


Calcium Signaling Dynamics in Vascular Cells and Their Dysregulation in Vascular Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ca2+ Mobilization Pathways
3. Ca2+ Release from Intracellular Stores
3.1. IP3-Induced Ca2+ Mobilization from SR
3.2. Ca2+-Induced Ca2+ Release (CICR)
4. Ca2+ Entry from the Extracellular Environment
4.1. Ca2+ Leak and Non-Specific Channels
4.2. Voltage-Dependent Ca2+ Channels (VDCCs)
4.3. Receptor-Operated Ca2+ Channels (ROCs)
4.4. Transient Receptor Potential (TRP) Channels
4.5. Store-Operated Channels (SOCs)
4.6. Stretch-Activated Ca2+ Channels
5. Mechanisms of Ca2+ Removal
5.1. Plasmalemmal Ca2+-ATPase (PMCA)
5.2. Sarco(endo)plasmic Reticulum Ca2+-ATPase (SERCA)
5.3. Sodium/Calcium Exchanger (NCX)
5.4. Superficial Ca2+ Gradient (Buffer Barrier)
5.5. Ca2+ Regulation by Mitochondria
6. Ca2+ Regulation of Cell Growth and Proliferation
7. Ca2+ Regulation of Endothelial Cell Function
7.1. Ca2+ Regulation of Endothelial Nitric Oxide (NO) Production
7.2. Ca2+ Regulation of Endothelial Prostacyclin Release
7.3. Ca2+ Regulation of Endothelin-1 (ET-1) Synthesis
7.4. Ca2+ Regulation of Mitochondrial Enzymes and Oxidative Stress
7.5. Ca2+ Regulation of Vascular Permeability
8. Ca2+ Regulation of VSMC Function
8.1. Ca2+-Dependent Myosin Light Chain Phosphorylation
8.2. Evidence for Other Mechanisms of VSMC Contraction
8.3. Ca2+ Regulation of Protein Kinase-C (PKC)
8.4. Ca2+ and Rho-Kinase (ROCK)
9. Ca2+ and Extracellular Matrix (ECM)
10. Ca2+ Dysregulation in Vascular Disease
10.1. Dysregulated Ca2+ Signaling in Hypertension (HTN)
10.2. Ca2+ Dysregulation in HTN-in-Pregnancy (HTN-Preg) and Preeclampsia
10.3. Ca2+ Dysregulation in Pulmonary Arterial Hypertension (PAH)
10.4. Ca2+ Dysregulation in Age-Related Cell Senescence and Arterial Stiffness
10.5. Ca2+ Dysregulation in Vascular Inflammation, Atherosclerosis, and Calcification
10.6. Ca2+ Dysregulation in Coronary Artery Disease
11. Restoration of Ca2+ Signaling Dynamics in Treatment of Vascular Disease
12. Discussion and Perspective
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ang II | angiotensin II |
| BKCa | large conductance Ca2+-activated K+ channel |
| [Ca2+]c | cytosolic free Ca2+ concentration |
| CaD | caldesmon |
| CAD | coronary artery disease |
| CaM | calmodulin |
| cAMP | cyclic adenosine monophosphate |
| CaP | calponin |
| cGMP | cyclic guanosine monophosphate |
| CICR | Ca2+-induced Ca2+ release |
| CPI-17 | PKC-potentiated phosphatase inhibitor protein-17 |
| DAG | diacylglycerol |
| EC | endothelial cell |
| ECM | extracellular matrix |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| ET-1 | endothelin-1 |
| GPCR | G-protein-coupled receptor |
| HTN | hypertension |
| IP3 | inositol 1,4,5-trisphosphate |
| Kv | voltage-gated K+ channel |
| LTCC | L-type CaV1.2 channel |
| MAPK | mitogen-activated protein kinase |
| MARCKS | myristoylated alanine-rich C kinase substrate |
| MLC | myosin light chain |
| MLCK | MLC kinase |
| NFAT | nuclear factor of activated T cells |
| NFκB | nuclear factor kappa B |
| NCX | Na+/Ca2+ exchanger |
| PDBu | phorbol 12,13-dibutyrate |
| PDGF | platelet-derived growth factor |
| PE | preeclampsia |
| PKA | cAMP-dependent protein kinase |
| PKC | protein kinase C |
| PKG | cGMP-dependent protein kinase |
| PMA | phorbol 12-myristate 13-acetate |
| PMCA | plasmalemmal Ca2+-ATPase |
| PLC | phospholipase C |
| PS | phosphatidylserine |
| RAAS | renin–angiotensin–aldosterone system |
| ROCK | Rho-kinase |
| ROS | reactive oxygen species |
| SERCA | sarco(endo)plasmic reticulum Ca2+-ATPase |
| SOC | store-operated Ca2+ channel |
| SR | sarcoplasmic reticulum |
| STIM1 | stromal-interacting molecule 1 |
| TRP | transient receptor potential |
| TTCC | T-type CaV3.1/3.2/3.3 |
| VDCC | voltage-dependent Ca2+ channel |
| VSMC | vascular smooth muscle cell |
References
- Grossman, D.C.; Curry, S.J.; Owens, D.K.; Barry, M.J.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Krist, A.H.; et al. Vitamin D, Calcium, or Combined Supplementation for the Primary Prevention of Fractures in Community-Dwelling Adults: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 1592–1599. [Google Scholar] [PubMed]
- Schwaller, B. Cytosolic Ca2+ Buffers Are Inherently Ca2+ Signal Modulators. Cold Spring Harb. Perspect. Biol. 2020, 12, a035543. [Google Scholar] [CrossRef] [PubMed]
- Burnett, R.W.; Christiansen, T.F.; Covington, A.K.; Fogh-Andersen, N.; Kulpmann, W.R.; Lewenstam, A.; Maas, A.H.J.; Muller-Plathe, O.; Sachs, C.; Siggaard-Andersen, O.; et al. IFCC recommended reference method for the determination of the substance concentration of ionized calcium in undiluted serum, plasma or whole blood. Clin. Chem. Lab. Med. 2000, 38, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Dubyak, G.R.; Scarpa, A. Sarcoplasmic Ca2+ transients during the contractile cycle of single barnacle muscle fibres: Measurements with arsenazo III-injected fibres. J. Muscle Res. Cell Motil. 1982, 3, 87–112. [Google Scholar] [CrossRef]
- Ashley, C.C. Calcium ion regulation in barnacle muscle fibers and its relation to force development. Ann. N. Y. Acad. Sci. 1978, 307, 308–329. [Google Scholar] [CrossRef]
- Blinks, J.R.; Wier, W.G.; Hess, P.; Prendergast, F.G. Measurement of Ca2+ concentrations in living cells. Prog. Biophys. Mol. Biol. 1982, 40, 1–114. [Google Scholar] [CrossRef]
- Gasser, R.; Frey, M.; Fleckenstein-Grun, G. Free calcium in rat papillary muscle at contraction assessed with Ca-selective microelectrodes. Angiology 1989, 40, 736–742. [Google Scholar] [CrossRef]
- Morgan, J.P.; Morgan, K.G. Stimulus-specific patterns of intracellular calcium levels in smooth muscle of ferret portal vein. J. Physiol. 1984, 351, 155–167. [Google Scholar] [CrossRef]
- Tsien, R.Y. Intracellular measurements of ion activities. Annu. Rev. Biophys. Bioeng. 1983, 12, 91–116. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kanaide, H.; Nakamura, M. Cytosolic-free calcium transients in cultured vascular smooth muscle cells: Microfluorometric measurements. Science 1985, 229, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Himpens, B.; Casteels, R. Measurement by Quin2 of changes of the intracellular calcium concentration in strips of the rabbit ear artery and of the guinea-pig ileum. Pflug. Arch. Eur. J. Physiol. 1987, 408, 32–37. [Google Scholar] [CrossRef]
- Sugiyama, T.; Yoshizumi, M.; Takaku, F.; Urabe, H.; Tsukakoshi, M.; Kasuya, T.; Yazaki, Y. The elevation of the cytoplasmic calcium ions in vascular smooth muscle cells in SHR—Measurement of the free calcium ions in single living cells by lasermicrofluorospectrometry. Biochem. Biophys. Res. Commun. 1986, 141, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Cheng, Z.Y.; Schmid, K.L. GABAB receptors are expressed in human aortic smooth muscle cells and regulate the intracellular Ca2+ concentration. Heart Vessel. 2015, 30, 249–257. [Google Scholar] [CrossRef]
- Boerman, E.M.; Segal, S.S. Aging alters spontaneous and neurotransmitter-mediated Ca2+ signaling in smooth muscle cells of mouse mesenteric arteries. Microcirculation 2020, 27, e12607. [Google Scholar] [CrossRef]
- Bronner, F. Extracellular and intracellular regulation of calcium homeostasis. Sci. World J. 2001, 1, 919–925. [Google Scholar] [CrossRef]
- Zhang, P.; Zheng, C.B.; Chen, Z.; Liu, X.Y. Editorial: The role of calcium channels in human health and disease—Volume II. Front. Mol. Biosci. 2023, 10, 1180456. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R.; Lodge, N.; Saida, K.; van Breemen, C. Mechanism of calcium activation in vascular smooth muscle. J. Hypertens. 1987, 5, S5–S15. [Google Scholar] [CrossRef]
- Kim, H.R.; Appel, S.; Vetterkind, S.; Gangopadhyay, S.S.; Morgan, K.G. Smooth muscle signalling pathways in health and disease. J. Cell. Mol. Med. 2008, 12, 2165–2180. [Google Scholar] [CrossRef]
- Salamanca, D.A.; Khalil, R.A. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem. Pharmacol. 2005, 70, 1537–1547. [Google Scholar] [CrossRef]
- Liu, Z.; Khalil, R.A. Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem. Pharmacol. 2018, 153, 91–122. [Google Scholar] [CrossRef] [PubMed]
- Ringer, S. A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. J. Physiol. 1883, 4, 29–42.3. [Google Scholar] [CrossRef] [PubMed]
- Heilbrunn, L.V.; Wiercinski, F.J. The action of various cations on muscle protoplasm. J. Cell. Comp. Physiol. 1947, 29, 15–32. [Google Scholar] [CrossRef]
- Reuter, H. On the effect of adrenaline on the cellular Ca-metabolism in the guinea pig atrium. Naunyn-Schmiedeb. Arch. Exp. Pathol. Pharmakol. 1965, 251, 401–412. [Google Scholar]
- Beeler, G.W., Jr.; Reuter, H. Membrane calcium current in ventricular myocardial fibres. J. Physiol. 1970, 207, 191–209. [Google Scholar] [CrossRef]
- Beeler, G.W., Jr.; Reuter, H. The relation between membrane potential, membrane currents and activation of contraction in ventricular myocardial fibres. J. Physiol. 1970, 207, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H.; Stevens, C.F.; Tsien, R.W.; Yellen, G. Properties of single calcium channels in cardiac cell culture. Nature 1982, 297, 501–504. [Google Scholar] [CrossRef]
- Khalil, R.A.; van Breemen, C. Sustained contraction of vascular smooth muscle: Calcium influx or C-kinase activation? J. Pharmacol. Exp. Ther. 1988, 244, 537–542. [Google Scholar] [CrossRef]
- Cauvin, C.; Loutzenhiser, R.; Van Breemen, C. Mechanisms of calcium antagonist-induced vasodilation. Annu. Rev. Pharmacol. Toxicol. 1983, 23, 373–396. [Google Scholar] [CrossRef]
- Deth, R.; van Breemen, C. Agonist induced release of intracellular Ca2+ in the rabbit aorta. J. Membr. Biol. 1977, 30, 363–380. [Google Scholar] [CrossRef]
- Hwang, K.S.; van Breemen, C. Ryanodine modulation of 45Ca efflux and tension in rabbit aortic smooth muscle. Pflug. Arch. Eur. J. Physiol. 1987, 408, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. The sarcoplasmic reticulum: Then and now. In Role of The Sarcoplasmic Reticulum in Smooth Muscle; Novartis Foundation Symposium; discussion 268–271, 272–276; John Wiley & Sons, Ltd.: Chichester, UK, 2002; Volume 246, pp. 258–268. [Google Scholar]
- Endo, M. Calcium release from the sarcoplasmic reticulum. Physiol. Rev. 1977, 57, 71–108. [Google Scholar] [CrossRef] [PubMed]
- Devine, C.E.; Somlyo, A.V.; Somlyo, A.P. Sarcoplasmic reticulum and mitochondria as cation accumulation sites in smooth muscle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1973, 265, 17–23. [Google Scholar]
- Bond, M.; Kitazawa, T.; Somlyo, A.P.; Somlyo, A.V. Release and recycling of calcium by the sarcoplasmic reticulum in guinea-pig portal vein smooth muscle. J. Physiol. 1984, 355, 677–695. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, B.E.; Watras, J. Inositol 1,4,5-trisphosphate activates a channel from smooth muscle sarcoplasmic reticulum. Nature 1988, 336, 583–586. [Google Scholar] [CrossRef]
- Saida, K.; Van Breemen, C. Cyclic AMP modulation of adrenoreceptor-mediated arterial smooth muscle contraction. J. Gen. Physiol. 1984, 84, 307–318. [Google Scholar] [CrossRef]
- Cassidy, P.; Hoar, P.E.; Kerrick, W.G. Irreversible thiophosphorylation and activation of tension in functionally skinned rabbit ileum strips by [35S]ATP gamma S. J. Biol. Chem. 1979, 254, 11148–11153. [Google Scholar] [CrossRef]
- Nishimura, J.; Kolber, M.; van Breemen, C. Norepinephrine and GTP-γ-S increase myofilament Ca2+ sensitivity in alpha-toxin permeabilized arterial smooth muscle. Biochem. Biophys. Res. Commun. 1988, 157, 677–683. [Google Scholar] [CrossRef]
- Kitazawa, T.; Kobayashi, S.; Horiuti, K.; Somlyo, A.V.; Somlyo, A.P. Receptor-coupled, permeabilized smooth muscle: Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+. J. Biol. Chem. 1989, 264, 5339–5342. [Google Scholar] [CrossRef]
- Berridge, M.J.; Irvine, R.F. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 1984, 312, 315–321. [Google Scholar] [CrossRef]
- Suematsu, E.; Hirata, M.; Hashimoto, T.; Kuriyama, H. Inositol 1,4,5-trisphosphate releases Ca2+ from intracellular store sites in skinned single cells of porcine coronary artery. Biochem. Biophys. Res. Commun. 1984, 120, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.V.; Bond, M.; Somlyo, A.P.; Scarpa, A. Inositol trisphosphate-induced calcium release and contraction in vascular smooth muscle. Proc. Natl. Acad. Sci. USA 1985, 82, 5231–5235. [Google Scholar] [CrossRef]
- Yamamoto, H.; van Breemen, C. Inositol-1,4,5-trisphosphate releases calcium from skinned cultured smooth muscle cells. Biochem. Biophys. Res. Commun. 1985, 130, 270–274. [Google Scholar] [CrossRef]
- Saida, K.; van Breemen, C. GTP requirement for inositol-1,4,5-trisphosphate-induced Ca2+ release from sarcoplasmic reticulum in smooth muscle. Biochem. Biophys. Res. Commun. 1987, 144, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Rittenhouse, S.E.; Brock, T.A.; Ekstein, L.S.; Gimbrone, M.A., Jr.; Alexander, R.W. Sustained diacylglycerol formation from inositol phospholipids in angiotensin II-stimulated vascular smooth muscle cells. J. Biol. Chem. 1986, 261, 5901–5906. [Google Scholar] [CrossRef]
- Ringvold, H.C.; Khalil, R.A. Protein Kinase C as Regulator of Vascular Smooth Muscle Function and Potential Target in Vascular Disorders. Adv. Pharmacol. 2017, 78, 203–301. [Google Scholar] [PubMed]
- Iino, M. Calcium dependent inositol trisphosphate-induced calcium release in the guinea-pig taenia caeci. Biochem. Biophys. Res. Commun. 1987, 142, 47–52. [Google Scholar] [CrossRef]
- Walker, J.W.; Somlyo, A.V.; Goldman, Y.E.; Somlyo, A.P.; Trentham, D.R. Kinetics of smooth and skeletal muscle activation by laser pulse photolysis of caged inositol 1,4,5-trisphosphate. Nature 1987, 327, 249–252. [Google Scholar] [CrossRef]
- Kobayashi, S.; Somlyo, A.V.; Somlyo, A.P. Heparin inhibits the inositol 1,4,5-trisphosphate-dependent, but not the independent, calcium release induced by guanine nucleotide in vascular smooth muscle. Biochem. Biophys. Res. Commun. 1988, 153, 625–631. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, G.; Fang, X.; Peng, X.; Tang, H.; Wang, H.; Jing, R.; Liu, J.; Lederer, W.J.; Chen, J.; et al. IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight 2016, 1, e89402. [Google Scholar] [CrossRef]
- Ford, L.E.; Podolsky, R.J. Regenerative calcium release within muscle cells. Science 1970, 167, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A.; Fabiato, F. Excitation-contraction coupling of isolated cardiac fibers with disrupted or closed sarcolemmas: Calcium-dependent cyclic and tonic contractions. Circ. Res. 1972, 31, 293–307. [Google Scholar] [CrossRef]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef]
- van Breemen, C.; Saida, K. Cellular mechanisms regulating [Ca2+]i smooth muscle. Annu. Rev. Physiol. 1989, 51, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Esfandiarei, M.; Fameli, N.; Choi, Y.Y.; Tehrani, A.Y.; Hoskins, J.G.; van Breemen, C. Waves of calcium depletion in the sarcoplasmic reticulum of vascular smooth muscle cells: An inside view of spatiotemporal Ca2+ regulation. PLoS ONE 2013, 8, e55333. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Structure and Function of IP3 Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef]
- Benham, C.D.; Tsien, R.W. A novel receptor-operated Ca2+-permeable channel activated by ATP in smooth muscle. Nature 1987, 328, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Bolton, T.B. Mechanisms of action of transmitters and other substances on smooth muscle. Physiol. Rev. 1979, 59, 606–718. [Google Scholar] [CrossRef]
- Van Breemen, C.; Aaronson, P.; Loutzenhiser, R. Sodium-calcium interactions in mammalian smooth muscle. Pharmacol. Rev. 1978, 30, 167–208. [Google Scholar] [CrossRef]
- Nelson, M.T.; Standen, N.B.; Brayden, J.E.; Worley, J.F., III. Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature 1988, 336, 382–385. [Google Scholar] [CrossRef]
- Timic Stamenic, T.; Todorovic, S.M. Thalamic T-Type Calcium Channels as Targets for Hypnotics and General Anesthetics. Int. J. Mol. Sci. 2022, 23, 2349. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Hanna, M.G. The Role of Calcium Channels in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022723. [Google Scholar] [CrossRef]
- de Amorim Ferreira, M.; Ferreira, J. Role of Cav2.3 (R-type) Calcium Channel in Pain and Analgesia: A Scoping Review. Curr. Neuropharmacol. 2024, 22, 1909–1922. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.; Gomez-Manjon, I.; Marco-Hernandez, A.V.; Fernandez-Martinez, F.J.; Camacho, A.; Martinez, F. N-Type Ca Channel in Epileptic Syndromes and Epilepsy: A Systematic Review of Its Genetic Variants. Int. J. Mol. Sci. 2023, 24, 6100. [Google Scholar] [CrossRef]
- Benham, C.D.; Hess, P.; Tsien, R.W. Two types of calcium channels in single smooth muscle cells from rabbit ear artery studied with whole-cell and single-channel recordings. Circ. Res. 1987, 61, I10–I16. [Google Scholar] [PubMed]
- Yatani, A.; Seidel, C.L.; Allen, J.; Brown, A.M. Whole-cell and single-channel calcium currents of isolated smooth muscle cells from saphenous vein. Circ. Res. 1987, 60, 523–533. [Google Scholar] [CrossRef]
- Sturek, M.; Hermsmeyer, K. Calcium and sodium channels in spontaneously contracting vascular muscle cells. Science 1986, 233, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Loirand, G.; Pacaud, P.; Mironneau, C.; Mironneau, J. Evidence for two distinct calcium channels in rat vascular smooth muscle cells in short-term primary culture. Pflug. Arch. Eur. J. Physiol. 1986, 407, 566–568. [Google Scholar] [CrossRef]
- Bean, B.P.; Sturek, M.; Puga, A.; Hermsmeyer, K. Calcium channels in muscle cells isolated from rat mesenteric arteries: Modulation by dihydropyridine drugs. Circ. Res. 1986, 59, 229–235. [Google Scholar] [CrossRef]
- Benham, C.D.; Tsien, R.W. Noradrenaline modulation of calcium channels in single smooth muscle cells from rabbit ear artery. J. Physiol. 1988, 404, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Ruth, P.; Bosse, E.; Hullin, R.; Stuhmer, W.; Flockerzi, V.; Hofmann, F. Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett. 1990, 269, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Syed, A.U.; Prada, M.P.; Nystoriak, M.A.; Santana, L.F.; Nieves-Cintron, M.; Navedo, M.F. Calcium Channels in Vascular Smooth Muscle. Adv. Pharmacol. 2017, 78, 49–87. [Google Scholar]
- Bezanilla, F. How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 2008, 9, 323–332. [Google Scholar] [CrossRef]
- Knot, H.J.; Nelson, M.T. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 1998, 508 Pt 1, 199–209. [Google Scholar] [CrossRef]
- VanBavel, E.; Sorop, O.; Andreasen, D.; Pfaffendorf, M.; Jensen, B.L. Role of T-type calcium channels in myogenic tone of skeletal muscle resistance arteries. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H2239–H2243. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Visser, F.; Brett, S.E.; Goldman, D.; Zechariah, A.; Hashad, A.M.; Menon, B.K.; Watson, T.; Starreveld, Y.; Welsh, D.G. CaV1.2/CaV3.x channels mediate divergent vasomotor responses in human cerebral arteries. J. Gen. Physiol. 2015, 145, 405–418. [Google Scholar] [CrossRef]
- Lee, N.; Jeong, S.; Kim, K.C.; Kim, J.A.; Park, J.Y.; Kang, H.W.; Perez-Reyes, E.; Lee, J.H. Ca2+ Regulation of Cav3.3 T-type Ca2+ Channel Is Mediated by Calmodulin. Mol. Pharmacol. 2017, 92, 347–357. [Google Scholar] [CrossRef]
- Meisheri, K.D.; Hwang, O.; van Breemen, C. Evidence for two separate Ca2+ pathways in smooth muscle plasmalemma. J. Membr. Biol. 1981, 59, 19–25. [Google Scholar] [CrossRef]
- Reuter, H.; Sigel, E. Ionic channels: Modulation by G proteins and by phosphorylation. Curr. Opin. Neurobiol. 1991, 1, 27–31. [Google Scholar] [CrossRef]
- Martinsen, A.; Dessy, C.; Morel, N. Regulation of calcium channels in smooth muscle: New insights into the role of myosin light chain kinase. Channels 2014, 8, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ma, Y.; Ye, X.; Zhang, N.; Pan, L.; Wang, B. TRP (transient receptor potential) ion channel family: Structures, biological functions and therapeutic interventions for diseases. Signal Transduct. Target. Ther. 2023, 8, 261. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.P. Gating mechanisms of canonical transient receptor potential channel proteins: Role of phosphoinositols and diacylglycerol. Adv. Exp. Med. Biol. 2011, 704, 391–411. [Google Scholar]
- Putney, J.W., Jr.; Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S. Mechanisms of capacitative calcium entry. J. Cell Sci. 2001, 114, 2223–2229. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Leung, F.P.; Yung, L.M.; Yao, X.; Laher, I.; Huang, Y. Store-operated calcium entry in vascular smooth muscle. Br. J. Pharmacol. 2008, 153, 846–857. [Google Scholar] [CrossRef]
- Parekh, A.B. Functional consequences of activating store-operated CRAC channels. Cell Calcium 2007, 42, 111–121. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Wang, O.L.; Whorton, A.R. Thapsigargin stimulates Ca2+ entry in vascular smooth muscle cells: Nicardipine-sensitive and -insensitive pathways. Am. J. Physiol.-Cell Physiol. 1992, 262, C1258–C1265. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Glass, P.S. Propofol regulation of calcium entry pathways in cultured A10 and rat aortic smooth muscle cells. Br. J. Pharmacol. 1996, 117, 5–12. [Google Scholar] [CrossRef]
- Tosun, M.; Paul, R.J.; Rapoport, R.M. Coupling of store-operated Ca++ entry to contraction in rat aorta. J. Pharmacol. Exp. Ther. 1998, 285, 759–766. [Google Scholar] [CrossRef]
- Golovina, V.A.; Platoshyn, O.; Bailey, C.L.; Wang, J.; Limsuwan, A.; Sweeney, M.; Rubin, L.J.; Yuan, J.X. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H746–H755. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Beech, D.J. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ. Res. 2001, 88, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Bergdahl, A.; Gomez, M.F.; Wihlborg, A.K.; Erlinge, D.; Eyjolfson, A.; Xu, S.Z.; Beech, D.J.; Dreja, K.; Hellstrand, P. Plasticity of TRPC expression in arterial smooth muscle: Correlation with store-operated Ca2+ entry. Am. J. Physiol.-Cell Physiol. 2005, 288, C872–C880. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Boulay, G.; Flemming, R.; Beech, D.J. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H2653–H2659. [Google Scholar] [CrossRef]
- Giamarchi, A.; Padilla, F.; Coste, B.; Raoux, M.; Crest, M.; Honore, E.; Delmas, P. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006, 7, 787–793. [Google Scholar] [CrossRef]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef]
- Zagranichnaya, T.K.; Wu, X.; Villereal, M.L. Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. J. Biol. Chem. 2005, 280, 29559–29569. [Google Scholar] [CrossRef]
- Song, T.; Hao, Q.; Zheng, Y.M.; Liu, Q.H.; Wang, Y.X. Inositol 1,4,5-trisphosphate activates TRPC3 channels to cause extracellular Ca2+ influx in airway smooth muscle cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 309, L1455–L1466. [Google Scholar] [CrossRef]
- Smani, T.; Zakharov, S.I.; Csutora, P.; Leno, E.; Trepakova, E.S.; Bolotina, V.M. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 2004, 6, 113–120. [Google Scholar] [CrossRef]
- Trepakova, E.S.; Gericke, M.; Hirakawa, Y.; Weisbrod, R.M.; Cohen, R.A.; Bolotina, V.M. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J. Biol. Chem. 2001, 276, 7782–7790. [Google Scholar] [CrossRef]
- Kim, H.Y.; Thomas, D.; Hanley, M.R. Chromatographic resolution of an intracellular calcium influx factor from thapsigargin-activated Jurkat cells. Evidence for multiple activities influencing calcium elevation in Xenopus oocytes. J. Biol. Chem. 1995, 270, 9706–9708. [Google Scholar] [CrossRef] [PubMed]
- Bolotina, V.M.; Csutora, P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem. Sci. 2005, 30, 378–387. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Lopez, J.J.; Jardin, I.; Bobe, R.; Pariente, J.A.; Enouf, J.; Salido, G.M.; Rosado, J.A. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochem. Pharmacol. 2008, 75, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Borelly, L.; Somasundaram, A.; Yamashita, M.; Ren, D.; Miller, R.J.; Prakriya, M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J. Physiol. 2008, 586, 5383–5401. [Google Scholar] [CrossRef]
- Soboloff, J.; Spassova, M.A.; Tang, X.D.; Hewavitharana, T.; Xu, W.; Gill, D.L. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef]
- Valadares, N.F.; d’ Muniz Pereira, H.; Ulian Araujo, A.P.; Garratt, R.C. Septin structure and filament assembly. Biophys. Rev. 2017, 9, 481–500. [Google Scholar] [CrossRef]
- Lopez, J.J.; Albarran, L.; Gomez, L.J.; Smani, T.; Salido, G.M.; Rosado, J.A. Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta 2016, 1863, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef]
- Lin, Y.P.; Bakowski, D.; Mirams, G.R.; Parekh, A.B. Selective recruitment of different Ca2+-dependent transcription factors by STIM1-Orai1 channel clusters. Nat. Commun. 2019, 10, 2516. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, W.M. On the local reactions of the arterial wall to changes of internal pressure. J. Physiol. 1902, 28, 220–231. [Google Scholar] [CrossRef]
- Bohr, D.F.; Webb, R.C. Vascular smooth muscle function and its changes in hypertension. Am. J. Med. 1984, 77, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Kirber, M.T.; Walsh, J.V., Jr.; Singer, J.J. Stretch-activated ion channels in smooth muscle: A mechanism for the initiation of stretch-induced contraction. Pflug. Arch. Eur. J. Physiol. 1988, 412, 339–345. [Google Scholar] [CrossRef]
- Harder, D.R. Pressure-induced myogenic activation of cat cerebral arteries is dependent on intact endothelium. Circ. Res. 1987, 60, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Aguettaz, E.; Bois, P.; Cognard, C.; Sebille, S. Stretch-activated TRPV2 channels: Role in mediating cardiopathies. Prog. Biophys. Mol. Biol. 2017, 130, 273–280. [Google Scholar] [CrossRef]
- Welsh, D.G.; Morielli, A.D.; Nelson, M.T.; Brayden, J.E. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ. Res. 2002, 90, 248–250. [Google Scholar] [CrossRef]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef]
- Douguet, D.; Patel, A.; Xu, A.; Vanhoutte, P.M.; Honore, E. Piezo Ion Channels in Cardiovascular Mechanobiology. Trends Pharmacol. Sci. 2019, 40, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, C.; Daniel, E.E. The influence of high potassium depolarization and acetylcholine on calcium exchange in the rat uterus. J. Gen. Physiol. 1966, 49, 1299–1317. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Vascular smooth muscle. II. Pharmacol. Norm. Hypotens. vessels. Pharmacol. Rev. 1970, 22, 249–353. [Google Scholar] [CrossRef]
- Casteels, R.; Van Breemen, C. Active and passive Ca2+ fluxes across cell membranes of the guinea-pig taenia coli. Pflug. Arch. Eur. J. Physiol. 1975, 359, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Raeymaekers, L.; Wuytack, F.; Casteels, R. Subcellular fractionation of pig stomach smooth muscle. A study of the distribution of the (Ca2+ + Mg2+)-ATPase activity in plasmalemma and endoplasmic reticulum. Biochim. Biophys. Acta 1985, 815, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Guerini, D.; Coletto, L.; Carafoli, E. Exporting calcium from cells. Cell Calcium 2005, 38, 281–289. [Google Scholar] [CrossRef]
- Popescu, L.M.; Ignat, P. Calmodulin-dependent Ca2+-pump ATPase of human smooth muscle sarcolemma. Cell Calcium 1983, 4, 219–235. [Google Scholar] [CrossRef]
- Rapp, J.P. Aortic responses to vanadate: Independence from (Na,K)-ATPase and comparison of Dahl salt-sensitive and salt-resistant rats. Hypertension 1981, 3, I168–I172. [Google Scholar] [CrossRef]
- Popescu, L.M.; Nutu, O.; Panoiu, C. Oxytocin contracts the human uterus at term by inhibiting the myometrial Ca2+-extrusion pump. Biosci. Rep. 1985, 5, 21–28. [Google Scholar] [CrossRef]
- Eggermont, J.A.; Vrolix, M.; Raeymaekers, L.; Wuytack, F.; Casteels, R. Ca2+-transport ATPases of vascular smooth muscle. Circ. Res. 1988, 62, 266–278. [Google Scholar] [CrossRef]
- Heim, R.; Iwata, T.; Zvaritch, E.; Adamo, H.P.; Rutishauser, B.; Strehler, E.E.; Guerini, D.; Carafoli, E. Expression, purification, and properties of the plasma membrane Ca2+ pump and of its N-terminally truncated 105-kDa fragment. J. Biol. Chem. 1992, 267, 24476–24484. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Haugen, J.D.; Penniston, J.T. Molecular cloning of a plasma membrane calcium pump from human osteoblasts. J. Bone Miner. Res. 1993, 8, 505–513. [Google Scholar] [CrossRef]
- De Jaegere, S.; Wuytack, F.; Eggermont, J.A.; Verboomen, H.; Casteels, R. Molecular cloning and sequencing of the plasma-membrane Ca2+ pump of pig smooth muscle. Biochem. J. 1990, 271, 655–660. [Google Scholar] [CrossRef]
- Chandan, K.; Gupta, M.; Ahmad, A.; Sarwat, M. P-type calcium ATPases play important roles in biotic and abiotic stress signaling. Planta 2024, 260, 37. [Google Scholar] [CrossRef] [PubMed]
- Ambesi, A.; Miranda, M.; Petrov, V.V.; Slayman, C.W. Biogenesis and function of the yeast plasma-membrane H+-ATPase. J. Exp. Biol. 2000, 203, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Sobolczyk, M.; Mackiewicz, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. Crosstalk among Calcium ATPases: PMCA, SERCA and SPCA in Mental Diseases. Int. J. Mol. Sci. 2021, 22, 2785. [Google Scholar] [CrossRef]
- Nitsche, J.; Josts, I.; Heidemann, J.; Mertens, H.D.; Maric, S.; Moulin, M.; Haertlein, M.; Busch, S.; Forsyth, V.T.; Svergun, D.I.; et al. Structural basis for activation of plasma-membrane Ca2+-ATPase by calmodulin. Commun. Biol. 2018, 1, 206. [Google Scholar] [CrossRef]
- Villalobo, A.; Gonzalez-Munoz, M.; Berchtold, M.W. Proteins with calmodulin-like domains: Structures and functional roles. Cell. Mol. Life Sci. 2019, 76, 2299–2328. [Google Scholar] [CrossRef]
- Inesi, G. Mechanism of calcium transport. Annu. Rev. Physiol. 1985, 47, 573–601. [Google Scholar] [CrossRef]
- Treves, S.; Jungbluth, H.; Voermans, N.; Muntoni, F.; Zorzato, F. Ca2+ handling abnormalities in early-onset muscle diseases: Novel concepts and perspectives. Semin. Cell Dev. Biol. 2017, 64, 201–212. [Google Scholar] [CrossRef]
- Makio, T.; Chen, J.; Simmen, T. ER stress as a sentinel mechanism for ER Ca2+ homeostasis. Cell Calcium 2024, 124, 102961. [Google Scholar] [CrossRef]
- Borle, A.B. Control, Modulation, and regulation of cell calcium. Rev. Physiol. Biochem. Pharmacol. 1981, 90, 13–153. [Google Scholar]
- MacLennan, D.H.; Wong, P.T. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1971, 68, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Wuytack, F.; Raeymaekers, L.; Verbist, J.; Jones, L.R.; Casteels, R. Smooth-muscle endoplasmic reticulum contains a cardiac-like form of calsequestrin. Biochim. Biophys. Acta 1987, 899, 151–158. [Google Scholar] [CrossRef]
- Haghighi, K.; Bidwell, P.; Kranias, E.G. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 2014, 77, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Chambers, P.J.; Juracic, E.S.; Fajardo, V.A.; Tupling, A.R. Role of SERCA and sarcolipin in adaptive muscle remodeling. Am. J. Physiol.-Cell Physiol. 2022, 322, C382–C394. [Google Scholar] [CrossRef]
- Vangheluwe, P.; Raeymaekers, L.; Dode, L.; Wuytack, F. Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: Cell biological implications. Cell Calcium 2005, 38, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.K.; Kwan, C.Y.; Rangachari, P.K.; Daniel, E.E. Na-Ca exchange in a smooth muscle plasma membrane-enriched fraction. Am. J. Physiol.-Cell Physiol. 1983, 244, C158–C165. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Barzilai, A.; Spanier, R.; Rahamimoff, H. Isolation, purification, and reconstitution of the Na+ gradient-dependent Ca2+ transporter (Na+-Ca2+ exchanger) from brain synaptic plasma membranes. Proc. Natl. Acad. Sci. USA 1984, 81, 6521–6525. [Google Scholar] [CrossRef]
- Matlib, M.A.; Reeves, J.P. Solubilization and reconstitution of the sarcolemmal Na+-Ca2+ exchange system of vascular smooth muscle. Biochim. Biophys. Acta 1987, 904, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Crompton, M. The regulation of intracellular calcium by mitochondria. Ann. N. Y. Acad. Sci. 1978, 307, 269–284. [Google Scholar] [CrossRef]
- Reeves, J.P.; Sutko, J.L. Sodium-calcium exchange activity generates a current in cardiac membrane vesicles. Science 1980, 208, 1461–1464. [Google Scholar] [CrossRef]
- Reeves, J.P.; Hale, C.C. The stoichiometry of the cardiac sodium-calcium exchange system. J. Biol. Chem. 1984, 259, 7733–7739. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J.; Aalkjaer, C.; Petersen, T.T. Intracellular sodium, membrane potential, and contractility of rat mesenteric small arteries. Circ. Res. 1984, 54, 740–749. [Google Scholar] [CrossRef]
- Ashida, T.; Blaustein, M.P. Regulation of cell calcium and contractility in mammalian arterial smooth muscle: The role of sodium-calcium exchange. J. Physiol. 1987, 392, 617–635. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Hamlyn, J.M. Sodium transport inhibition, cell calcium, and hypertension. The natriuretic hormone/Na+-Ca2+ exchange/hypertension hypothesis. Am. J. Med. 1984, 77, 45–59. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, C.; Lukeman, S.; Leijten, P.; Yamamoto, H.; Loutzenhiser, R. The role of superficial SR in modulating force development induced by Ca entry into arterial smooth muscle. J. Cardiovasc. Pharmacol. 1986, 8 (Suppl. S8), S111–S116. [Google Scholar] [CrossRef]
- Loutzenhiser, R.; van Breemen, C. The influence of receptor occupation on Ca++ influx-mediated vascular smooth muscle contraction. Circ. Res. 1983, 52, I97–I103. [Google Scholar]
- van Breemen, C.; Leijten, P.; Yamamoto, H.; Aaronson, P.; Cauvin, C. Calcium activation of vascular smooth muscle. State of the art lecture. Hypertension 1986, 8, II89–II95. [Google Scholar] [CrossRef]
- Zhang, W.B.; Kwan, C.Y. Pharmacological evidence that potentiation of plasmalemmal Ca2+-extrusion is functionally coupled to inhibition of SR Ca2+-ATPases in vascular smooth muscle cells. Naunyn-Schmiedeb. Arch. Pharmacol. 2016, 389, 447–455. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, C.; Fameli, N.; Evans, A.M. Pan-junctional sarcoplasmic reticulum in vascular smooth muscle: Nanospace Ca2+ transport for site- and function-specific Ca2+ signalling. J. Physiol. 2013, 591, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M. Nanojunctions of the Sarcoplasmic Reticulum Deliver Site- and Function-Specific Calcium Signaling in Vascular Smooth Muscles. In Advances in Pharmacology; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 78, pp. 1–47. [Google Scholar]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef]
- Waisman, D.M.; Gimble, J.M.; Goodman, D.B.; Rasmussen, H. Studies of the Ca2+ transport mechanism of human erythrocyte inside-out plasma membrane vesicles. II. Stimulation of the Ca2+ pump by phosphate. J. Biol. Chem. 1981, 256, 415–419. [Google Scholar] [CrossRef]
- Puskin, J.S.; Gunter, T.E.; Gunter, K.K.; Russell, P.R. Evidence for more than one Ca2+ transport mechanism in mitochondria. Biochemistry 1976, 15, 3834–3842. [Google Scholar] [CrossRef]
- Rasmussen, H.; Barrett, P.Q. Calcium messenger system: An integrated view. Physiol. Rev. 1984, 64, 938–984. [Google Scholar] [CrossRef]
- Carafoli, E. Intracellular calcium homeostasis. Annu. Rev. Biochem. 1987, 56, 395–433. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, D.; Vecellio Reane, D.; Raffaello, A. Neither too much nor too little: Mitochondrial calcium concentration as a balance between physiological and pathological conditions. Front. Mol. Biosci. 2023, 10, 1336416. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; Gonzalez-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vazquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef]
- Lee, S.H.; Duron, H.E.; Chaudhuri, D. Beyond the TCA cycle: New insights into mitochondrial calcium regulation of oxidative phosphorylation. Biochem. Soc. Trans. 2023, 51, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; van Breemen, C. Ca2+ compartments in saponin-skinned cultured vascular smooth muscle cells. J. Gen. Physiol. 1986, 87, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Keizer, J.; Smith, G.D. Spark-to-wave transition: Saltatory transmission of calcium waves in cardiac myocytes. Biophys. Chem. 1998, 72, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Wieder, N.; Fink, R.H.A.; von Wegner, F. Simulation Strategies for Calcium Microdomains and Calcium Noise. Adv. Exp. Med. Biol. 2020, 1131, 771–797. [Google Scholar]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium homeostasis and cancer: Insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol. 2023, 33, 312–323. [Google Scholar] [CrossRef]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Sharma, S.; Findlay, G.M.; Bandukwala, H.S.; Oberdoerffer, S.; Baust, B.; Li, Z.; Schmidt, V.; Hogan, P.G.; Sacks, D.B.; Rao, A. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proc. Natl. Acad. Sci. USA 2011, 108, 11381–11386. [Google Scholar] [CrossRef]
- Takemoto-Kimura, S.; Suzuki, K.; Horigane, S.I.; Kamijo, S.; Inoue, M.; Sakamoto, M.; Fujii, H.; Bito, H. Calmodulin kinases: Essential regulators in health and disease. J. Neurochem. 2017, 141, 808–818. [Google Scholar] [CrossRef]
- Kreusser, M.M.; Backs, J. Integrated mechanisms of CaMKII-dependent ventricular remodeling. Front. Pharmacol. 2014, 5, 36. [Google Scholar] [CrossRef]
- Cabrera-Orefice, A.; Ibarra-Garcia-Padilla, R.; Maldonado-Guzman, R.; Guerrero-Castillo, S.; Luevano-Martinez, L.A.; Perez-Vazquez, V.; Gutierrez-Aguilar, M.; Uribe-Carvajal, S. The Saccharomyces cerevisiae mitochondrial unselective channel behaves as a physiological uncoupling system regulated by Ca2+, Mg2+, phosphate and ATP. J. Bioenerg. Biomembr. 2015, 47, 477–491. [Google Scholar] [CrossRef]
- Boehm, M.; Nabel, E.G. The cell cycle and cardiovascular diseases. Prog. Cell Cycle Res. 2003, 5, 19–30. [Google Scholar] [PubMed]
- Schultz, K.; Fanburg, B.L.; Beasley, D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H2528–H2534. [Google Scholar] [CrossRef]
- Yang, M.; Huang, H.L.; Zhu, B.Y.; Tuo, Q.H.; Liao, D.F. Onychin inhibits proliferation of vascular smooth muscle cells by regulating cell cycle. Acta Pharmacol. Sin. 2005, 26, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Dudits, D.; Abraham, E.; Miskolczi, P.; Ayaydin, F.; Bilgin, M.; Horvath, G.V. Cell-cycle control as a target for calcium, hormonal and developmental signals: The role of phosphorylation in the retinoblastoma-centred pathway. Ann. Bot. 2011, 107, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Major, J.L.; Salih, M.; Tuana, B.S. Interplay between the E2F pathway and beta-adrenergic signaling in the pathological hypertrophic response of myocardium. J. Mol. Cell Cardiol. 2015, 84, 179–190. [Google Scholar] [CrossRef]
- Baran, I. Exit from mitosis induced by a calcium transient: The relation to the MPF and InsP3 dynamics. Biosystems 1994, 33, 203–214. [Google Scholar] [CrossRef]
- Umemura, M.; Nakakaji, R.; Ishikawa, Y. Physiological functions of calcium signaling via Orai1 in cancer. J. Physiol. Sci. 2023, 73, 21. [Google Scholar] [CrossRef]
- Marini, M.; Titiz, M.; de Araujo, D.S.M.; Geppetti, P.; Nassini, R.; De Logu, F. TRP Channels in Cancer: Signaling Mechanisms and Translational Approaches. Biomolecules 2023, 13, 1557. [Google Scholar] [CrossRef]
- Paoletti, R.; Bernini, F.; Corsini, A.; Soma, M.R. The antiatherosclerotic effects of calcium antagonists. J. Cardiovasc. Pharmacol. 1995, 25 (Suppl. S3), S6–S10. [Google Scholar] [CrossRef]
- Herrman, J.P.; Hermans, W.R.; Vos, J.; Serruys, P.W. Pharmacological approaches to the prevention of restenosis following angioplasty: The search for the Holy Grail? (Part I). Drugs 1993, 46, 18–52. [Google Scholar] [CrossRef]
- Russell, K.S.; Haynes, M.P.; Sinha, D.; Clerisme, E.; Bender, J.R. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 5930–5935. [Google Scholar] [CrossRef] [PubMed]
- Hisamoto, K.; Ohmichi, M.; Kurachi, H.; Hayakawa, J.; Kanda, Y.; Nishio, Y.; Adachi, K.; Tasaka, K.; Miyoshi, E.; Fujiwara, N.; et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 2001, 276, 3459–3467. [Google Scholar] [CrossRef]
- Tokiwa, H.; Ueda, K.; Takimoto, E. The emerging role of estrogen’s non-nuclear signaling in the cardiovascular disease. Front. Cardiovasc. Med. 2023, 10, 1127340. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef]
- Mordi, I.; Mordi, N.; Delles, C.; Tzemos, N. Endothelial dysfunction in human essential hypertension. J. Hypertens. 2016, 34, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. 2013, 64, 409–421. [Google Scholar]
- Wang, L.; Cheng, C.K.; Yi, M.; Lui, K.O.; Huang, Y. Targeting endothelial dysfunction and inflammation. J. Mol. Cell. Cardiol. 2022, 168, 58–67. [Google Scholar] [CrossRef]
- Park, Y.J.; Yoo, S.A.; Kim, M.; Kim, W.U. The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef]
- Gutierrez, A.; Gomez Del Val, A.; Contreras, C.; Olmos, L.; Sanchez, A.; Prieto, D. Calcium handling coupled to the endothelin ETA and ETB receptor-mediated vasoconstriction in resistance arteries: Differential regulation by PI3K, PKC and RhoK. Eur. J. Pharmacol. 2023, 956, 175948. [Google Scholar] [CrossRef] [PubMed]
- Adu-Gyamfi, M.; Goettsch, C.; Kamhieh-Milz, J.; Chen, L.; Pfefferkorn, A.M.; Hofmann, A.; Brunssen, C.; Muller, G.; Walther, T.; Ashraf, M.I.; et al. The Role of NOX2-Derived Reactive Oxygen Species in the Induction of Endothelin-Converting Enzyme-1 by Angiotensin II. Antioxidants 2024, 13, 500. [Google Scholar] [CrossRef] [PubMed]
- D’Orleans-Juste, P.; Plante, M.; Honore, J.C.; Carrier, E.; Labonte, J. Synthesis and degradation of endothelin-1. Can. J. Physiol. Pharmacol. 2003, 81, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Wicinski, M.; Szadujkis-Szadurska, K.; Weclewicz, M.M.; Malinowski, B.; Matusiak, G.; Walczak, M.; Wodkiewicz, E.; Grzesk, G.; Pawlak-Osinska, K. The role of Rho-kinase and calcium ions in constriction triggered by ET-1. Microvasc. Res. 2018, 119, 84–90. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Ling, H.; Li, Y.; Fu, S.; Xu, M.; Li, B.; Liu, X.; Wang, Q.; Li, A.; et al. Navigating the Landscape of Coronary Microvascular Research: Trends, Triumphs, and Challenges Ahead. Rev. Cardiovasc. Med. 2024, 25, 288. [Google Scholar] [CrossRef]
- Palacios-Ramirez, R.; Hernanz, R.; Martin, A.; Perez-Giron, J.V.; Barrus, M.T.; Gonzalez-Carnicero, Z.; Aguado, A.; Jaisser, F.; Briones, A.M.; Salaices, M.; et al. Pioglitazone Modulates the Vascular Contractility in Hypertension by Interference with ET-1 Pathway. Sci. Rep. 2019, 9, 16461. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol.-Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Cui, X.L.; Brockman, D.; Campos, B.; Myatt, L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: Involvement in preeclampsia. Placenta 2006, 27, 422–431. [Google Scholar] [CrossRef]
- Inoue, M. Role of cross talk between superoxide and NO in the energy metabolism and pathogenesis of vasogenic tissue injury. J. Toxicol. Sci. 1996, 21, 289. [Google Scholar]
- Bates, D.O. Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 2010, 87, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Mehta, D.; Malik, A.B. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 2016, 60, 163–171. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, J.A.; Weinrauch, L.A. Calcium Ion Channels: Roles in Infection and Sepsis Mechanisms of Calcium Channel Blocker Benefits in Immunocompromised Patients at Risk for Infection. Int. J. Mol. Sci. 2018, 19, 2465. [Google Scholar] [CrossRef]
- Ikebe, M.; Hartshorne, D.J. The role of myosin phosphorylation in the contraction-relaxation cycle of smooth muscle. Experientia 1985, 41, 1006–1010. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Regulation of smooth muscle contractile elements by second messengers. Annu. Rev. Physiol. 1989, 51, 299–313. [Google Scholar] [CrossRef]
- Khalil, R.A.; van Breemen, C. Intracellular free calcium concentration/force relationship in rabbit inferior vena cava activated by norepinephrine and high K+. Pflug. Arch. Eur. J. Physiol. 1990, 416, 727–734. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 1984, 220, 345–360. [Google Scholar] [CrossRef]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [CrossRef]
- Takai, Y.; Kishimoto, A.; Inoue, M.; Nishizuka, Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Purification and characterization of an active enzyme from bovine cerebellum. J. Biol. Chem. 1977, 252, 7603–7609. [Google Scholar] [CrossRef]
- Kishimoto, A.; Mikawa, K.; Hashimoto, K.; Yasuda, I.; Tanaka, S.; Tominaga, M.; Kuroda, T.; Nishizuka, Y. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain). J. Biol. Chem. 1989, 264, 4088–4092. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef]
- Poli, A.; Mongiorgi, S.; Cocco, L.; Follo, M.Y. Protein kinase C involvement in cell cycle modulation. Biochem. Soc. Trans. 2014, 42, 1471–1476. [Google Scholar] [CrossRef]
- Rozengurt, E. Protein kinase D signaling: Multiple biological functions in health and disease. Physiology 2011, 26, 23–33. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Ying, Y.S.; Chapline, C.; Jaken, S.; Anderson, R.G. Targeting of protein kinase Cα to caveolae. J. Cell Biol. 1998, 141, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Dubois, T.; Oudinet, J.P.; Mira, J.P.; Russo-Marie, F. Annexins and protein kinases C. Biochim. Biophys. Acta 1996, 1313, 290–294. [Google Scholar] [CrossRef]
- Xu, T.R.; Rumsby, M.G. Phorbol ester-induced translocation of PKC epsilon to the nucleus in fibroblasts: Identification of nuclear PKC epsilon-associating proteins. FEBS Lett. 2004, 570, 20–24. [Google Scholar] [CrossRef]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [CrossRef]
- Parekh, D.B.; Ziegler, W.; Parker, P.J. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000, 19, 496–503. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.H.; Thelen, M.; Rosen, A.; Janmey, P.A.; Nairn, A.C.; Aderem, A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature 1992, 356, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Woodsome, T.P.; Eto, M.; Everett, A.; Brautigan, D.L.; Kitazawa, T. Expression of CPI-17 and myosin phosphatase correlates with Ca2+ sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J. Physiol. 2001, 535, 553–564. [Google Scholar] [CrossRef] [PubMed]
- El-Yazbi, A.F.; Abd-Elrahman, K.S.; Moreno-Dominguez, A. PKC-mediated cerebral vasoconstriction: Role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem. Pharmacol. 2015, 95, 263–278. [Google Scholar] [CrossRef]
- Parker, C.A.; Takahashi, K.; Tao, T.; Morgan, K.G. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. Am. J. Physiol.-Cell Physiol. 1994, 267, C1262–C1270. [Google Scholar] [CrossRef]
- Je, H.D.; Gangopadhyay, S.S.; Ashworth, T.D.; Morgan, K.G. Calponin is required for agonist-induced signal transduction—Evidence from an antisense approach in ferret smooth muscle. J. Physiol. 2001, 537, 567–577. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Igarashi, M.; Hirata, A.; Sugae, N.; Tsuchiya, H.; Jimbu, Y.; Tominaga, M.; Kato, T. Altered PDGF-BB-induced p38 MAP kinase activation in diabetic vascular smooth muscle cells: Roles of protein kinase C-δ. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2095–2101. [Google Scholar] [CrossRef]
- Ginnan, R.; Singer, H.A. PKC-δ-dependent pathways contribute to PDGF-stimulated ERK1/2 activation in vascular smooth muscle. Am. J. Physiol.-Cell Physiol. 2005, 288, C1193–C1201. [Google Scholar] [CrossRef]
- Mii, S.; Khalil, R.A.; Morgan, K.G.; Ware, J.A.; Kent, K.C. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H142–H150. [Google Scholar] [CrossRef]
- Khalil, R.A.; Menice, C.B.; Wang, C.L.; Morgan, K.G. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ. Res. 1995, 76, 1101–1108. [Google Scholar] [CrossRef]
- Kim, H.R.; Gallant, C.; Morgan, K.G. Regulation of PKC autophosphorylation by calponin in contractile vascular smooth muscle tissue. BioMed Res. Int. 2013, 2013, 358643. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Sohn, U.D.; Bitar, K.N.; Behar, J.; Biancani, P.; Harnett, K.M. MAPK mediates PKC-dependent contraction of cat esophageal and lower esophageal sphincter circular smooth muscle. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 285, G86–G95. [Google Scholar] [CrossRef]
- Chen, Y.L.; Daneva, Z.; Kuppusamy, M.; Ottolini, M.; Baker, T.M.; Klimentova, E.; Shah, S.A.; Sokolowski, J.D.; Park, M.S.; Sonkusare, S.K. Novel Smooth Muscle Ca2+-Signaling Nanodomains in Blood Pressure Regulation. Circulation 2022, 146, 548–564. [Google Scholar] [CrossRef] [PubMed]
- Quayle, J.M.; Nelson, M.T.; Standen, N.B. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev. 1997, 77, 1165–1232. [Google Scholar] [CrossRef]
- Zhu, R.; Xiao, D.; Zhang, L. Potassium channels and uterine vascular adaptation to pregnancy and chronic hypoxia. Curr. Vasc. Pharmacol. 2013, 11, 737–747. [Google Scholar] [CrossRef]
- Cole, W.C.; Malcolm, T.; Walsh, M.P.; Light, P.E. Inhibition by protein kinase C of the KNDP subtype of vascular smooth muscle ATP-sensitive potassium channel. Circ. Res. 2000, 87, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Resta, T.C. Calcium homeostasis and sensitization in pulmonary arterial smooth muscle. Microcirculation 2014, 21, 259–271. [Google Scholar] [CrossRef]
- Taguchi, K.; Kaneko, K.; Kubo, T. Protein kinase C modulates Ca2+-activated K+ channels in cultured rat mesenteric artery smooth muscle cells. Biol. Pharm. Bull. 2000, 23, 1450–1454. [Google Scholar] [CrossRef]
- Barman, S.A.; Zhu, S.; White, R.E. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L149–L155. [Google Scholar] [CrossRef]
- Kizub, I.V.; Pavlova, O.O.; Ivanova, I.V.; Soloviev, A.I. Protein kinase C-dependent inhibition of BKCa current in rat aorta smooth muscle cells following gamma-irradiation. Int. J. Radiat. Biol. 2010, 86, 291–299. [Google Scholar] [CrossRef]
- Novokhatska, T.; Tishkin, S.; Dosenko, V.; Boldyriev, A.; Ivanova, I.; Strielkov, I.; Soloviev, A. Correction of vascular hypercontractility in spontaneously hypertensive rats using shRNAs-induced delta protein kinase C gene silencing. Eur. J. Pharmacol. 2013, 718, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Fukuzawa, K.; Nakaya, Y. Protein kinase C inhibits the Ca2+-activated K+ channel of cultured porcine coronary artery smooth muscle cells. Biochem. Biophys. Res. Commun. 1993, 190, 263–269. [Google Scholar] [CrossRef]
- Lange, A.; Gebremedhin, D.; Narayanan, J.; Harder, D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J. Biol. Chem. 1997, 272, 27345–27352. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Q.; Xiao, D.; Zhu, R.; Huang, X.; Yang, S.; Wilson, S.; Zhang, L. Pregnancy upregulates large-conductance Ca2+-activated K+ channel activity and attenuates myogenic tone in uterine arteries. Hypertension 2011, 58, 1132–1139. [Google Scholar] [CrossRef]
- Crozatier, B. Central role of PKCs in vascular smooth muscle cell ion channel regulation. J. Mol. Cell. Cardiol. 2006, 41, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, J.; Werner, M.E.; Brayden, J.E.; Nelson, M.T. Calcium-activated potassium channels and the regulation of vascular tone. Physiology 2006, 21, 69–78. [Google Scholar] [CrossRef]
- Brueggemann, L.I.; Mackie, A.R.; Cribbs, L.L.; Freda, J.; Tripathi, A.; Majetschak, M.; Byron, K.L. Differential protein kinase C-dependent modulation of Kv7.4 and Kv7.5 subunits of vascular Kv7 channels. J. Biol. Chem. 2014, 289, 2099–2111. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: Role of protein kinase Cζ. Circ. Res. 2003, 93, 656–663. [Google Scholar] [CrossRef]
- Park, W.S.; Han, J.; Kim, N.; Youm, J.B.; Joo, H.; Kim, H.K.; Ko, J.H.; Earm, Y.E. Endothelin-1 inhibits inward rectifier K+ channels in rabbit coronary arterial smooth muscle cells through protein kinase C. J. Cardiovasc. Pharmacol. 2005, 46, 681–689. [Google Scholar] [CrossRef]
- Park, W.S.; Kim, N.; Youm, J.B.; Warda, M.; Ko, J.H.; Kim, S.J.; Earm, Y.E.; Han, J. Angiotensin II inhibits inward rectifier K+ channels in rabbit coronary arterial smooth muscle cells through protein kinase Cα. Biochem. Biophys. Res. Commun. 2006, 341, 728–735. [Google Scholar] [CrossRef]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995, 268, C799–C822. [Google Scholar] [CrossRef] [PubMed]
- Bonev, A.D.; Nelson, M.T. Vasoconstrictors inhibit ATP-sensitive K+ channels in arterial smooth muscle through protein kinase C. J. Gen. Physiol. 1996, 108, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Wang, X.D.; Huang, K.; Wang, L.; Jiang, Z.L.; Qi, Y.X. Temporal phosphoproteomics to investigate the mechanotransduction of vascular smooth muscle cells in response to cyclic stretch. J. Biomech. 2014, 47, 3622–3629. [Google Scholar] [CrossRef]
- Li, C.; Wernig, F.; Leitges, M.; Hu, Y.; Xu, Q. Mechanical stress-activated PKCδ regulates smooth muscle cell migration. FASEB J. 2003, 17, 2106–2108. [Google Scholar] [CrossRef]
- Slish, D.F.; Welsh, D.G.; Brayden, J.E. Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H2196–H2201. [Google Scholar] [CrossRef]
- Albert, A.P.; Large, W.A. Signal transduction pathways and gating mechanisms of native TRP-like cation channels in vascular myocytes. J. Physiol. 2006, 570, 45–51. [Google Scholar] [CrossRef]
- Austin, C.; Wray, S. Interactions between Ca2+ and H+ and functional consequences in vascular smooth muscle. Circ. Res. 2000, 86, 355–363. [Google Scholar] [CrossRef]
- Wray, S.; Smith, R.D. Mechanisms of action of pH-induced effects on vascular smooth muscle. Mol. Cell. Biochem. 2004, 263, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Aviv, A. Cytosolic Ca2+, Na+/H+ antiport, protein kinase C trio in essential hypertension. Am. J. Hypertens. 1994, 7, 205–212. [Google Scholar] [CrossRef]
- Rosoff, P.M.; Stein, L.F.; Cantley, L.C. Phorbol esters induce differentiation in a pre-B-lymphocyte cell line by enhancing Na+/H+ exchange. J. Biol. Chem. 1984, 259, 7056–7060. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, H.; Wu, B.; Zhou, Q.; Cui, D.; Wang, L. Protein Kinase C Isoforms Distinctly Regulate Propofol-induced Endothelium-dependent and Endothelium-independent Vasodilation. J. Cardiovasc. Pharmacol. 2015, 66, 276–284. [Google Scholar] [CrossRef]
- Johnson, J.A.; Barman, S.A. Protein kinase C modulation of cyclic GMP in rat neonatal pulmonary vascular smooth muscle. Lung 2004, 182, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Mori, E.; Mori, Y.; Mori, M.; Li, J.; Ito, Y.; Inoue, R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J. Physiol. 2004, 561, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Large, W.A.; Saleh, S.N.; Albert, A.P. Role of phosphoinositol 4,5-bisphosphate and diacylglycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium 2009, 45, 574–582. [Google Scholar] [CrossRef]
- Inagaki, M.; Yokokura, H.; Itoh, T.; Kanmura, Y.; Kuriyama, H.; Hidaka, H. Purified rabbit brain protein kinase C relaxes skinned vascular smooth muscle and phosphorylates myosin light chain. Arch. Biochem. Biophys. 1987, 254, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; MacAllister, R.J.; Hobbs, A.J. Vascular actions of natriuretic peptides: Cyclic GMP-dependent and -independent mechanisms. Basic. Res. Cardiol. 2004, 99, 83–89. [Google Scholar] [CrossRef]
- Mendonca, M.C.; Doi, S.Q.; Glerum, S.; Sellitti, D.F. Increase of C-type natriuretic peptide expression by serum and platelet-derived growth factor-BB in human aortic smooth muscle cells is dependent on protein kinase C activation. Endocrinology 2006, 147, 4169–4178. [Google Scholar] [CrossRef]
- Mendonca, M.C.; Koles, N.; Sellitti, D.F. Protein kinase C-δ (PKC-δ) and PKC-α mediate Ca2+-dependent increases in CNP mRNA in human vascular cells. Vasc. Pharmacol. 2012, 57, 98–104. [Google Scholar] [CrossRef]
- Limas, C.J. Phosphorylation of cardiac sarcoplasmic reticulum by a calcium-activated, phospholipid-dependent protein kinase. Biochem. Biophys. Res. Commun. 1980, 96, 1378–1383. [Google Scholar] [CrossRef]
- Bertorello, A.M.; Aperia, A.; Walaas, S.I.; Nairn, A.C.; Greengard, P. Phosphorylation of the catalytic subunit of Na+,K(+)-ATPase inhibits the activity of the enzyme. Proc. Natl. Acad. Sci. USA 1991, 88, 11359–11362. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, R.H.; Webb, R.C. Molecular aspects of arterial smooth muscle contraction: Focus on Rho. Exp. Biol. Med. 2005, 230, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Budzyn, K.; Sobey, C.G. Vascular rho kinases and their potential therapeutic applications. Curr. Opin. Drug Discov. Devel 2007, 10, 590–596. [Google Scholar] [PubMed]
- Gong, M.C.; Fujihara, H.; Somlyo, A.V.; Somlyo, A.P. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J. Biol. Chem. 1997, 272, 10704–10709. [Google Scholar] [CrossRef]
- Loirand, G.; Guerin, P.; Pacaud, P. Rho kinases in cardiovascular physiology and pathophysiology. Circ. Res. 2006, 98, 322–334. [Google Scholar] [CrossRef]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Amano, M.; Yamamoto, T.; Chihara, K.; Nakafuku, M.; Ito, M.; Nakano, T.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996, 15, 2208–2216. [Google Scholar] [CrossRef]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef]
- Szasz, T.; Webb, R.C. Rho-Mancing to Sensitize Calcium Signaling for Contraction in the Vasculature: Role of Rho Kinase. Adv. Pharmacol. 2017, 78, 303–322. [Google Scholar]
- Wibberley, A.; Chen, Z.; Hu, E.; Hieble, J.P.; Westfall, T.D. Expression and functional role of Rho-kinase in rat urinary bladder smooth muscle. Br. J. Pharmacol. 2003, 138, 757–766. [Google Scholar] [CrossRef]
- Hiroki, J.; Shimokawa, H.; Higashi, M.; Morikawa, K.; Kandabashi, T.; Kawamura, N.; Kubota, T.; Ichiki, T.; Amano, M.; Kaibuchi, K.; et al. Inflammatory stimuli upregulate Rho-kinase in human coronary vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2004, 37, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Blumenstein, L.; Ahmadian, M.R. Models of the cooperative mechanism for Rho effector recognition: Implications for RhoA-mediated effector activation. J. Biol. Chem. 2004, 279, 53419–53426. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.K.; Mack, H.; Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Kitazawa, T.; Eto, M.; Woodsome, T.P.; Brautigan, D.L. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J. Biol. Chem. 2000, 275, 9897–9900. [Google Scholar] [CrossRef]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef]
- Kaneko, T.; Amano, M.; Maeda, A.; Goto, H.; Takahashi, K.; Ito, M.; Kaibuchi, K. Identification of calponin as a novel substrate of Rho-kinase. Biochem. Biophys. Res. Commun. 2000, 273, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Riddick, N.; Ohtani, K.; Surks, H.K. Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J. Cell Biochem. 2008, 103, 1158–1170. [Google Scholar] [CrossRef]
- Nunes, K.P.; Rigsby, C.S.; Webb, R.C. RhoA/Rho-kinase and vascular diseases: What is the link? Cell Mol. Life Sci. 2010, 67, 3823–3836. [Google Scholar] [CrossRef]
- de Godoy, M.A.; Rattan, S. Role of rho kinase in the functional and dysfunctional tonic smooth muscles. Trends Pharmacol. Sci. 2011, 32, 384–393. [Google Scholar] [CrossRef]
- Feng, J.; Ito, M.; Kureishi, Y.; Ichikawa, K.; Amano, M.; Isaka, N.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K.; Hartshorne, D.J.; et al. Rho-associated kinase of chicken gizzard smooth muscle. J. Biol. Chem. 1999, 274, 3744–3752. [Google Scholar] [CrossRef]
- Shirao, S.; Kashiwagi, S.; Sato, M.; Miwa, S.; Nakao, F.; Kurokawa, T.; Todoroki-Ikeda, N.; Mogami, K.; Mizukami, Y.; Kuriyama, S.; et al. Sphingosylphosphorylcholine is a novel messenger for Rho-kinase-mediated Ca2+ sensitization in the bovine cerebral artery: Unimportant role for protein kinase C. Circ. Res. 2002, 91, 112–119. [Google Scholar] [CrossRef]
- Amano, M.; Fukata, Y.; Kaibuchi, K. Regulation and functions of Rho-associated kinase. Exp. Cell Res. 2000, 261, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.C.; Fuglsang, A.; Alessi, D.; Kobayashi, S.; Cohen, P.; Somlyo, A.V.; Somlyo, A.P. Arachidonic acid inhibits myosin light chain phosphatase and sensitizes smooth muscle to calcium. J. Biol. Chem. 1992, 267, 21492–21498. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef]
- Street, C.A.; Bryan, B.A. Rho kinase proteins—Pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 2011, 31, 3645–3657. [Google Scholar] [PubMed]
- Kawano, Y.; Fukata, Y.; Oshiro, N.; Amano, M.; Nakamura, T.; Ito, M.; Matsumura, F.; Inagaki, M.; Kaibuchi, K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 1999, 147, 1023–1038. [Google Scholar] [CrossRef]
- Amano, T.; Tanabe, K.; Eto, T.; Narumiya, S.; Mizuno, K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem. J. 2001, 354, 149–159. [Google Scholar] [CrossRef]
- Matsui, T.; Maeda, M.; Doi, Y.; Yonemura, S.; Amano, M.; Kaibuchi, K.; Tsukita, S.; Tsukita, S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J. Cell Biol. 1998, 140, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Oshiro, N.; Kinoshita, N.; Kawano, Y.; Matsuoka, Y.; Bennett, V.; Matsuura, Y.; Kaibuchi, K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J. Cell Biol. 1999, 145, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Vahebi, S.; Kobayashi, T.; Warren, C.M.; de Tombe, P.P.; Solaro, R.J. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ. Res. 2005, 96, 740–747. [Google Scholar] [CrossRef]
- Riento, K.; Guasch, R.M.; Garg, R.; Jin, B.; Ridley, A.J. RhoE binds to ROCK I and inhibits downstream signaling. Mol. Cell. Biol. 2003, 23, 4219–4229. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. In Progress in Molecular Biology and Translational Science; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 147, pp. 1–73. [Google Scholar]
- Amalinei, C.; Caruntu, I.D.; Balan, R.A. Biology of metalloproteinases. Rom. J. Morphol. Embryol. 2007, 48, 323–334. [Google Scholar]
- Sreesada, P.; Vandana; Krishnan, B.; Amrutha, R.; Chavan, Y.; Alfia, H.; Jyothis, A.; Venugopal, P.; Aradhya, R.; Suravajhala, P.; et al. Matrix metalloproteinases: Master regulators of tissue morphogenesis. Gene 2025, 933, 148990. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef]
- Bode, W.; Fernandez-Catalan, C.; Grams, F.; Gomis-Ruth, F.X.; Nagase, H.; Tschesche, H.; Maskos, K. Insights into MMP-TIMP interactions. Ann. N. Y. Acad. Sci. 1999, 878, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Togliatto, G.; Lombardo, G.; Brizzi, M.F. The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. Int. J. Mol. Sci. 2017, 18, 1988. [Google Scholar] [CrossRef]
- Daghbouche-Rubio, N.; Lopez-Lopez, J.R.; Perez-Garcia, M.T.; Cidad, P. Vascular smooth muscle ion channels in essential hypertension. Front. Physiol. 2022, 13, 1016175. [Google Scholar] [CrossRef]
- Manolis, A.J.; Poulimenos, L.E.; Kallistratos, M.S.; Gavras, I.; Gavras, H. Sympathetic overactivity in hypertension and cardiovascular disease. Curr. Vasc. Pharmacol. 2014, 12, 4–15. [Google Scholar] [PubMed]
- Grassi, G.; Drager, L.F. Sympathetic overactivity, hypertension and cardiovascular disease: State of the art. Curr. Med. Res. Opin. 2024, 40, 5–13. [Google Scholar] [CrossRef]
- Rey-Garcia, J.; Townsend, R.R. Renal Denervation: A Review. Am. J. Kidney Dis. 2022, 80, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R.A.; Lajoie, C.; Morgan, K.G. In situ determination of [Ca2+]i threshold for translocation of the alpha-protein kinase C isoform. Am. J. Physiol.-Cell Physiol. 1994, 266, C1544–C1551. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.M.; Morgan, K.G. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. Am. J. Physiol.-Cell Physiol. 1994, 267, C980–C989. [Google Scholar] [CrossRef]
- Okazaki, J.; Mawatari, K.; Liu, B.; Kent, K.C. The effect of protein kinase C and its alpha subtype on human vascular smooth muscle cell proliferation, migration and fibronectin production. Surgery 2000, 128, 192–197. [Google Scholar] [CrossRef]
- Campbell, M.; Trimble, E.R. Modification of PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth muscle cells by protein kinase CβII. Circ. Res. 2005, 96, 197–206. [Google Scholar] [CrossRef]
- Liu, B.; Ryer, E.J.; Kundi, R.; Kamiya, K.; Itoh, H.; Faries, P.L.; Sakakibara, K.; Kent, K.C. Protein kinase C-δ regulates migration and proliferation of vascular smooth muscle cells through the extracellular signal-regulated kinase 1/2. J. Vasc. Surg. 2007, 45, 160–168. [Google Scholar] [CrossRef]
- Kim, J.; Min, G.; Bae, Y.S.; Min, D.S. Phospholipase D is involved in oxidative stress-induced migration of vascular smooth muscle cells via tyrosine phosphorylation and protein kinase C. Exp. Mol. Med. 2004, 36, 103–109. [Google Scholar] [CrossRef]
- Ding, Q.; Chai, H.; Mahmood, N.; Tsao, J.; Mochly-Rosen, D.; Zhou, W. Matrix metalloproteinases modulated by protein kinase Cε mediate resistin-induced migration of human coronary artery smooth muscle cells. J. Vasc. Surg. 2011, 53, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Newby, A.C. Effect of matrix metalloproteinase-9 knockout on vein graft remodelling in mice. J. Vasc. Res. 2010, 47, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pla, A.; Bosch-Gil, J.A.; Rossello-Urgell, J.; Huguet-Redecilla, P.; Stone, J.H.; Vilardell-Tarres, M. Metalloproteinase-2 and -9 in giant cell arteritis: Involvement in vascular remodeling. Circulation 2005, 112, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Little, P.J.; Williams, M.R.; Dai, A.; Hashimura, K.; Liu, J.P.; Komesaroff, P.A.; Sudhir, K. High glucose abolishes the antiproliferative effect of 17β-estradiol in human vascular smooth muscle cells. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E746–E751. [Google Scholar] [CrossRef]
- Ding, R.Q.; Tsao, J.; Chai, H.; Mochly-Rosen, D.; Zhou, W. Therapeutic potential for protein kinase C inhibitor in vascular restenosis. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 160–167. [Google Scholar] [CrossRef]
- Deuse, T.; Koyanagi, T.; Erben, R.G.; Hua, X.; Velden, J.; Ikeno, F.; Reichenspurner, H.; Robbins, R.C.; Mochly-Rosen, D.; Schrepfer, S. Sustained inhibition of ε protein kinase C inhibits vascular restenosis after balloon injury and stenting. Circulation 2010, 122, S170–S178. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, Y.; Zhou, Q.; Wu, B.; Wang, A.; Jiang, W.; Wang, L. Down-Regulation of Protein Kinase C-ε by Prolonged Incubation with PMA Inhibits the Proliferation of Vascular Smooth Muscle Cells. Cell. Physiol. Biochem. 2016, 40, 379–390. [Google Scholar] [CrossRef]
- Huang, H.; Lee, D.H.; Zabolotny, J.M.; Kim, Y.B. Metabolic actions of Rho-kinase in periphery and brain. Trends Endocrinol. Metab. 2013, 24, 506–514. [Google Scholar] [CrossRef]
- Knipe, R.S.; Tager, A.M.; Liao, J.K. The Rho kinases: Critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacol. Rev. 2015, 67, 103–117. [Google Scholar] [CrossRef]
- Sawada, N.; Liao, J.K. Rho/Rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid. Redox Signal. 2014, 20, 1251–1267. [Google Scholar] [CrossRef]
- Seko, T.; Ito, M.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Onishi, K.; Isaka, N.; Hartshorne, D.J.; Nakano, T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 2003, 92, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef]
- Mukai, Y.; Shimokawa, H.; Matoba, T.; Kandabashi, T.; Satoh, S.; Hiroki, J.; Kaibuchi, K.; Takeshita, A. Involvement of Rho-kinase in hypertensive vascular disease: A novel therapeutic target in hypertension. FASEB J. 2001, 15, 1062–1064. [Google Scholar]
- Kataoka, C.; Egashira, K.; Inoue, S.; Takemoto, M.; Ni, W.; Koyanagi, M.; Kitamoto, S.; Usui, M.; Kaibuchi, K.; Shimokawa, H.; et al. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension 2002, 39, 245–250. [Google Scholar] [CrossRef]
- Higashi, M.; Shimokawa, H.; Hattori, T.; Hiroki, J.; Mukai, Y.; Morikawa, K.; Ichiki, T.; Takahashi, S.; Takeshita, A. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: Effect on endothelial NAD(P)H oxidase system. Circ. Res. 2003, 93, 767–775. [Google Scholar] [CrossRef]
- Funakoshi, Y.; Ichiki, T.; Shimokawa, H.; Egashira, K.; Takeda, K.; Kaibuchi, K.; Takeya, M.; Yoshimura, T.; Takeshita, A. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension 2001, 38, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ichiki, T.; Tokunou, T.; Iino, N.; Fujii, S.; Kitabatake, A.; Shimokawa, H.; Takeshita, A. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, K.; Nakaki, T.; Marumo, T.; Hayashi, M.; Suzuki, H.; Kato, R.; Saruta, T. Pressure promotes DNA synthesis in rat cultured vascular smooth muscle cells. J. Clin. Investig. 1994, 93, 1975–1980. [Google Scholar] [CrossRef]
- Numaguchi, K.; Eguchi, S.; Yamakawa, T.; Motley, E.D.; Inagami, T. Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circ. Res. 1999, 85, 5–11. [Google Scholar] [CrossRef]
- Zeidan, A.; Nordstrom, I.; Albinsson, S.; Malmqvist, U.; Sward, K.; Hellstrand, P. Stretch-induced contractile differentiation of vascular smooth muscle: Sensitivity to actin polymerization inhibitors. Am. J. Physiol.-Cell Physiol. 2003, 284, C1387–C1396. [Google Scholar] [CrossRef]
- Castro, M.M.; Rizzi, E.; Figueiredo-Lopes, L.; Fernandes, K.; Bendhack, L.M.; Pitol, D.L.; Gerlach, R.F.; Tanus-Santos, J.E. Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis 2008, 198, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Prado, A.F.; Batista, R.I.M.; Tanus-Santos, J.E.; Gerlach, R.F. Matrix Metalloproteinases and Arterial Hypertension: Role of Oxidative Stress and Nitric Oxide in Vascular Functional and Structural Alterations. Biomolecules 2021, 11, 585. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.C.; Wynne, B.M.; Yu, L.; Tomilin, V.; Yue, Q.; Zhou, Y.; Al-Khalili, O.; Mallick, R.; Cai, H.; Alli, A.A.; et al. The sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC) associate. Biochem. J. 2016, 473, 3237–3252. [Google Scholar] [CrossRef] [PubMed]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia-Pathophysiology and Clinical Presentations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef] [PubMed]
- Burwick, R.M.; Rodriguez, M.H. Angiogenic Biomarkers in Preeclampsia. Obs. Gynecol. 2024, 143, 515–523. [Google Scholar] [CrossRef]
- Allerkamp, H.H.; Bondarenko, A.I.; Tawfik, I.; Kamali-Simsek, N.; Horvat Mercnik, M.; Madreiter-Sokolowski, C.T.; Wadsack, C. In vitro examination of Piezo1-TRPV4 dynamics: Implications for placental endothelial function in normal and preeclamptic pregnancies. Am. J. Physiol.-Cell Physiol. 2025, 328, C227–C244. [Google Scholar] [CrossRef]
- Qu, H.; Khalil, R.A. Vascular mechanisms and molecular targets in hypertensive pregnancy and preeclampsia. Am. J. Physiol.-Heart Circ. Physiol. 2020, 319, H661–H681. [Google Scholar] [CrossRef]
- Hosking, D.J. Calcium homeostasis in pregnancy. Clin. Endocrinol. 1996, 45, 1–6. [Google Scholar] [CrossRef]
- Chappell, L.C.; Cluver, C.A.; Kingdom, J.; Tong, S. Pre-eclampsia. Lancet 2021, 398, 341–354. [Google Scholar] [CrossRef]
- Wu, P.; Green, M.; Myers, J.E. Hypertensive disorders of pregnancy. BMJ 2023, 381, e071653. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Xiao, D.; Huang, X.; Longo, L.D.; Zhang, L. Chronic hypoxia increases pressure-dependent myogenic tone of the uterine artery in pregnant sheep: Role of ERK/PKC pathway. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H1840–H1849. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhu, R.; Zhang, L. Gestational hypoxia up-regulates protein kinase C and inhibits calcium-activated potassium channels in ovine uterine arteries. Int. J. Med. Sci. 2014, 11, 886–892. [Google Scholar] [CrossRef]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jupner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Investig. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Li, C.; Ansari, R.; Yu, Z.; Shah, D. Definitive molecular evidence of renin-angiotensin system in human uterine decidual cells. Hypertension 2000, 36, 159–164. [Google Scholar] [CrossRef]
- Shah, D.M. Role of the renin-angiotensin system in the pathogenesis of preeclampsia. Am. J. Physiol.-Ren. Physiol. 2005, 288, F614–F625. [Google Scholar] [CrossRef]
- Murphy, J.G.; Herrington, J.N.; Granger, J.P.; Khalil, R.A. Enhanced [Ca2+]i in renal arterial smooth muscle cells of pregnant rats with reduced uterine perfusion pressure. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H393–H403. [Google Scholar] [CrossRef]
- Hsu, C.H.; Huang, W.C.; Chang, W.T. Future Perspectives of Pulmonary Hypertension Treatment. Acta Cardiol. Sin. 2022, 38, 435–442. [Google Scholar] [PubMed]
- Dorris, S.L.; Peebles, R.S., Jr. PGI2 as a regulator of inflammatory diseases. Mediat. Inflamm. 2012, 2012, 926968. [Google Scholar] [CrossRef]
- Mouratoglou, S.A.; Giannakoulas, G.; Deftereos, S.; Giannopoulos, G.; Angelidis, C.; Cleman, M.W.; Vassilikos, V.P. Intra--and Intercellular Calcium Handling in Pulmonary Arterial Hypertension. Med. Chem. 2016, 12, 162–169. [Google Scholar] [CrossRef]
- Liao, J.; Lu, W.; Chen, Y.; Duan, X.; Zhang, C.; Luo, X.; Lin, Z.; Chen, J.; Liu, S.; Yan, H.; et al. Upregulation of Piezo1 (Piezo Type Mechanosensitive Ion Channel Component 1) Enhances the Intracellular Free Calcium in Pulmonary Arterial Smooth Muscle Cells From Idiopathic Pulmonary Arterial Hypertension Patients. Hypertension 2021, 77, 1974–1989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Babicheva, A.; Jain, P.P.; Rodriguez, M.; Ayon, R.J.; Ravellette, K.S.; Wu, L.; Balistrieri, F.; Tang, H.; et al. Endothelial upregulation of mechanosensitive channel Piezo1 in pulmonary hypertension. Am. J. Physiol.-Cell Physiol. 2021, 321, C1010–C1027. [Google Scholar] [CrossRef]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.P.; Quignard, J.F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef]
- Kuhr, F.K.; Smith, K.A.; Song, M.Y.; Levitan, I.; Yuan, J.X. New mechanisms of pulmonary arterial hypertension: Role of Ca2+ signaling. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H1546–H1562. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Ayon, R.J.; Tang, H.; Makino, A.; Yuan, J.X. Calcium-Sensing Receptor Regulates Cytosolic [Ca2+] and Plays a Major Role in the Development of Pulmonary Hypertension. Front. Physiol. 2016, 7, 517. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Wu, D.; Tian, L.; Xiong, P.Y.; Dunham-Snary, K.J.; Chen, K.H.; Alizadeh, E.; Motamed, M.; Potus, F.; Hindmarch, C.C.T.; et al. Mitochondria in the Pulmonary Vasculature in Health and Disease: Oxygen-Sensing, Metabolism, and Dynamics. Compr. Physiol. 2020, 10, 713–765. [Google Scholar] [CrossRef]
- He, R.L.; Wu, Z.J.; Liu, X.R.; Gui, L.X.; Wang, R.X.; Lin, M.J. Calcineurin/NFAT Signaling Modulates Pulmonary Artery Smooth Muscle Cell Proliferation, Migration and Apoptosis in Monocrotaline-Induced Pulmonary Arterial Hypertension Rats. Cell. Physiol. Biochem. 2018, 49, 172–189. [Google Scholar] [CrossRef]
- Wang, Y.; Patti, G.J. The Warburg effect: A signature of mitochondrial overload. Trends Cell Biol. 2023, 33, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, Y.H.; Gou, X.; Li, F.Y.; Yang, X.Y.; Li, Y.M.; Chen, F. Oxidative Stress and Antioxidative Therapy in Pulmonary Arterial Hypertension. Molecules 2022, 27, 3724. [Google Scholar] [CrossRef]
- Campen, M.J.; Shimoda, L.A.; O’Donnell, C.P. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J. Appl. Physiol. 2005, 99, 2028–2035. [Google Scholar] [CrossRef]
- Snow, J.B.; Gonzalez Bosc, L.V.; Kanagy, N.L.; Walker, B.R.; Resta, T.C. Role for PKCβ in enhanced endothelin-1-induced pulmonary vasoconstrictor reactivity following intermittent hypoxia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 301, L745–L754. [Google Scholar] [CrossRef] [PubMed]
- Allahdadi, K.J.; Duling, L.C.; Walker, B.R.; Kanagy, N.L. Eucapnic intermittent hypoxia augments endothelin-1 vasoconstriction in rats: Role of PKCδ. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H920–H927. [Google Scholar] [CrossRef]
- Snow, J.B.; Kitzis, V.; Norton, C.E.; Torres, S.N.; Johnson, K.D.; Kanagy, N.L.; Walker, B.R.; Resta, T.C. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J. Appl. Physiol. 2008, 104, 110–118. [Google Scholar] [CrossRef]
- Zhu, S.; White, R.E.; Barman, S.A. Role of phosphodiesterases in modulation of BKCa channels in hypertensive pulmonary arterial smooth muscle. Ther. Adv. Respir. Dis. 2008, 2, 119–127. [Google Scholar] [CrossRef]
- Dakshinamurti, S.; Mellow, L.; Stephens, N.L. Regulation of pulmonary arterial myosin phosphatase activity in neonatal circulatory transition and in hypoxic pulmonary hypertension: A role for CPI-17. Pediatr. Pulmonol. 2005, 40, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Sauzeau, V.; Rolli-Derkinderen, M.; Guerin, P.; Sagan, C.; Pacaud, P.; Loirand, G. Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br. J. Pharmacol. 2005, 146, 1010–1018. [Google Scholar] [CrossRef]
- Nagaoka, T.; Fagan, K.A.; Gebb, S.A.; Morris, K.G.; Suzuki, T.; Shimokawa, H.; McMurtry, I.F.; Oka, M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2005, 171, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Mandegar, M.; Fung, Y.C.; Huang, W.; Remillard, C.V.; Rubin, L.J.; Yuan, J.X. Cellular and molecular mechanisms of pulmonary vascular remodeling: Role in the development of pulmonary hypertension. Microvasc. Res. 2004, 68, 75–103. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Chronic hypoxia augments protein kinase G-mediated Ca2+ desensitization in pulmonary vascular smooth muscle through inhibition of RhoA/Rho kinase signaling. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L1220–L1229. [Google Scholar] [CrossRef]
- Fagan, K.A.; Oka, M.; Bauer, N.R.; Gebb, S.A.; Ivy, D.D.; Morris, K.G.; McMurtry, I.F. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L656–L664. [Google Scholar] [CrossRef]
- Nagaoka, T.; Morio, Y.; Casanova, N.; Bauer, N.; Gebb, S.; McMurtry, I.; Oka, M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L665–L672. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Shimokawa, H.; Morikawa, K.; Uwatoku, T.; Oi, K.; Matsumoto, Y.; Hattori, T.; Nakashima, Y.; Kaibuchi, K.; Sueishi, K.; et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ. Res. 2004, 94, 385–393. [Google Scholar] [CrossRef]
- Harraz, O.F.; Jensen, L.J. Vascular calcium signalling and ageing. J. Physiol. 2021, 599, 5361–5377. [Google Scholar] [CrossRef]
- Lee, H.Y.; Zeeshan, H.M.A.; Kim, H.R.; Chae, H.J. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 2017, 113, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Chirkov, Y.Y.; Nguyen, T.H.; Horowitz, J.D. Impairment of Anti-Aggregatory Responses to Nitric Oxide and Prostacyclin: Mechanisms and Clinical Implications in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 1042. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- Kassmann, M.; Fan, G.; Gollasch, M. Arterial elementary calcium signaling in aging. Aging 2020, 12, 24476–24478. [Google Scholar] [CrossRef] [PubMed]
- Palomo, I.; Wehinger, S.; Andres, V.; Garcia-Garcia, F.J.; Fuentes, E. RhoA/rho kinase pathway activation in age-associated endothelial cell dysfunction and thrombosis. J. Cell. Mol. Med. 2024, 28, e18153. [Google Scholar] [CrossRef]
- Meschiari, C.A.; Ero, O.K.; Pan, H.; Finkel, T.; Lindsey, M.L. The impact of aging on cardiac extracellular matrix. Geroscience 2017, 39, 7–18. [Google Scholar] [CrossRef]
- Freitas-Rodriguez, S.; Folgueras, A.R.; Lopez-Otin, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Maurer, M.S.; Roh, J. Aging in Heart Failure: Embracing Biology Over Chronology: JACC Family Series. JACC Heart Fail. 2024, 12, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The role of cellular senescence in cardiac disease: Basic biology and clinical relevance. Nat. Rev. Cardiol. 2022, 19, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tapa, S.; Francis Stuart, S.D.; Wang, L.; Bossuyt, J.; Delisle, B.P.; Ripplinger, C.M. Aging Disrupts Normal Time-of-Day Variation in Cardiac Electrophysiology. Circ. Arrhythm. Electrophysiol. 2020, 13, e008093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transduct. Target. Ther. 2023, 8, 239. [Google Scholar] [CrossRef]
- Bautista-Nino, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.; Roks, A.J. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef]
- Thompson, J.; Maceyka, M.; Chen, Q. Targeting ER stress and calpain activation to reverse age-dependent mitochondrial damage in the heart. Mech. Ageing Dev. 2020, 192, 111380. [Google Scholar] [CrossRef]
- Chen, Q.; Thompson, J.; Hu, Y.; Lesnefsky, E.J. Reversing mitochondrial defects in aged hearts: Role of mitochondrial calpain activation. Am. J. Physiol.-Cell Physiol. 2022, 322, C296–C310. [Google Scholar] [CrossRef]
- Yan, B.; Yu, X.; Cai, X.; Huang, X.; Xie, B.; Lian, D.; Chen, J.; Li, W.; Lin, Y.; Ye, J.; et al. A Review: The Significance of Toll-Like Receptors 2 and 4, and NF-κB Signaling in Endothelial Cells during Atherosclerosis. Front. Biosci. 2024, 29, 161. [Google Scholar] [CrossRef]
- Cheng, W.; Cui, C.; Liu, G.; Ye, C.; Shao, F.; Bagchi, A.K.; Mehta, J.L.; Wang, X. NF-κB, A Potential Therapeutic Target in Cardiovascular Diseases. Cardiovasc. Drugs Ther. 2023, 37, 571–584. [Google Scholar] [CrossRef]
- Olsen, M.B.; Gregersen, I.; Sandanger, O.; Yang, K.; Sokolova, M.; Halvorsen, B.E.; Gullestad, L.; Broch, K.; Aukrust, P.; Louwe, M.C. Targeting the Inflammasome in Cardiovascular Disease. JACC Basic Transl. Sci. 2021, 7, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Doran, A.C. Inflammation Resolution: Implications for Atherosclerosis. Circ. Res. 2022, 130, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Gollentz, C.; Chinetti, G. Macrophage death in atherosclerosis: Potential role in calcification. Front. Immunol. 2023, 14, 1215612. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C. Oxidation of LDL, atherogenesis, and apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 698–709. [Google Scholar] [CrossRef]
- Bostrom, K.I. DNA Damage Response, Runx2 (Runt-Related Transcription Factor 2), and Vascular Calcification. Arter. Thromb. Vasc. Biol. 2021, 41, 1358–1359. [Google Scholar] [CrossRef]
- Frostegard, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Burgmaier, M.; Willems, B.; Heeneman, S.; Stadler, N.; Gremse, F.; Reith, S.; Burgmaier, K.; Kahles, F.; Marx, N.; et al. Nicotine promotes vascular calcification via intracellular Ca2+-mediated, Nox5-induced oxidative stress, and extracellular vesicle release in vascular smooth muscle cells. Cardiovasc. Res. 2022, 118, 2196–2210. [Google Scholar] [CrossRef]
- Mulangala, J.; Akers, E.J.; Solly, E.L.; Bamhare, P.M.; Wilsdon, L.A.; Wong, N.K.P.; Tan, J.T.M.; Bursill, C.A.; Nicholls, S.J.; Di Bartolo, B.A. Pro-Calcific Environment Impairs Ischaemia-Driven Angiogenesis. Int. J. Mol. Sci. 2022, 23, 3363. [Google Scholar] [CrossRef]
- Laera, N.; Malerba, P.; Vacanti, G.; Nardin, S.; Pagnesi, M.; Nardin, M. Impact of Immunity on Coronary Artery Disease: An Updated Pathogenic Interplay and Potential Therapeutic Strategies. Life 2023, 13, 2128. [Google Scholar] [CrossRef]
- Dokumacioglu, E.; Duzcan, I.; Iskender, H.; Sahin, A. RhoA/ROCK-1 Signaling Pathway and Oxidative Stress in Coronary Artery Disease Patients. Braz. J. Cardiovasc. Surg. 2022, 37, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Hearon, C.M., Jr.; Dinenno, F.A. Escape, lysis, and feedback: Endothelial modulation of sympathetic vasoconstriction. Curr. Opin. Pharmacol. 2019, 45, 81–86. [Google Scholar] [CrossRef]
- Oparil, S.; Schmieder, R.E. New approaches in the treatment of hypertension. Circ. Res. 2015, 116, 1074–1095. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef]
- Roffey, J.; Rosse, C.; Linch, M.; Hibbert, A.; McDonald, N.Q.; Parker, P.J. Protein kinase C intervention: The state of play. Curr. Opin. Cell Biol. 2009, 21, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Dodson, P.M. PKC inhibition and diabetic microvascular complications. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 573–586. [Google Scholar] [CrossRef]
- House, C.; Kemp, B.E. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science 1987, 238, 1726–1728. [Google Scholar] [CrossRef]
- Eichholtz, T.; de Bont, D.B.; de Widt, J.; Liskamp, R.M.; Ploegh, H.L. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J. Biol. Chem. 1993, 268, 1982–1986. [Google Scholar] [CrossRef] [PubMed]
- Bogard, A.S.; Tavalin, S.J. Protein Kinase C (PKC)ζ Pseudosubstrate Inhibitor Peptide Promiscuously Binds PKC Family Isoforms and Disrupts Conventional PKC Targeting and Translocation. Mol. Pharmacol. 2015, 88, 728–735. [Google Scholar] [CrossRef]
- Churchill, E.N.; Qvit, N.; Mochly-Rosen, D. Rationally designed peptide regulators of protein kinase C. Trends Endocrinol. Metab. 2009, 20, 25–33. [Google Scholar] [CrossRef]
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calo, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular-Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328. [Google Scholar] [CrossRef]
- Budzyn, K.; Paull, M.; Marley, P.D.; Sobey, C.G. Segmental differences in the roles of rho-kinase and protein kinase C in mediating vasoconstriction. J. Pharmacol. Exp. Ther. 2006, 317, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Kandabashi, T.; Shimokawa, H.; Miyata, K.; Kunihiro, I.; Eto, Y.; Morishige, K.; Matsumoto, Y.; Obara, K.; Nakayama, K.; Takahashi, S.; et al. Evidence for protein kinase C-mediated activation of Rho-kinase in a porcine model of coronary artery spasm. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2209–2214. [Google Scholar] [CrossRef]
- Noma, K.; Goto, C.; Nishioka, K.; Jitsuiki, D.; Umemura, T.; Ueda, K.; Kimura, M.; Nakagawa, K.; Oshima, T.; Chayama, K.; et al. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J. Am. Coll. Cardiol. 2007, 49, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 99, 1426–1432. [Google Scholar] [CrossRef]
- Kishi, T.; Hirooka, Y.; Masumoto, A.; Ito, K.; Kimura, Y.; Inokuchi, K.; Tagawa, T.; Shimokawa, H.; Takeshita, A.; Sunagawa, K. Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation 2005, 111, 2741–2747. [Google Scholar] [CrossRef]
- Jacobs, M.; Hayakawa, K.; Swenson, L.; Bellon, S.; Fleming, M.; Taslimi, P.; Doran, J. The structure of dimeric ROCK I reveals the mechanism for ligand selectivity. J. Biol. Chem. 2006, 281, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.R.; Venitz, J.; Figg, W.D.; Sparreboom, A. Chemically modified tetracyclines as inhibitors of matrix metalloproteinases. Drug Resist. Updates 2004, 7, 195–208. [Google Scholar] [CrossRef]
- Renkiewicz, R.; Qiu, L.; Lesch, C.; Sun, X.; Devalaraja, R.; Cody, T.; Kaldjian, E.; Welgus, H.; Baragi, V. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003, 48, 1742–1749. [Google Scholar] [CrossRef]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef] [PubMed]
- Singer, H.A. Ca2+/calmodulin-dependent protein kinase II function in vascular remodelling. J. Physiol. 2012, 590, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Katsuki, S.; Ohashi, M.; Miyagawa, Y.; Tanaka, Y.; Takeya, K.; Kitazawa, T. Possible roles of N- and C-terminal unstructured tails of CPI-17 in regulating Ca2+ sensitization force of smooth muscle. J. Smooth Muscle Res. 2022, 58, 22–33. [Google Scholar] [CrossRef]
- Porras-Gonzalez, C.; Ordonez, A.; Castellano, A.; Urena, J. Regulation of RhoA/ROCK and sustained arterial contraction by low cytosolic Ca2+ levels during prolonged depolarization of arterial smooth muscle. Vasc. Pharmacol. 2017, 93–95, 33–41. [Google Scholar] [CrossRef]
- Morgado, M.; Cairrao, E.; Santos-Silva, A.J.; Verde, I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell. Mol. Life Sci. 2012, 69, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Balistrieri, A.; Makino, A.; Yuan, J.X. Pathophysiology and pathogenic mechanisms of pulmonary hypertension: Role of membrane receptors, ion channels, and Ca2+ signaling. Physiol. Rev. 2023, 103, 1827–1897. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef]
- Adachi, T. Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. Adv. Pharmacol. 2010, 59, 165–195. [Google Scholar]
- Crews, J.K.; Murphy, J.G.; Khalil, R.A. Gender differences in Ca2+ entry mechanisms of vasoconstriction in Wistar-Kyoto and spontaneously hypertensive rats. Hypertension 1999, 34, 931–936. [Google Scholar] [CrossRef]
- Sueta, D.; Tabata, N.; Hokimoto, S. Clinical roles of calcium channel blockers in ischemic heart diseases. Hypertens. Res. 2017, 40, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Takuwa, Y.; Johshita, H.; Yamashita, K.; Asano, T. Possible role of protein kinase C-dependent smooth muscle contraction in the pathogenesis of chronic cerebral vasospasm. J. Cereb. Blood Flow Metab. 1991, 11, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Shimokawa, H.; Nakaike, R.; Fukai, T.; Sakata, M.; Takayanagi, T.; Egashira, K.; Takeshita, A. Role of protein kinase C-mediated pathway in the pathogenesis of coronary artery spasm in a swine model. Circulation 1994, 90, 2425–2431. [Google Scholar] [CrossRef] [PubMed]
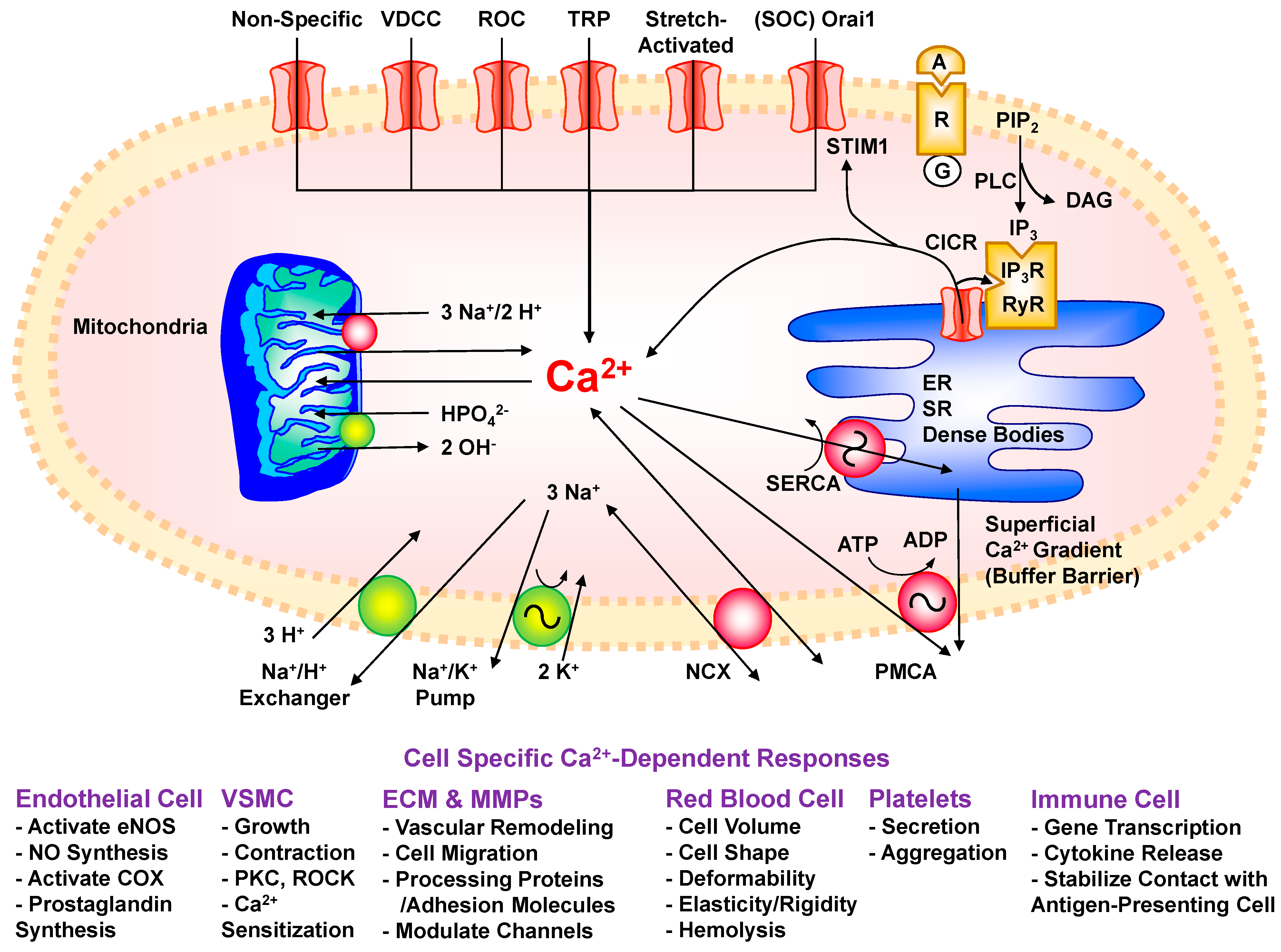





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, C.; Khalil, R.A. Calcium Signaling Dynamics in Vascular Cells and Their Dysregulation in Vascular Disease. Biomolecules 2025, 15, 892. https://doi.org/10.3390/biom15060892
Dai C, Khalil RA. Calcium Signaling Dynamics in Vascular Cells and Their Dysregulation in Vascular Disease. Biomolecules. 2025; 15(6):892. https://doi.org/10.3390/biom15060892
Chicago/Turabian StyleDai, Chang, and Raouf A. Khalil. 2025. "Calcium Signaling Dynamics in Vascular Cells and Their Dysregulation in Vascular Disease" Biomolecules 15, no. 6: 892. https://doi.org/10.3390/biom15060892
APA StyleDai, C., & Khalil, R. A. (2025). Calcium Signaling Dynamics in Vascular Cells and Their Dysregulation in Vascular Disease. Biomolecules, 15(6), 892. https://doi.org/10.3390/biom15060892