
Could Horizontal Gene Transfer Explain 5S rDNA Similarities Between Frogs and Worm Parasites?

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Phylogenetic and Similarity Analysis of 5S rRNA Genes
2.3. Analysis of 5S rDNA Non-Transcribing Spacers in Frogs and Worms
2.4. Assessment of DNA Contamination Hypothesis
3. Results
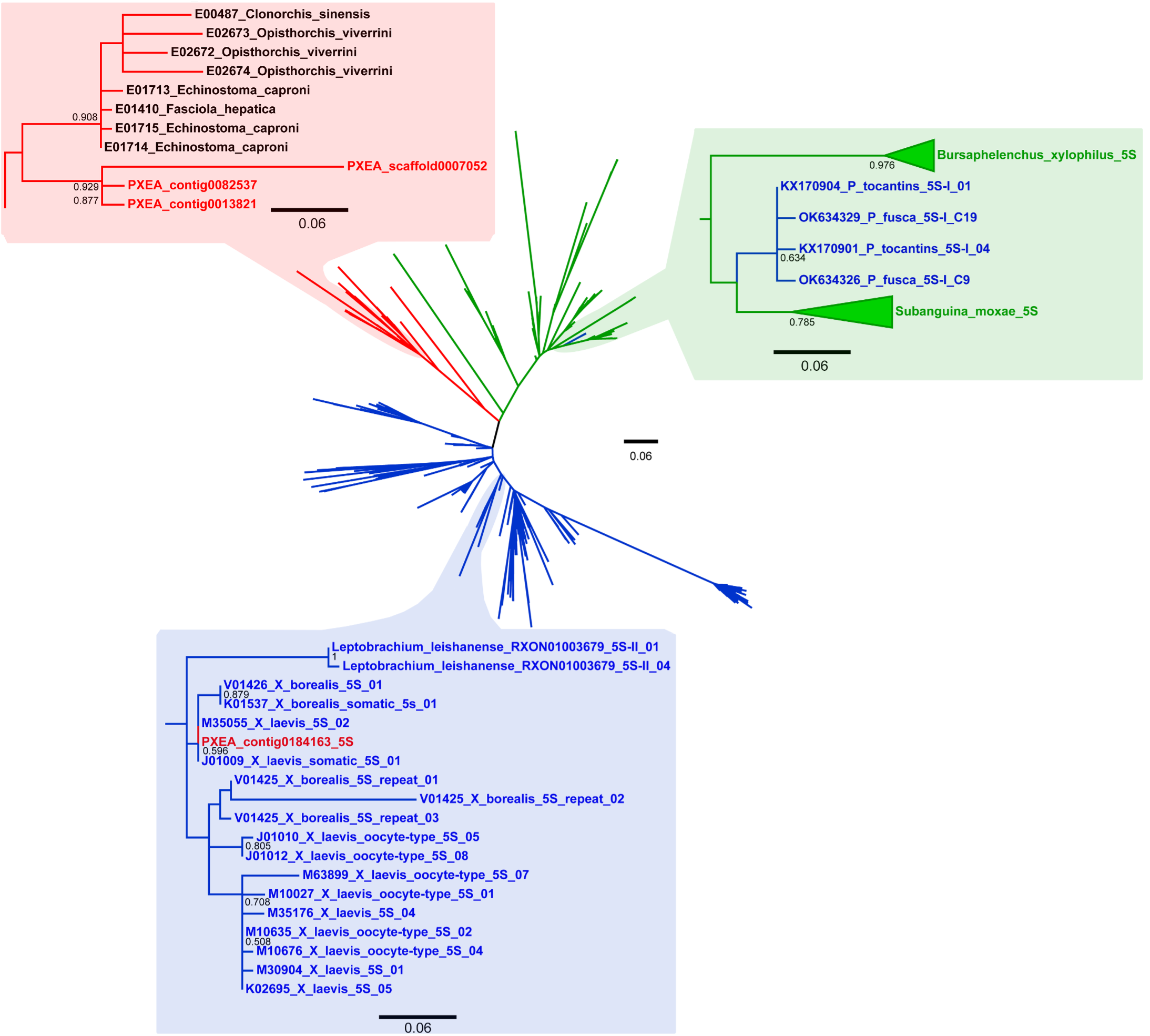
3.1. Incongruities in the ML Analysis
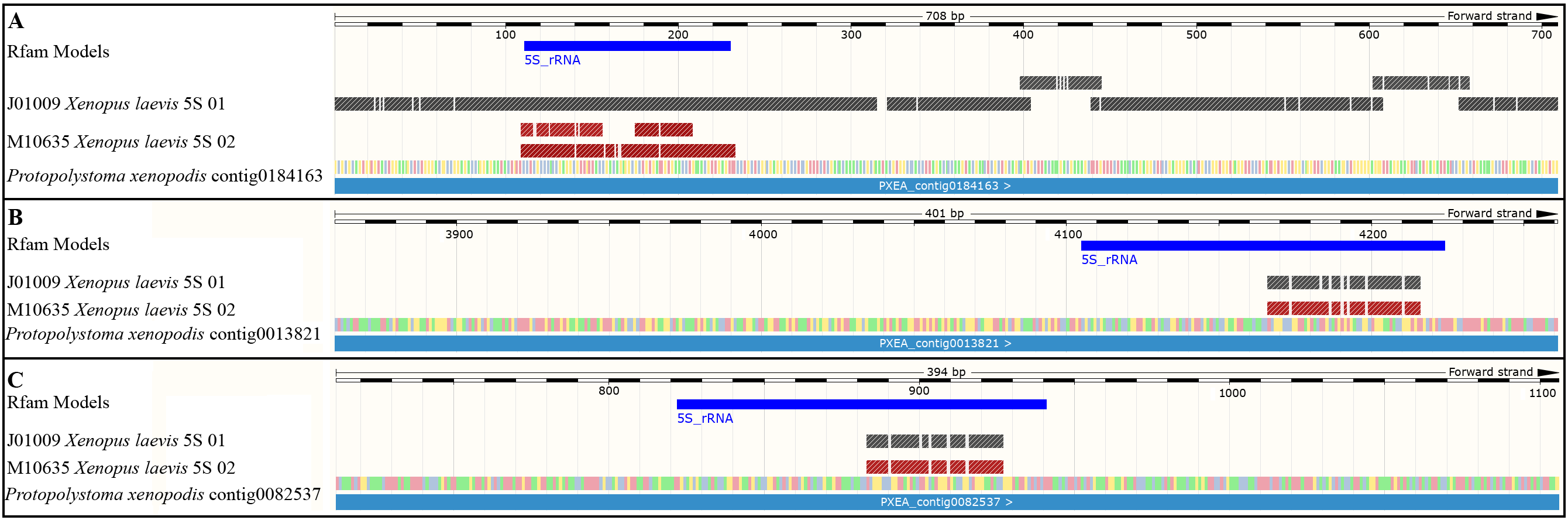
3.2. An Extended Comparison Between the 5S rDNA of Pr. xenopodis and Xenopus Species
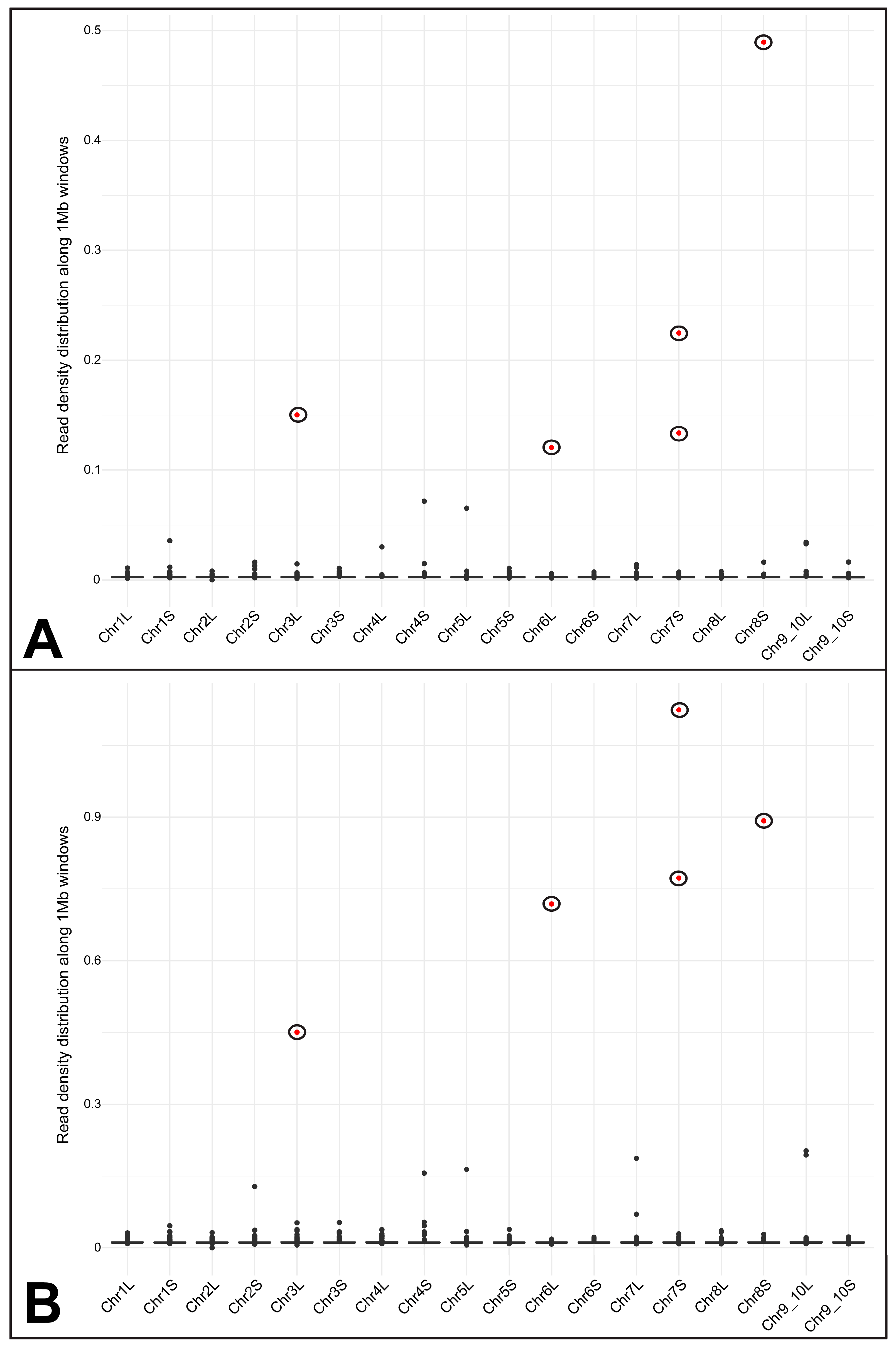
3.3. Assessing the Hypothesis of Frog DNA Contamination in the Pr. xenopodis Genome Assembly
3.4. Comparison of the Type I 5S rDNA of Pseudis tocantins and Pseudis fusca with the 5S rDNA of Bursaphelenchus xilophilus and Subanguina moxae
4. Discussion
4.1. Evidence of a Possible HGT Between Frogs and Worm Parasites
4.2. HGT and the Diversity of 5S rDNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hotopp, J.C.D.; Clark, M.E.; Oliveira, D.C.S.G.; Foster, J.M.; Fischer, P.; Torres, M.C.M.; Giebel, J.D.; Kumar, N.; Ishmael, N.; Wang, S.; et al. Widespread Lateral Gene Transfer from Intracellular Bacteria to Multicellular Eukaryotes. Science 2007, 317, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J. Functional and Ecological Impacts of Horizontal Gene Transfer in Eukaryotes. Curr. Opin. Genet. Dev. 2009, 19, 613–619. [Google Scholar] [CrossRef]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal Transfer of Transposable Elements and Why It Matters for Eukaryotic Evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef]
- Wijayawardena, B.K.; Minchella, D.J.; DeWoody, J.A. Hosts, Parasites, and Horizontal Gene Transfer. Trends Parasitol. 2013, 29, 329–338. [Google Scholar] [CrossRef]
- Boto, L. Horizontal Gene Transfer in the Acquisition of Novel Traits by Metazoans. Proc. R. Soc. B Biol. Sci. 2014, 281, 20132450. [Google Scholar] [CrossRef]
- Dunning Hotopp, J.C. Grafting or Pruning in the Animal Tree: Lateral Gene Transfer and Gene Loss? BMC Genom. 2018, 19, 470. [Google Scholar] [CrossRef] [PubMed]
- Beiko, R.G.; Harlow, T.J.; Ragan, M.A. Highways of Gene Sharing in Prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14332–14337. [Google Scholar] [CrossRef]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal Gene Transfer: Building the Web of Life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Lerminiaux, N.A.; Cameron, A.D.S. Horizontal Transfer of Antibiotic Resistance Genes in Clinical Environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [CrossRef]
- Dunning Hotopp, J.C. Horizontal Gene Transfer between Bacteria and Animals. Trends Genet. 2011, 27, 157–163. [Google Scholar] [CrossRef]
- Husnik, F. Host–Symbiont–Pathogen Interactions in Blood-Feeding Parasites: Nutrition, Immune Cross-Talk and Gene Exchange. Parasitology 2018, 145, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Quispe-Huamanquispe, D.G.; Gheysen, G.; Kreuze, J.F. Horizontal Gene Transfer Contributes to Plant Evolution: The Case of Agrobacterium T-DNAs. Front. Plant Sci. 2017, 8, 2015. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Palmer, J.D. Horizontal Gene Transfer in Eukaryotic Evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef]
- Martin, W.F. Too Much Eukaryote LGT. BioEssays 2017, 39, 1700115. [Google Scholar] [CrossRef]
- Leger, M.M.; Eme, L.; Stairs, C.W.; Roger, A.J. Demystifying Eukaryote Lateral Gene Transfer (Response to Martin 2017 DOI: 10.1002/bies.201700115). BioEssays 2018, 40, 1700242. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nowell, R.W.; Arakawa, K.; Blaxter, M. Horizontal Gene Transfer in Metazoa: Examples and Methods. In Horizontal Gene Transfer; Villa, T., Viñas, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 203–226. [Google Scholar]
- Jensen, L.; Grant, J.R.; Laughinghouse, H.D.; Katz, L.A. Assessing the Effects of a Sequestered Germline on Interdomain Lateral Gene Transfer in Metazoa. Evolution 2016, 70, 1322–1333. [Google Scholar] [CrossRef]
- Walsh, A.M.; Kortschak, R.D.; Gardner, M.G.; Bertozzi, T.; Adelson, D.L. Widespread Horizontal Transfer of Retrotransposons. Proc. Natl. Acad. Sci. USA 2013, 110, 1012–1016. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Xu, C.; Zhao, H.; Zhang, X.; Zeng, G.; Qian, Y.; Liu, R.; Guo, N.; Mi, W.; et al. Horizontal Gene Transfer Allowed the Emergence of Broad Host Range Entomopathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 7982–7989. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.A. Genome Evolution: Horizontal Movements in the Fungi. Curr. Biol. 2011, 21, R166–R168. [Google Scholar] [CrossRef]
- Moran, N.A.; Jarvik, T. Lateral Transfer of Genes from Fungi Underlies Carotenoid Production in Aphids. Science 2010, 328, 624–627. [Google Scholar] [CrossRef]
- Wang, C.-F.; Sun, W.; Zhang, Z. Functional Characterization of the Horizontally Transferred 4,5-DOPA Extradiol Dioxygenase Gene in the Domestic Silkworm, Bombyx Mori. Insect Mol. Biol. 2019, 28, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-W.; Villarreal, J.C.; Kelly, S.; Rothfels, C.J.; Melkonian, M.; Frangedakis, E.; Ruhsam, M.; Sigel, E.M.; Der, J.P.; Pittermann, J.; et al. Horizontal Transfer of an Adaptive Chimeric Photoreceptor from Bryophytes to Ferns. Proc. Natl. Acad. Sci. USA 2014, 111, 6672–6677. [Google Scholar] [CrossRef]
- Zarlenga, D.S.; Mitreva, M.; Thompson, P.; Tyagi, R.; Tuo, W.; Hoberg, E.P. A Tale of Three Kingdoms: Members of the Phylum Nematoda Independently Acquired the Detoxifying Enzyme Cyanase through Horizontal Gene Transfer from Plants and Bacteria. Parasitology 2019, 146, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wafula, E.K.; Kim, G.; Shahid, S.; McNeal, J.R.; Ralph, P.E.; Timilsena, P.R.; Yu, W.; Kelly, E.A.; Zhang, H.; et al. Convergent Horizontal Gene Transfer and Cross-Talk of Mobile Nucleic Acids in Parasitic Plants. Nat. Plants 2019, 5, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.A.; Lougheed, S.C.; Ewart, K.V.; Davies, P.L. Lateral Transfer of a Lectin-Like Antifreeze Protein Gene in Fishes. PLoS ONE 2008, 3, e2616. [Google Scholar] [CrossRef]
- Gilbert, C.; Schaack, S.; Pace II, J.K.; Brindley, P.J.; Feschotte, C. A Role for Host–Parasite Interactions in the Horizontal Transfer of Transposons across Phyla. Nature 2010, 464, 1347–1350. [Google Scholar] [CrossRef]
- Kuraku, S.; Qiu, H.; Meyer, A. Horizontal Transfers of Tc1 Elements between Teleost Fishes and Their Vertebrate Parasites, Lampreys. Genome Biol. Evol. 2012, 4, 929–936. [Google Scholar] [CrossRef]
- Wallau, G.L.; Vieira, C.; Loreto, É.L.S. Genetic Exchange in Eukaryotes through Horizontal Transfer: Connected by the Mobilome. Mob. DNA 2018, 9, 6. [Google Scholar] [CrossRef]
- Graham, L.A.; Li, J.; Davidson, W.S.; Davies, P.L. Smelt Was the Likely Beneficiary of an Antifreeze Gene Laterally Transferred between Fishes. BMC Evol. Biol. 2012, 12, 190. [Google Scholar] [CrossRef]
- Merlo, M.A.; Cross, I.; Palazon, J.L.; Ubeda-Manzanaro, M.; Sarasquete, C.; Rebordinos, L. Evidence for 5S RDNA Horizontal Transfer in the Toadfish Halobatrachus Didactylus (Schneider, 1801) Based on the Analysis of Three Multigene Families. BMC Evol. Biol. 2012, 12, 201. [Google Scholar] [CrossRef]
- Ageitos, J.M.; Viñas, M.; Villa, T.G. Horizontal Gene Transfer in Obligate Parasites. In Horizontal Gene Transfer; Villa, T., Viñas, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 235–255. [Google Scholar]
- Vierna, J.; Wehner, S.; Höner zu Siederdissen, C.; Martínez-Lage, A.; Marz, M. Systematic Analysis and Evolution of 5S Ribosomal DNA in Metazoans. Heredity 2013, 111, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Gornung, E.; Colangelo, P.; Annesi, F. 5S Ribosomal RNA Genes in Six Species of Mediterranean Grey Mullets: Genomic Organization and Phylogenetic Inference. Genome 2007, 50, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Perina, A.; Seoane, D.; González-Tizón, A.M.; Rodríguez-Fariña, F.; Martínez-Lage, A. Molecular Organization and Phylogenetic Analysis of 5S RDNA in Crustaceans of the Genus Pollicipes Reveal Birth-and-Death Evolution and Strong Purifying Selection. BMC Evol. Biol. 2011, 11, 304. [Google Scholar] [CrossRef]
- Sultana, S.; Bang, J.-W.; Choi, H.-W. Organization of the 5S RRNA Gene Units in Korean Lilium Species. Genes Genom. 2011, 33, 251–257. [Google Scholar] [CrossRef]
- Rodrigues, D.; Rivera, M.; Lourenço, L. Molecular Organization and Chromosomal Localization of 5S RDNA in Amazonian Engystomops (Anura, Leiuperidae). BMC Genet. 2012, 13, 17. [Google Scholar] [CrossRef]
- Rebordinos, L.; Cross, I.; Merlo, A. High Evolutionary Dynamism in 5S RDNA of Fish: State of the Art. Cytogenet. Genome Res. 2013, 141, 103–113. [Google Scholar] [CrossRef]
- Szymanski, M.; Zielezinski, A.; Barciszewski, J.; Erdmann, V.A.; Karlowski, W.M. 5SRNAdb: An Information Resource for 5S Ribosomal RNAs. Nucleic Acids Res. 2016, 44, D180–D183. [Google Scholar] [CrossRef]
- Coghlan, A.; Tyagi, R.; Cotton, J.A.; Holroyd, N.; Rosa, B.A.; Tsai, I.J.; Laetsch, D.R.; Beech, R.N.; Day, T.A.; Hallsworth-Pepin, K.; et al. Comparative Genomics of the Major Parasitic Worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef]
- Targueta, C.P.; Gatto, K.P.; Vittorazzi, S.E.; Recco-Pimentel, S.M.; Lourenço, L.B. High Diversity of 5S Ribosomal DNA and Evidence of Recombination with the Satellite DNA PcP190 in Frogs. Gene 2023, 851, 147015. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program from Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Gatto, K.P.; Busin, C.S.; Lourenço, L.B. Unraveling the Sex Chromosome Heteromorphism of the Paradoxical Frog Pseudis Tocantins. PLoS ONE 2016, 11, e0156176. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup, 1000 Genome Projetc Data Processing The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Gel, B.; Serra, E. KaryoploteR: An R/Bioconductor Package to Plot Customizable Genomes Displaying Arbitrary Data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 2 July 2024).
- Rangwala, S.H.; Rudnev, D.V.; Ananiev, V.V.; Oh, D.-H.; Asztalos, A.; Benica, B.; Borodin, E.A.; Bouk, N.; Evgeniev, V.I.; Kodali, V.K.; et al. The NCBI Comparative Genome Viewer (CGV) Is an Interactive Visualization Tool for the Analysis of Whole-Genome Eukaryotic Alignments. PLOS Biol. 2024, 22, e3002405. [Google Scholar] [CrossRef]
- Brown, D.D.; Wensink, P.C.; Jordan, E. Purification and Some Characteristics of 5S DNA from Xenopus Laevis. Proc. Natl. Acad. Sci. USA 1971, 68, 3175–3179. [Google Scholar] [CrossRef]
- Brown, D.D.; Sugimoto, K. 5S DNAs of Xenopus Laevis and Xenopus Mulleri: Evolution of a Gene Family. J. Mol. Biol. 1973, 78, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N.V.; Brown, D.D. The Nucleotide Sequence of Oocyte 5S DNA in Xenopus Laevis. II. The GC-Rich Region. Cell 1978, 13, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.C.; Doering, J.L.; Brown, D.D. Characterization of Two Xenopus Somatic 5S DNAs and One Minor Oocyte-Specific 5S DNA. Cell 1980, 20, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Nietfeld, W.; Digweed, M.; Mentzel, H.; Meyerhof, W.; Köster, M.; Knöchel, W.; Erdmann, V.A.; Pieler, T. Oocyte and Somatic 5S Ribosomal RNA and 5S RNA Enconding Genes in Xenopus Tropicalis. Nucleic Acids Res. 1988, 18, 8803–8815. [Google Scholar] [CrossRef]
- Ravenhall, M.; Škunca, N.; Lassalle, F.; Dessimoz, C. Inferring Horizontal Gene Transfer. PLoS Comput. Biol. 2015, 11, e1004095. [Google Scholar] [CrossRef]
- Long, E.O.; Dawid, I.B. Repeated Genes in Eukaryotes. Annu. Rev. Biochem. 1980, 49, 727–764. [Google Scholar] [CrossRef]
- Dover, G. Molecular Drive: A Cohesive Mode of Species Evolution. Nature 1982, 299, 111–117. [Google Scholar] [CrossRef]
- Wijayawardena, B.K.; Minchella, D.J.; DeWoody, J.A. Horizontal Gene Transfer in Schistosomes: A Critical Assessment. Mol. Biochem. Parasitol. 2015, 201, 57–65. [Google Scholar] [CrossRef]
- Sibbald, S.J.; Eme, L.; Archibald, J.M.; Roger, A.J. Lateral Gene Transfer Mechanisms and Pan-Genomes in Eukaryotes. Trends Parasitol. 2020, 36, 927–941. [Google Scholar] [CrossRef]
- Thurston, J.P. The Morphology and Life Cycle of Protopolystoma Xenopi (Price) Bychovsky in Uganda. Parasitology 1964, 54, 441. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, R.C.; Owen, R.W. Studies on the Biology of Protopolystoma Xenopodis (Monogenoidea): The Oncomiracidium and Life-Cycle. Parasitology 1975, 71, 445. [Google Scholar] [CrossRef]
- Tinsley, R.C. Platyhelminth Parasite Reproduction: Some General Principles Derived from Monogeneans. Can. J. Zool. 2004, 82, 270–291. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; González-Tizón, A.M.; Martínez, A.; Méndez, J. Birth-and-Death Evolution with Strong Purifying Selection in the Histone H1 Multigene Family and the Origin of Orphon H1 Genes. Mol. Biol. Evol. 2004, 21, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M.; Dixon, M.T. Ribosomal DNA: Molecular Evolution and Phylogenetic Inference. Q. Rev. Biol. 1991, 66, 411–453. [Google Scholar] [CrossRef]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome Evolution in the Allotetraploid Frog Xenopus Laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef]
- Birnstiel, M.L.; Grunstein, M.; Speirs, J.; Hennig, W. Family of Ribosomal Genes of Xenopus Laevis. Nature 1969, 223, 1265–1267. [Google Scholar] [CrossRef]
- Birnstiel, M.L.; Chipchease, M.; Speirs, J. The Ribosomal RNA Cistrons. In Progress in Nucleic Acids Research and Molecular Biology; Davidson, J., Cohn, W., Eds.; Academic Press: New York, NY, USA, 1971; pp. 351–389. [Google Scholar]
- Pardue, M.L.; Brown, D.D.; Birnstiel, M.L. Location of the Genes for 5S Ribosomal RNA in Xenopus Laevis. Chromosoma 1973, 42, 191–203. [Google Scholar] [CrossRef]
- Roger, B.; Moisand, A.; Amalric, F.; Bouvet, P. RDNA Transcription during Xenopus Laevis Oogenesis. Biochem. Biophys. Res. Commun. 2002, 290, 1151–1160. [Google Scholar] [CrossRef]
- Kupriyanova, N.S. Conservation and Variation of Ribosomal DNA in Eukaryotes. Mol. Biol. 2000, 34, 637–647. [Google Scholar] [CrossRef]
- Weider, L.J.; Elser, J.J.; Crease, T.J.; Mateos, M.; Cotner, J.B.; Markow, T.A. The Functional Significance of Ribosomal (r)DNA Variation: Impacts on the Evolutionary Ecology of Organisms. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 219–242. [Google Scholar] [CrossRef]
- Ambrose, C.D.; Crease, T.J. Evolution of Repeated Sequences in the Ribosomal DNA Intergenic Spacer of 32 Arthropod Species. J. Mol. Evol. 2010, 70, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Korn, L.J.; Brown, D.D. Nucleotide Sequence of Xenopus borealis Oocyte 5S DNA: Comparison of Sequences that Flank Several Related Eukaryotic Genes. Cell 1978, 15, 1145–1156. [Google Scholar] [CrossRef]
- Nederby-Nielsen, J.; Hallenberg, C.; Frederiksen, S.; Sorensen, P.D.; Lomholt, B. Transcription of human 5S rRNA genes is influenced by an upstream DNA sequence. Nucleic Acids Res. 1993, 21, 3631–3636. [Google Scholar] [CrossRef]
- Hallenberg, C.; Frederiksen, S. Effect of Mutations In The Upstream Promoter On The Transcription Of Human 5S rRNA Genes. Biochim. Biophys. Acta 2001, 1520, 169–173. [Google Scholar] [CrossRef]
- Pieler, T.; Hamm, J.; Roeder, R.G. The 5S Gene Internal Control Region Is Composed Of Three Distinct Sequence Elements, Organized As Two Functional Domains With Variable Spacing. Cell 1987, 48, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.A.; Cross, I.; Manchado, M.; Cárdenas, S.; Rebordinos, L. The 5S RDNA High Dynamism in Diplodus Sargus Is a Transposon-Mediated Mechanism. Comparison with Other Multigene Families and Sparidae Species. J. Mol. Evol. 2013, 76, 83–97. [Google Scholar] [CrossRef]
- Emamalipour, M.; Seidi, K.; Vahed, S.Z.; Jahanban-Esfahlan, A.; Jaymand, M.; Majdi, H.; Amoozgar, Z.; Chitkushev, L.T.; Javaheri, T.; Jahanban-Esfahlan, R.; et al. Horizontal Gene Transfer: From Evolutionary Flexibility to Disease Progression. Front. Cell Dev. Biol. 2020, 8, 229. [Google Scholar] [CrossRef]
- Keeling, P.J. Horizontal Gene Transfer in Eukaryotes: Aligning Theory With Data. Nature Rev. Genet. 2024, 25, 416–430. [Google Scholar] [CrossRef]
- Doolittle, W.F. You Are What You Eat: A Gene Transfer Ratchet Could Account For Bacterial Genes In Eukaryotic Nuclear Genomes. Trends Genet. 1998, 14, 307–311. [Google Scholar] [CrossRef]
- Huang, J. Horizontal Gene Transfer in Eukaryotes: The Weak-link Model. BioEssays 2013, 35, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, R.C.; Jackson, J.A. Speciation of Protopolystoma Bychowsky, 1957 (Monogenea: Polystomatidae) in Hosts of the Genus Shape Xenopus (Anura: Pipidae). Syst. Parasitol. 1998, 40, 93–142. [Google Scholar] [CrossRef]
- Subbotin, S.A.; Krall, E.L.; Riley, I.T.; Chizhov, V.N.; Staelens, A.; De Loose, M.; Moens, M. Evolution of the Gall-Forming Plant Parasitic Nematodes (Tylenchida: Anguinidae) and Their Relationships with Hosts as Inferred from Internal Transcribed Spacer Sequences of Nuclear Ribosomal DNA. Mol. Phylogenet. Evol. 2004, 30, 226–235. [Google Scholar] [CrossRef]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.J.; Gaur, H.S.; Helder, J.; Jones, M.G.K.; Kikuchi, T.; Manzanilla-López, R.; Palomares-Rius, J.E.; Wesemael, W.M.L.; et al. Top 10 Plant-parasitic Nematodes in Molecular Plant Pathology. Mol. Plant Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Campião, K.M.; Morais, D.H.; Dias, O.T.; Aguiar, A.; Toledo, G.; Tavares, L.E.R.; da Silva, R.J. Checklist of Helminth Parasites of Amphibians from South America. Zootaxa 2014, 3843, 1–93. [Google Scholar] [CrossRef]
- Campião, K.M.; Ribas, A.C.d.A.; Morais, D.H.; da Silva, R.J.; Tavares, L.E.R. How Many Parasites Species a Frog Might Have? Determinants of Parasite Diversity in South American Anurans. PLoS ONE 2015, 10, e0140577. [Google Scholar] [CrossRef]
- Martins, C.; Wasko, A.P. Organization and Evolution of 5S Ribosomal DNA in the Fish Genome. In Focus on Genome Research; Williams, C.R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2004; pp. 335–363. ISBN 1590339606. [Google Scholar]
- Dover, G.A. Molecular Drive in Multigene Families: How Biological Novelties Arise, Spread and Are Assimilated. Trends Genet. 1986, 168, 159–165. [Google Scholar] [CrossRef]
- Nei, M.; Rooney, A.P. Concerted and Birth-and-Death Evolution of Multigene Families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar] [CrossRef]
- Rooney, A.P.; Ward, T.J. Evolution of a Large Ribosomal RNA Multigene Family in Filamentous Fungi: Birth and Death of a Concerted Evolution Paradigm. Proc. Natl. Acad. Sci. USA 2005, 102, 5084–5089. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; Rebordinos, L.; Rooney, A.P.; Rozas, J. The Birth-and-Death Evolution of Multigene Families Revisited. In Genome Dynamics; Garrido-Ramos, M.A., Ed.; Karger Publishers: Basel, Switzerland, 2012; Volume 7, pp. 170–196. [Google Scholar]
- Freire, R.; Arias, A.; Ínsua, A.M.; Méndez, J.; Eirín-López, J.M. Evolutionary Dynamics of the 5S RDNA Gene Family in the Mussel Mytilus: Mixed Effects of Birth-and-Death and Concerted Evolution. J. Mol. Evol. 2010, 70, 413–426. [Google Scholar] [CrossRef]
- Úbeda-Manzanaro, M.; Merlo, M.A.; Palazón, J.L.; Sarasquete, C.; Rebordinos, L. Sequence Characterization and Phylogenetic Analysis of the 5S Ribosomal DNA in Species of the Family Batrachoididae. Genome 2010, 53, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Pinhal, D.; Yoshimura, T.S.; Araki, C.S.; Martins, C. The 5S RDNA Family Evolves through Concerted and Birth-and-Death Evolution in Fish Genomes: An Example from Freshwater Stingrays. BMC Evol. Biol. 2011, 11, 151. [Google Scholar] [CrossRef] [PubMed]
- Targueta, C.P.; Vittorazzi, S.E.; Gatto, K.P.; Bruschi, D.P.; Veiga-Menoncello, A.C.P.; Recco-Pimentel, S.M.; Lourenço, L.B. Anuran Cytogenetics: An Overview. In An Essential Guide to Cytogenetics; Norris, N., Miller, C., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2018; pp. 1–64. ISBN 978-1-53613-370-7. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| 1. Anura | 80.00 | |||||
| 2. Platyhelminthes | 67.94 | 85.50 | ||||
| 3. Nematoda | 68.45 | 68.73 | 82.35 | |||
| 4. Pseudis 5S-I | 70.16 | 68.14 | 84.57 | 98.03 | ||
| 5. Pr. xenopodis contig-0184163 | 84.55 | 73.51 | 75.29 | 81.91 | - | |
| 6. Pr. xenopodis 5S * | 63.37 | 80.27 | 64.24 | 64.58 | 67.70 | 86.83 |
| 5S rDNA (Query) | P. xenopodis Contigs | Annotation | Query Cover (%) | E-Value |
|---|---|---|---|---|
| Xenopus laevis 5S (J01009) | contig0184163 | rRNA 5S | 82 | 5 × 10−139 |
| contig0082537 | rRNA 5S | 6 | 7.4 × 10−6 | |
| contig0013821 | rRNA 5S | 2 | 1.2 × 10−4 | |
| Xenopus laevis 5S (M10635) | contig0184163 | rRNA 5S | 31 | 5 × 10−49 |
| contig0082537 | rRNA 5S | 6 | 0.0013 | |
| contig0013821 | rRNA 5S | 2 | 0.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatto, K.P.; Targueta, C.P.; Vittorazzi, S.E.; Lourenço, L.B. Could Horizontal Gene Transfer Explain 5S rDNA Similarities Between Frogs and Worm Parasites? Biomolecules 2025, 15, 1001. https://doi.org/10.3390/biom15071001
Gatto KP, Targueta CP, Vittorazzi SE, Lourenço LB. Could Horizontal Gene Transfer Explain 5S rDNA Similarities Between Frogs and Worm Parasites? Biomolecules. 2025; 15(7):1001. https://doi.org/10.3390/biom15071001
Chicago/Turabian StyleGatto, Kaleb Pretto, Cintia Pelegrineti Targueta, Stenio Eder Vittorazzi, and Luciana Bolsoni Lourenço. 2025. "Could Horizontal Gene Transfer Explain 5S rDNA Similarities Between Frogs and Worm Parasites?" Biomolecules 15, no. 7: 1001. https://doi.org/10.3390/biom15071001
APA StyleGatto, K. P., Targueta, C. P., Vittorazzi, S. E., & Lourenço, L. B. (2025). Could Horizontal Gene Transfer Explain 5S rDNA Similarities Between Frogs and Worm Parasites? Biomolecules, 15(7), 1001. https://doi.org/10.3390/biom15071001