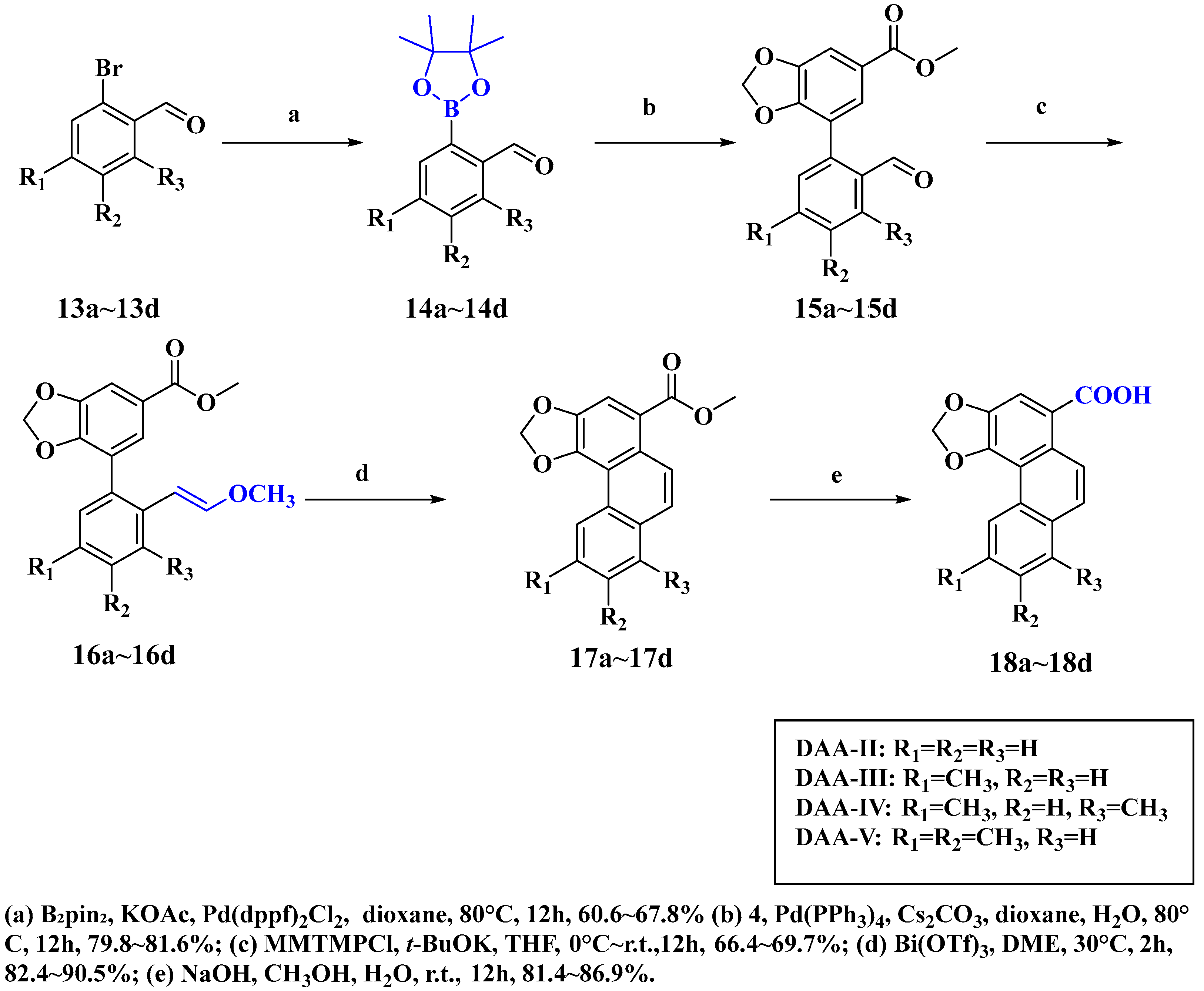
2.1.7. General Procedure for the Synthesis of 2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde Derivatives (14a–14d)
2.1.8. General Procedure for the Synthesis of Methyl 7-(2-Formylphenyl)benzo[d][1,3]dioxole-5-carboxylate Derivatives (15a–15d)
2.1.9. General Procedure for the Synthesis of Methyl (E)-7-[2-(2-Methoxyvinyl)phenyl]benzo[d][1,3]dioxole-5-carboxylate Derivatives (16a–16d)
2.1.10. General Procedure for the Synthesis of Methyl Phenanthro [3,4-d][1,3]dioxole-5-Carboxylate Derivatives (17a–17d)
2.1.11. General Procedure for the Synthesis of Phenanthro [3,4-d][1,3]dioxole-5-Carboxylic Acid Derivatives (18a–18d)
2.2. Cytotoxicity of Denitroaristolochic Acids in HK2 Cells
3. Results
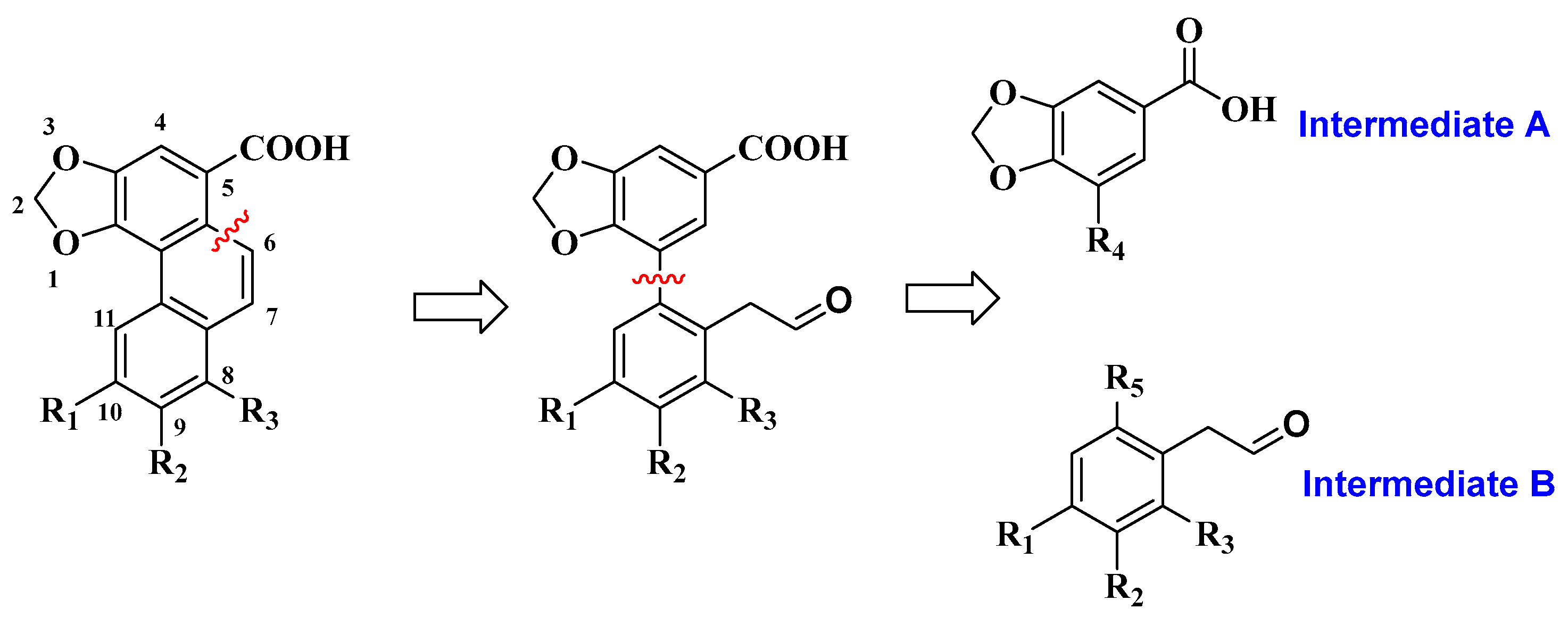
3.1. Synthesis of Four Denitroaristolochic Acids
3.2. Study on the Toxicity of Denitroaristolochic Acids
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | aristolochic acid |
| B2pin2 | bis(pinacolato)diboron |
| Bi(OTf)3 | bismuth triflate |
| CCK-8 | cell counting kit-8 |
| DAA | denitroaristolochic acid |
| DCM | dichloromethane |
| DME | dimethoxyethane |
| DMEM | dulbecco’s modified eagle medium |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| ESI | electrospray ionization |
| HK2 | human renal cortical proximal tubular epithelial cell |
| HPLC | high performance liquid chromatography |
| HRMS | high-resolution mass spectrometry |
| IARC | International Agency for Research on Cancer |
| LC-MS | liquid chromatography–mass spectrometry |
| LD50 | median lethal dose |
| m-CPBA | m-chloroperbenzoic acid |
| MMTPCl | methoxymethyltriphenylphosphonium chloride |
| NBS | N-bromosuccinimide |
| ND | not detected |
| NMR | nuclear magnetic resonance |
| Pd(dppf)Cl2 | dichloro [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) |
| Ph(PPh3)4 | tetrakis(triphenylphosphine)palladium |
| r.t. | room temperature |
| SAR | structure–activity relationships |
| t-BuOK | potassium tert-butoxide |
| t-BuONa | sodium tert-butoxide |
| TEA | triethylamine |
| THF | tetrahydrofuran |
| TLC | thin layer chromatography |
| TMS | tetramethylsilane |
Appendix A
| No. | Chemical Name | Chemical Structure | CAS No. |
| compound 2 | 3-bromo-4,5-dihydroxybenzaldehyde | 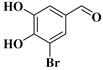 | 16414-34-9 |
| compound 3 | 7-bromobenzo[d][1,3]dioxole-5-carbaldehyde | 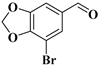 | 19522-96-4 |
| compound 4 | methyl 7-bromobenzo[d][1,3]dioxole-5-carboxylate | 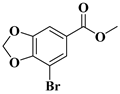 | 103095-80-3 |
| compound 6 | (E)-1-bromo-3,5-dimethoxy-2-(2-nitrovinyl)benzene | 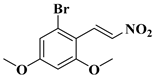 | 2229645-24-1 |
| compound 13a | 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde | 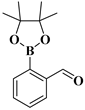 | 380151-85-9 |
| compound 13b | 4-methoxy-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde | 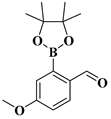 | 1196474-59-5 |
| compound 13c | 2,4-dimethoxy-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde | 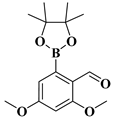 | 1265360-45-9 |
| compound 13d | 4,5-dimethoxy-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde | 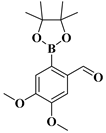 | 1098755-60-2 |
| compound 17a | methyl phenanthro [3,4-d][1,3]dioxole-5-carboxylate | 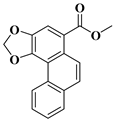 | 310396-78-2 |
| compound 17c | methyl 8,10-dimethoxyphenanthro [3,4-d][1,3]dioxole-5-carboxylate | 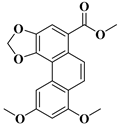 | 64543-60-8 |
| compound 18a | phenanthro [3,4-d][1,3]dioxole-5-carboxylic acid | 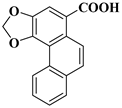 | 70195-23-2 |
| compound 18c | 8,10-dimethoxyphenanthro [3,4-d][1,3]dioxole-5-carboxylic acid | 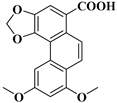 | 64543-59-5 |
References
- Abolhassanzadeh, Z.; Ansari, S.; Lorigooini, Z.; Anjomshoa, M.; Bijad, E.; Ramezannezhad, P.; Zarei, M.H. The Nephrotoxicity of aristolochia Rotunda L. in rats: Mitochondrion as a target for renal toxicity of aristolochic acids-containing plants. Heliyon 2023, 9, e21848. [Google Scholar] [CrossRef]
- Tian, J.; Liu, C.; Wang, L.; Xian, Z.; Zhao, Y.; Qin, S.; Yi, Y.; Li, C.; Han, J.; Pan, C.; et al. Study on the difference and correlation between the contents and toxicity of aristolochic acid analogues in Aristolochia plants. J. Ethnopharmacol. 2023, 315, 116568. [Google Scholar] [CrossRef] [PubMed]
- Dechbumroong, P.; Aumnouypol, S.; Denduangboripant, J.; Sukrong, S. DNA barcoding of Aristolochia plants and development of species-specific multiplex PCR to aid HPTLC in ascertainment of Aristolochia herbal materials. PLoS ONE 2018, 13, e0202625. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Chan, J.; Wanke, S.; Neinhuis, C.; Simmonds, M.S. Local uses of aristolochia species and content of nephrotoxic aristolochic acid 1 and 2–A global assessment based on bibliographic sources. J. Ethnopharmacol. 2009, 125, 108–144. [Google Scholar] [CrossRef]
- Frei, H.; Würgler, F.E.; Juon, H.; Hall, C.B.; Graf, U. Aristolochic acid is mutagenic and recombinogenic in Drosophila genotoxicity tests. Arch. Toxicol. 1985, 56, 158–166. [Google Scholar] [CrossRef]
- Chen, C.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef]
- Pakrashi, A.; Chakrabarty, B. Antifertility effect of aristolic acid from Aristolochia indica (Linn) in female albino rabbits. Experientia 1978, 34, 1377. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Barnes, C.J.; Cornett, C.; Holmfred, E.; Hansen, S.H.; Persson, C.; Antonelli, A.; Rønsted, N. Phylogeny predicts the quantity of antimalarial alkaloids within the iconic yellow cinchona bark (Rubiaceae: Cinchona calisaya). Front. Plant Sci. 2017, 8, 391. [Google Scholar] [CrossRef]
- Sivarajan, V.V. A New species of Thottea (Aristolochiaceae) from India. Pl. Syst. Evol. 1985, 150, 201–204. [Google Scholar] [CrossRef]
- Zhang, S.-H.; Wang, Y.; Yang, J.; Zhang, D.-D.; Wang, Y.-L.; Li, S.-H.; Pan, Y.-N.; Zhang, H.-M.; Sun, Y. Comparative analysis of aristolochic acids in Aristolochia medicinal herbs and evaluation of their toxicities. Toxins 2022, 14, 879. [Google Scholar] [CrossRef]
- Li, X.L.; Guo, X.Q.; Wang, H.R.; Chen, T.; Mei, N. Aristolochic acid-induced genotoxicity and toxicogenomic changes in rodents. World J. Tradit. Chin. Med. 2020, 6, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, R.; Zhan, C.; Wu, H.; Fan, J.; Li, Z.; Yang, X. Aristolochic acid induces acute kidney injury through ferroptosis. Front. Pharmacol. 2024, 15, 1330376. [Google Scholar] [CrossRef]
- Tankeu, S.; Vermaak, I.; Chen, W.; Sandasi, M.; Viljoen, A. Differentiation between two “fang ji” herbal medicines, Stephania tetrandra and the nephrotoxic Aristolochia fangchi, using hyperspectral imaging. Phytochemistry 2016, 122, 213–222. [Google Scholar] [CrossRef]
- Cosyns, J.P.; Goebbels, R.M.; Liberton, V. Chinese herbs nephropathy-associated slimming regimen induces tumours in the forestomach but no interstitial nephropathy in rats. Arch. Toxicol. 1998, 72, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Bastek, H.; Zubel, T.; Stemmer, K.; Mangerich, A.; Beneke, S.; Dietrich, D.R. Comparison of aristolochic acid I derived DNA adduct levels in human renal toxicity models. Toxicology 2019, 420, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M.; Slade, N.; Brdar, B.; Medverec, Z.; Tomic, K.; Jelakovic, B.; Wu, L.; Truong, S.; Fernandes, A.; Grollman, A.P. TP53 mutational signature for aristolochic acid: An environmental carcinogen. Int. J. Cancer 2011, 129, 1532–1536. [Google Scholar] [CrossRef]
- Au, C.-K.; Chan, C.-K.; Tung, K.-K.; Zhang, J.; Chan, W. Quantitation of DNA adducts of aristolochic acids in repair-deficient cells: A mechanistic study of the DNA repair mechanism. Chem. Res. Toxicol. 2020, 33, 1323–1327. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, P.; Chen, J.; Yang, C.; Xia, F.; Zhang, J.; Tang, H.; Liu, D.; Gu, L.; Shi, Q.; et al. Dissection of targeting molecular mechanisms of aristolochic acid-induced nephrotoxicity via a combined deconvolution strategy of chemoproteomics and metabolomics. Int. J. Biol. Sci. 2022, 18, 2003–2017. [Google Scholar] [CrossRef]
- Hou, C.; Suo, Y.; Lv, P.; Yuan, H.; Zhao, L.; Wang, Y.; Zhang, H.; Sun, J.; Sun, L.; Lu, W.; et al. Aristolochic acids-hijacked P53 promotes liver cancer cell growth by inhibiting ferroptosis. Acta Pharmacol. Sin. 2025, 46, 208–221. [Google Scholar] [CrossRef]
- Li, W.; Hu, Q.; Chan, W. Mass Spectrometric and spectrofluorometric studies of the interaction of aristolochic acids with proteins. Sci. Rep. 2015, 5, 15192. [Google Scholar] [CrossRef]
- Tung, N.H.; Song, G.Y.; Kim, J.-A.; Hyun, J.-H.; Kang, H.-K.; Kim, Y.H. Dammarane-Type Saponins from the Flower Buds of Panax Ginseng and Their Effects on Human Leukemia Cells. Bioorg. Med. Chem. Lett. 2010, 20, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xian, Z.; Zhao, Y.; Wang, L.; Tian, J.; Pan, C.; Han, J.; Zhang, Y.; Li, C.; Yi, Y.; et al. Quantitative determination and toxicity evaluation of aristolochic acid analogues in Asarum heterotropoides F. Schmidt (Xixin) and traditional Chinese patent medicines. Front. Pharmacol. 2021, 12, 761593. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, H.-C. Chemical constituents from Aristolochia tagala and their chemotaxonomic significance. Biochem. Syst. Ecol. 2020, 90, 104037. [Google Scholar] [CrossRef]
- Schaneberg, B.T.; Khan, I.A. Analysis of products suspected of containing Aristolochia or Asarum species. J. Ethnopharmacol. 2004, 94, 245–249. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Shang, M.; Yu, J.; Xu, Y.; Li, Z.; Lei, L.; Li, X.; Cai, S.; Namba, T. Simultaneous determination of five aristolochic acids and two aristololactams in Aristolochia plants by high-performance liquid chromatography. Biomed. Chromatogr. 2006, 20, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Achari, B.; Bandyopadhyay, S.; Chakravarty, A.K.; Pakrashi, S.C. Carbon-13 NMR spectra of some phenanthrene derivatives from Aristolochia indica and their analogues. Org. Magn. Reson. 1984, 22, 741–746. [Google Scholar] [CrossRef]
- Duan, W.; Li, Y.; Dong, H.; Yang, G.; Wang, W.; Wang, X. Isolation and purification of six aristolochic acids with similar structures from Aristolochia manshuriensis kom stems by pH-zone-refining counter-current chromatography. J. Chromatogr. A 2020, 1613, 460657. [Google Scholar] [CrossRef]
- Houghton, P.J.; Ogutveren, M. Aristolochic acids and aristolactams from Aristolochia auricularia. Phytochemistry 1991, 30, 253–254. [Google Scholar] [CrossRef]
- Whale, G.F.; Dawick, J.; Hughes, C.B.; Lyon, D.; Boogaard, P.J. Toxicological and ecotoxicological properties of gas-to-liquid (GTL) products. 2. Ecotoxicology. Crit. Rev. Toxicol. 2018, 48, 273–296. [Google Scholar] [CrossRef]
- Murai, M.; Hosokawa, N.; Roy, D.; Takai, K. Bismuth-catalyzed synthesis of polycyclic aromatic hydrocarbons (PAHs) with a phenanthrene backbone via cyclization and aromatization of 2-(2-arylphenyl)vinyl ethers. Org. Lett. 2014, 16, 4134–4137. [Google Scholar] [CrossRef]

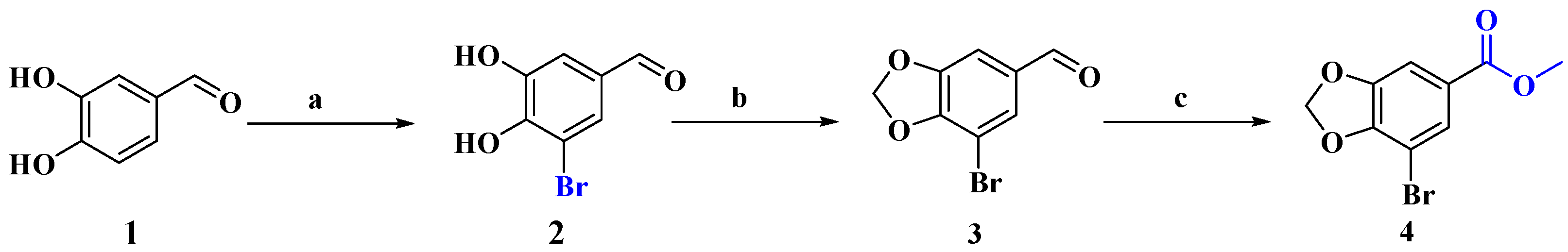
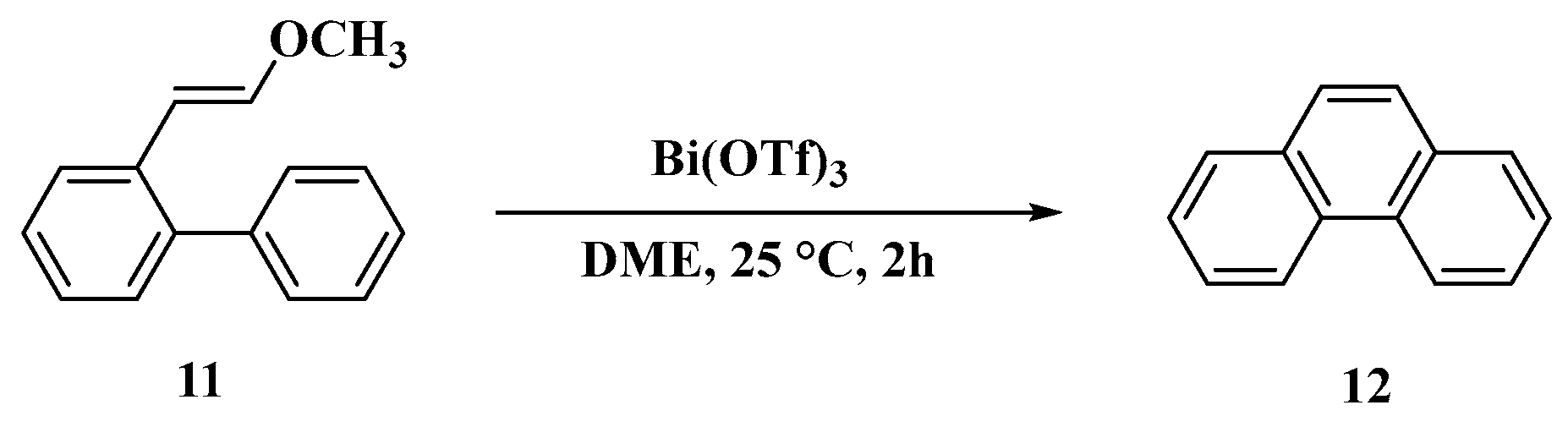

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
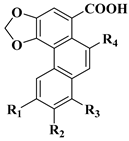
| No. | General Name | Abbreviation | Substituents | CAS No. |
|---|---|---|---|---|
| 1 | aristolochic acid I (aristolochic acid A) | AA-I | R1=R2=H, R3=OCH3, R4=NO2 | 313-67-7 |
| 2 | aristolochi acid IA | AA-IA | R1=R2=H, R3=OH, R4=NO2 | 38965-71-8 |
| 3 | aristolochi acid II (aristolochi acid B) | AA-II | R1=R2=R3=H, R4=NO2 | 475-80-9 |
| 4 | denitroaristolochic acid II | DAA-II | R1=R2=R3=R4=H | 70195-23-2 |
| 5 | aristolochi acid III | AA-III | R2=R3=H, R1=OCH3, R4=NO2 | 7267-92-7 |
| 6 | aristolochi acid IIIA (aristolochi acid C) | AA-IIIA | R2=R3=H, R1=OH, R4=NO2 | 4849-90-5 |
| 7 | denitroaristolochic acid III | DAA-III | R2=R3=H, R1=OCH3, R4=H | / |
| 8 | aristolochi acid IV | AA-IV | R2=H, R1=R3=OCH3, R4=NO2 | 15918-62-4 |
| 9 | aristolochi acid IVA (aristolochi acid D) | AA-IVA | R2=H, R1=OH, R3=OCH3, R4=NO2 | 17413-38-6 |
| 10 | aristolochi acid IVB | AA-IVB | R2=H, R1=OCH3, R3=OH, R4=NO2 | 1374601-32-7 |
| 11 | denitroaristolochic acid IV | DAA-IV | R2=R4=H, R1=R3=OCH3 | 64543-59-5 |
| 12 | aristolochi acid V | AA-V | R3=H, R1=R2=OCH3, R4=NO2 | 108766-28-5 |
| 13 | aristolochi acid VA | AA-VA | R3=H, R1=OH, R2=OCH3, R4=NO2 | 108779-46-0 |
| 14 | denitroaristolochic acid V | DAA-V | R3=R4=H, R1=R2=OCH3 | / |
| Entry | Reagent | Catalyst | Solvent | Time | Temperature | Yield |
|---|---|---|---|---|---|---|
| 1 | NBS (1.1 eq) | m-CPBA (0.1 eq) | methanol | 12 h | r.t./65 °C | — |
| 2 | NBS (1.1 eq) | m-CPBA (0.1 eq) | DCM | 12 h | r.t./40 °C | — |
| 3 | NBS (1.1 eq) | m-CPBA (0.1 eq) | THF | 12 h | r.t./66 °C | — |
| 4 | dibromohydantoin (1.1 eq) | m-CPBA (0.1 eq) | methanol | 12 h | r.t./65 °C | — |
| 5 | dibromohydantoin (1.1 eq) | m-CPBA (0.1 eq) | DCM | 12 h | r.t./65 °C | — |
| 6 | dibromohydantoin (1.1 eq) | m-CPBA (0.1 eq) | THF | 12 h | r.t./40 °C | — |
| 7 | dibromohydantoin (1.1 eq) | / | acetic acid | 12 h | r.t. | — |
| 8 | NBS (1.1 eq) | / | acetic acid | 12 h | r.t. | — |
| 9 | Br2 (1.1 eq) | / | acetic acid | 6 h | r.t. | 50.8% |
| Entry | Reagent | pH | Solvent | Time | Temperature | Yield |
|---|---|---|---|---|---|---|
| 1 | H2SO4 | 0.5 | THF/DCM | overnight | Reflux | <10% |
| 2 | H2SO4 | 1.5 | THF/DCM | overnight | Reflux | <10% |
| 3 | H2SO4 | 2.5 | THF/DCM | overnight | Reflux | <10% |
| 4 | HCl | 0.5 | THF/DCM | overnight | Reflux | <10% |
| 5 | HCl | 1.5 | THF/DCM | overnight | Reflux | <10% |
| 6 | HCl | 2.5 | THF/DCM | overnight | Reflux | <10% |
| 7 | H3PO4 | 0.5 | THF/DCM | overnight | Reflux | <10% |
| 8 | H3PO4 | 1.5 | THF/DCM | overnight | Reflux | <10% |
| 9 | H3PO4 | 2.5 | THF/DCM | overnight | Reflux | <10% |
| 10 | H3PO4 | 1.5 | diphenyl ether | 1 h | 260 °C | —— |
| No. | Compounds | Abbreviation | IC50 (μM) * |
|---|---|---|---|
| 1 | denitroaristolochic acid II | DAA-II | ND |
| 2 | denitroaristolochic acid III | DAA-III | 371 |
| 3 | denitroaristolochic acid IV | DAA-IV | 515 |
| 4 | denitroaristolochic acid V | DAA-V | ND |
| 5 | aristolochic acid I | AA-I | 270 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Zhao, M.; Su, J.; Gao, Y.; Zhang, X.; Ding, Y.; Liu, X.; Luan, Y.; Hu, C. Synthesis and Cytotoxicity Evaluation of Denitroaristolochic Acids: Structural Insights and Mechanistic Implications in Nephrotoxicity. Biomolecules 2025, 15, 1014. https://doi.org/10.3390/biom15071014
Gao J, Zhao M, Su J, Gao Y, Zhang X, Ding Y, Liu X, Luan Y, Hu C. Synthesis and Cytotoxicity Evaluation of Denitroaristolochic Acids: Structural Insights and Mechanistic Implications in Nephrotoxicity. Biomolecules. 2025; 15(7):1014. https://doi.org/10.3390/biom15071014
Chicago/Turabian StyleGao, Jianfei, Mengtong Zhao, Jianhua Su, Yi Gao, Xiaofeng Zhang, Yongzhao Ding, Xiaoping Liu, Yang Luan, and Chun Hu. 2025. "Synthesis and Cytotoxicity Evaluation of Denitroaristolochic Acids: Structural Insights and Mechanistic Implications in Nephrotoxicity" Biomolecules 15, no. 7: 1014. https://doi.org/10.3390/biom15071014
APA StyleGao, J., Zhao, M., Su, J., Gao, Y., Zhang, X., Ding, Y., Liu, X., Luan, Y., & Hu, C. (2025). Synthesis and Cytotoxicity Evaluation of Denitroaristolochic Acids: Structural Insights and Mechanistic Implications in Nephrotoxicity. Biomolecules, 15(7), 1014. https://doi.org/10.3390/biom15071014