


The Role and Pathogenesis of Tau Protein in Alzheimer’s Disease
 ,
, 
Abstract
1. Introduction
2. Search Strategy
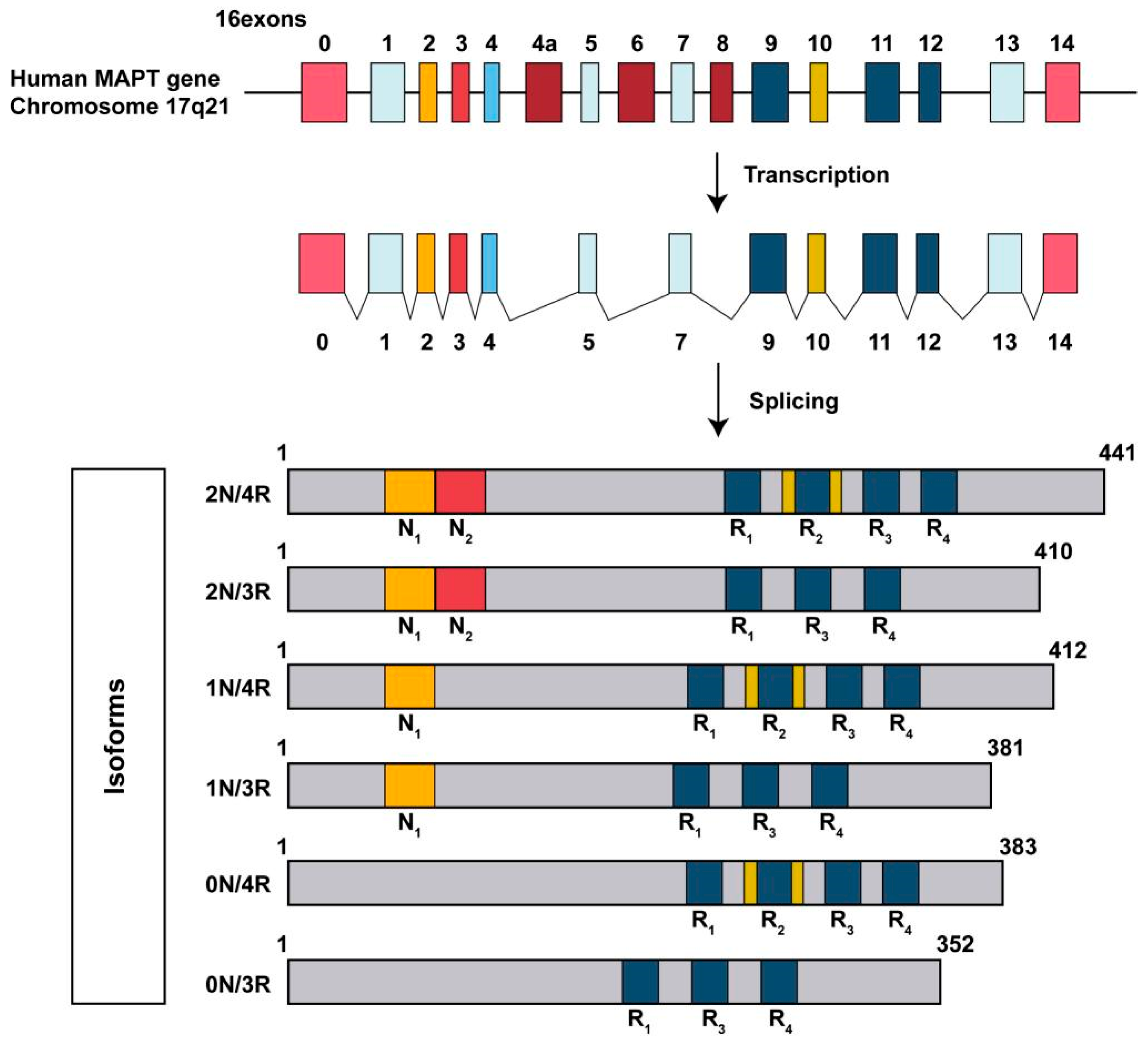
3. Basic Structure of Tau Protein
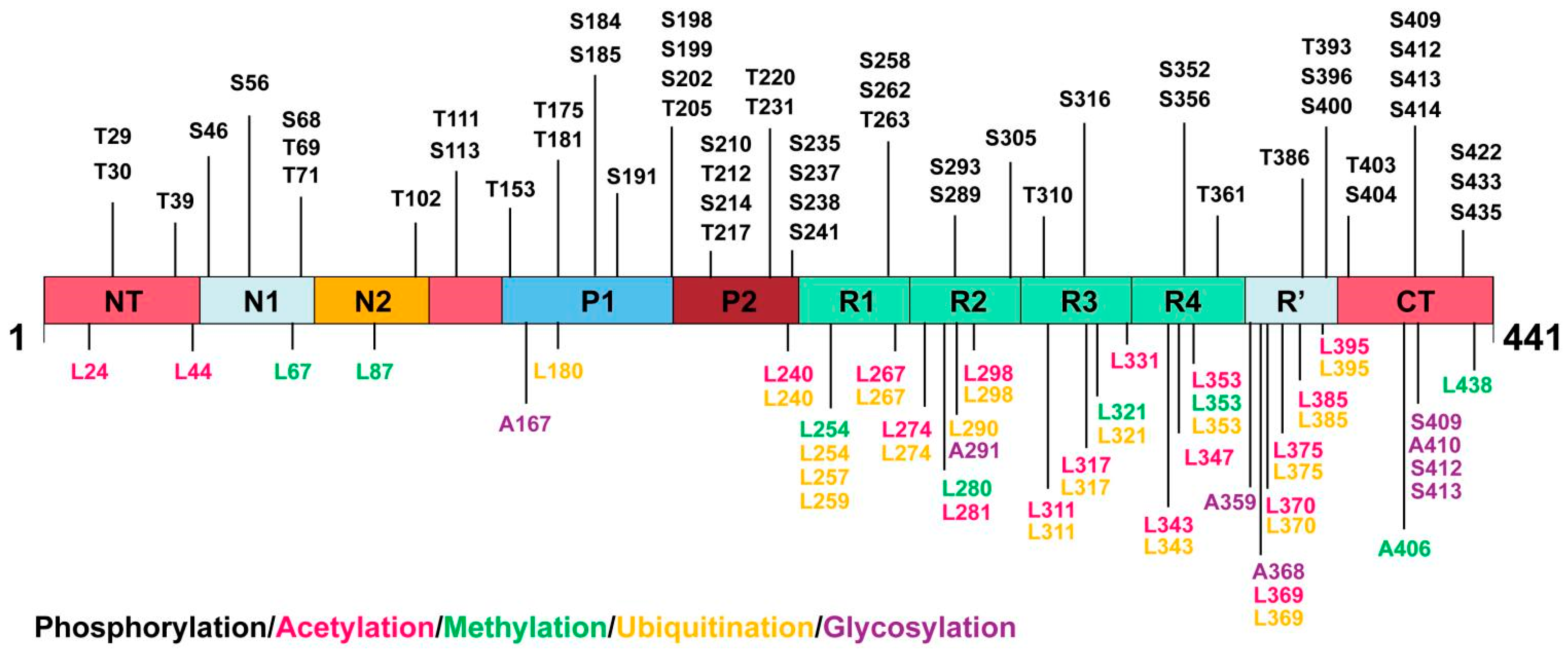
4. PTMs of Tau Protein
4.1. Tau Phosphorylation
4.2. Tau Acetylation
4.3. Tau Methylation
4.4. Tau Ubiquitination
4.5. Tau Glycosylation
4.6. Tau Truncation
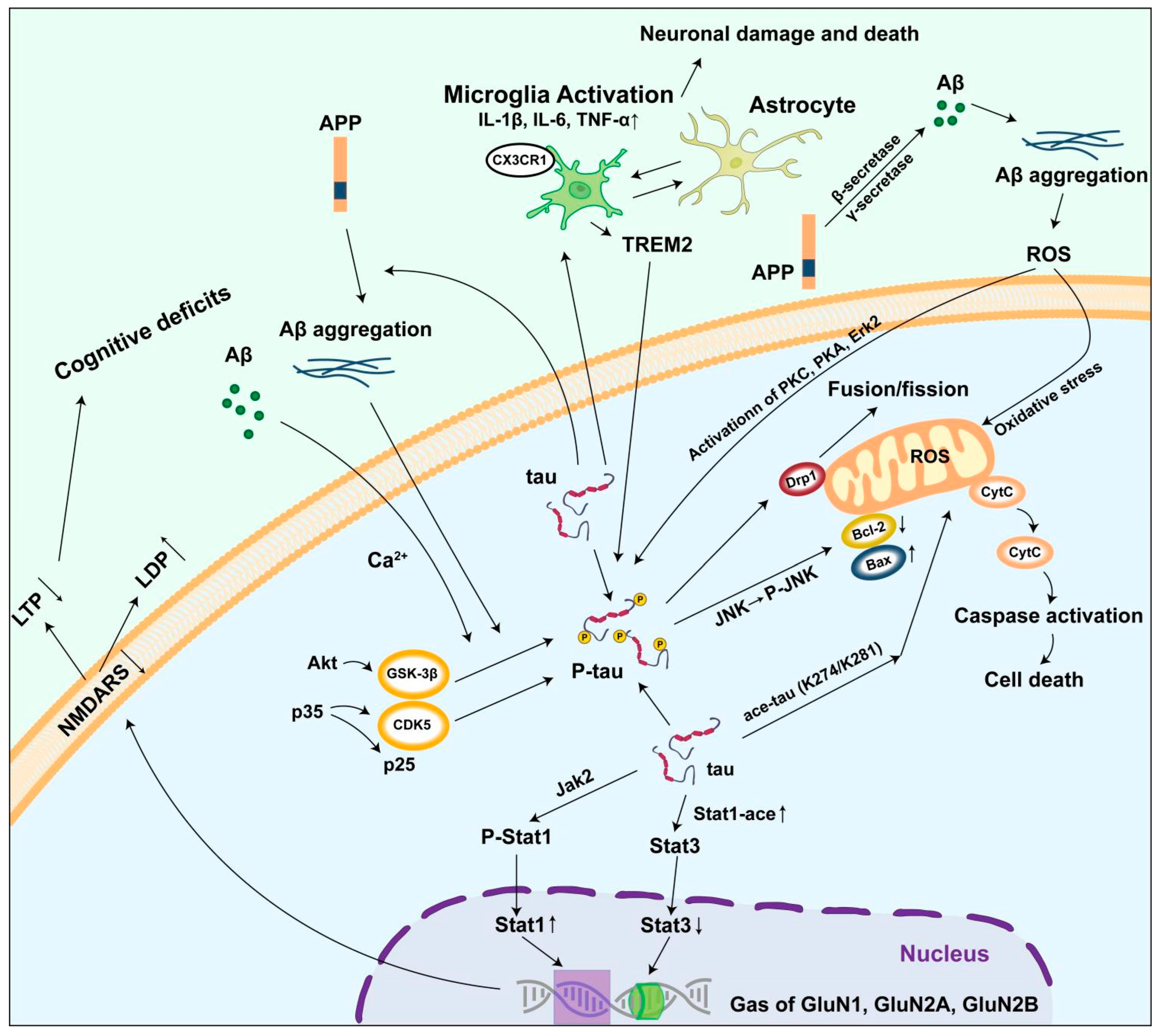
5. Mechanism of Abnormal Tau Protein Accumulation Triggering AD
5.1. Damage to the Neural Microtubule System
5.2. Inducing Mitochondrial Damage
5.3. Causing Synaptic Dysfunction
5.4. Activating the Neuroinflammatory Response
5.5. Interaction with β-Amyloid Protein
6. Conclusions and Future Perspectives
7. Reflexivity Statement
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | amyloid beta |
| PTMs | post-translational modifications |
| MAPT | Microtubule-associated protein tau |
| CNS | central nervous system |
| PDPKs | proline-directed protein kinases |
| non-PDPKs | non-proline-directed protein kinases |
| PTKs | protein tyrosine kinases |
| GSK-3β | Glycogen synthetase kinase-3β |
| CDK5 | cyclin-dependent kinase 5 |
| p38 MAPK | p38 mitogen-activated protein kinase |
| JNK | c-Jun kinase family |
| CaMKII | Calmodulin-dependent protein kinase II |
| PKA | protein kinase A |
| Pyk2 | proline-rich tyrosine kinase 2 |
| PCAF | p300/CBP-associated factor |
| p300/CBP | p300/CREB-binding protein |
| HDACs | histone deacetylases |
| SIRT1 | sirtuin 1 |
| SIRT2 | sirtuin 2 |
| MBD | microtubule-binding domain |
| PHFs | paired helical filaments |
| UPS | ubiquitin proteasome system |
| ALP | autophagy lysosome pathway |
| TRAF6 | tumor necrosis factor receptor-associated factor 6 |
| NEDD4 | neural precursor cell expressed, developmentally downregulated 4 |
| AEP | asparagine endopeptidase |
| ROS | reactive oxygen species |
| PSD-95 | postsynaptic density protein 95 |
| TNF-α | tumor necrosis factor-alpha |
| IL-1β | interleukin-1 beta |
| LTP | long-term potentiation |
| LTD | long-term depression |
| NMDAR | N-methyl-D-aspartate receptor |
| STAT | signal transducer and activator of transcription |
| CSF | cerebrospinal fluid |
| PGE2 | prostaglandin E2 |
| APP | amyloid precursor protein |
References
- Guerreiro, R.J.; Schymick, J.C.; Crews, C.; Singleton, A.; Hardy, J.; Traynor, B.J. TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS ONE 2008, 3, e2450. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 173–185. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Opare, S.K.; Xu, X.; Ganesan, A.; Rao, P.P.N. Post-Translational Modifications in Tau and Their Roles in Alzheimer’s Pathology. Curr. Alzheimer Res. 2024, 21, 24–49. [Google Scholar] [CrossRef] [PubMed]
- Himmler, A. Structure of the bovine tau gene: Alternatively spliced transcripts generate a protein family. Mol. Cell. Biol. 1989, 9, 1389–1396. [Google Scholar] [CrossRef]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W., Jr. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar] [CrossRef]
- O’Conner, S.E.; Imperiali, B. A molecular basis for glycosylation-induced conformational switching. Chem. Biol. 1998, 5, 427–437. [Google Scholar] [CrossRef]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human tau gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Ahmad, W.D.; Shabbiri, K.D.; Ahmad, I. Prediction of human tau 3D structure, and interplay between O-β-GlcNAc and phosphorylation modifications in Alzheimer’s disease: C. elegans as a suitable model to study these interactions in vivo. Biochem. Biophys. Res. Commun. 2020, 528, 466–472. [Google Scholar] [CrossRef]
- Nuzzo, T.; Feligioni, M.; Cristino, L.; Pagano, I.; Marcelli, S.; Iannuzzi, F.; Imperatore, R.; D’Angelo, L.; Petrella, C.; Carella, M.; et al. Free d-aspartate triggers NMDA receptor-dependent cell death in primary cortical neurons and perturbs JNK activation, Tau phosphorylation, and protein SUMOylation in the cerebral cortex of mice lacking d-aspartate oxidase activity. Exp. Neurol. 2019, 317, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, C.; Sarad, N.; DeCrumpe, A.; Goswami, D.; Herrmann, S.; Morales, J.; Patel, P.; Osborne, J. Biomarkers in the Diagnosis and Prognosis of Alzheimer’s Disease. J. Lab. Autom. 2015, 20, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhang, L.; Su, Y.; Liang, X.; Yang, J.; Luo, Q.; Luo, H. Sensitive detection of multiple blood biomarkers via immunomagnetic exosomal PCR for the diagnosis of Alzheimer’s disease. Sci. Adv. 2024, 10, eabm3088. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain A J. Neurol. 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Drepper, F.; Biernat, J.; Kaniyappan, S.; Meyer, H.E.; Mandelkow, E.M.; Warscheid, B.; Mandelkow, E. A combinatorial native MS and LC-MS/MS approach reveals high intrinsic phosphorylation of human Tau but minimal levels of other key modifications. J. Biol. Chem. 2020, 295, 18213–18225. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Han, F.; Cai, L.; Wang, L.; Niu, T.; Zhai, J.; Ji, B.; Gao, J.; Wang, J. Phosphorylation at Ser289 Enhances the Oligomerization of Tau Repeat R2. J. Chem. Inf. Model. 2023, 63, 1351–1361. [Google Scholar] [CrossRef]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Cohen, P.; Vanmechelen, E.; Vandermeeren, M.; Cras, P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: Identification of phosphorylation sites in tau protein. Biochem. J. 1994, 301 Pt 3, 871–877. [Google Scholar] [CrossRef]
- Yu, Y.; Run, X.; Liang, Z.; Li, Y.; Liu, F.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 2009, 108, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Barros-Miñones, L.; Martín-de-Saavedra, D.; Perez-Alvarez, S.; Orejana, L.; Suquía, V.; Goñi-Allo, B.; Hervias, I.; López, M.G.; Jordan, J.; Aguirre, N.; et al. Inhibition of calpain-regulated p35/cdk5 plays a central role in sildenafil-induced protection against chemical hypoxia produced by malonate. Biochim. Biophys. Acta 2013, 1832, 705–717. [Google Scholar] [CrossRef]
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. MMBR 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Benítez, M.J.; Cuadros, R.; Jiménez, J.S. Phosphorylation and Dephosphorylation of Tau Protein by the Catalytic Subunit of PKA, as Probed by Electrophoretic Mobility Retard. J. Alzheimer’s Dis. JAD 2021, 79, 1143–1156. [Google Scholar] [CrossRef]
- Bennecib, M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett. 2001, 490, 15–22. [Google Scholar] [CrossRef]
- Gong, C.X.; Lidsky, T.; Wegiel, J.; Zuck, L.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J. Biol. Chem. 2000, 275, 5535–5544. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Wegiel, J.; Gong, C.X. Decrease of protein phosphatase 2A and its association with accumulation and hyperphosphorylation of tau in Down syndrome. J. Alzheimer’s Dis. JAD 2008, 13, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Yin, X.; Hu, W.; Shi, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X.; Liu, F. Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 23, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Gong, C.X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3β-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef]
- Foster, K.; Manca, M.; McClure, K.; Koivula, P.; Trojanowski, J.Q.; Havas, D.; Chancellor, S.; Goldstein, L.; Brunden, K.R.; Kraus, A.; et al. Preclinical characterization and IND-enabling safety studies for PNT001, an antibody that recognizes cis-pT231 tau. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023, 19, 4662–4674. [Google Scholar] [CrossRef]
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R.; et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 2016, 90, 245–260. [Google Scholar] [CrossRef]
- Bai, H.; Wu, Y.; Li, H.; Zhu, Y.; Che, R.; Wang, F.; Zhang, C. Cerebral neurotoxicity of amino-modified polystyrene nanoplastics in mice and the protective effects of functional food Camellia pollen. Sci. Total Environ. 2024, 912, 169511. [Google Scholar] [CrossRef]
- Yang, J.; Zhi, W.; Wang, L. Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review. Molecules 2024, 29, 2812. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Shin, M.K.; Vázquez-Rosa, E.; Koh, Y.; Dhar, M.; Chaubey, K.; Cintrón-Pérez, C.J.; Barker, S.; Miller, E.; Franke, K.; Noterman, M.F.; et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021, 184, 2715–2732.E23. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef]
- Cohen, T.J.; Constance, B.H.; Hwang, A.W.; James, M.; Yuan, C.X. Intrinsic Tau Acetylation Is Coupled to Auto-Proteolytic Tau Fragmentation. PLoS ONE 2016, 11, e0158470. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.Y.; Wan, H.L.; Li, T.; Zhang, B.G.; Li, X.G.; Wang, X.; Li, X.; Liu, Q.; Chen, C.Y.; Yang, Y.; et al. STAT3 ameliorates cognitive deficits by positively regulating the expression of NMDARs in a mouse model of FTDP-17. Signal Transduct. Target. Ther. 2020, 5, 295. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.A.; Gestwicki, J.E.; Gan, L. SIRT1 Deacetylates Tau and Reduces Pathogenic Tau Spread in a Mouse Model of Tauopathy. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yan, Z.; Zhou, T.; Wang, G. SIRT1 Regulates Cognitive Performance and Ability of Learning and Memory in Diabetic and Nondiabetic Models. J. Diabetes Res. 2017, 2017, 7121827. [Google Scholar] [CrossRef]
- Silva, D.F.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondria: The common upstream driver of amyloid-β and tau pathology in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 563–572. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Dou, H.; Wang, S.; Qu, D.; Peng, X.; Zou, N.; Yang, L. Caffeine improves mitochondrial dysfunction in the white matter of neonatal rats with hypoxia-ischemia through deacetylation: A proteomic analysis of lysine acetylation. Front. Mol. Neurosci. 2024, 17, 1394886. [Google Scholar] [CrossRef]
- Yin, T.C.; Britt, J.K.; De Jesús-Cortés, H.; Lu, Y.; Genova, R.M.; Khan, M.Z.; Voorhees, J.R.; Shao, J.; Katzman, A.C.; Huntington, P.J.; et al. P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep. 2014, 8, 1731–1740. [Google Scholar] [CrossRef]
- Song, H.L.; Kim, N.Y.; Park, J.; Kim, M.I.; Jeon, Y.N.; Lee, S.J.; Cho, K.; Shim, Y.L.; Lee, K.H.; Mun, Y.S.; et al. Monoclonal antibody Y01 prevents tauopathy progression induced by lysine 280-acetylated tau in cell and mouse models. J. Clin. Investig. 2023, 133, e156537. [Google Scholar] [CrossRef]
- Didonna, A.; Cantó, E.; Shams, H.; Isobe, N.; Zhao, C.; Caillier, S.J.; Condello, C.; Yamate-Morgan, H.; Tiwari-Woodruff, S.K.; Mofrad, M.R.K.; et al. Sex-specific Tau methylation patterns and synaptic transcriptional alterations are associated with neural vulnerability during chronic neuroinflammation. J. Autoimmun. 2019, 101, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Balmik, A.A.; Chinnathambi, S. Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun. Signal. CCS 2021, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Huseby, C.J.; Hoffman, C.N.; Cooper, G.L.; Cocuron, J.C.; Alonso, A.P.; Thomas, S.N.; Yang, A.J.; Kuret, J. Quantification of Tau Protein Lysine Methylation in Aging and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2019, 71, 979–991. [Google Scholar] [CrossRef]
- Bichmann, M.; Prat Oriol, N.; Ercan-Herbst, E.; Schöndorf, D.C.; Gomez Ramos, B.; Schwärzler, V.; Neu, M.; Schlüter, A.; Wang, X.; Jin, L.; et al. SETD7-mediated monomethylation is enriched on soluble Tau in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 46. [Google Scholar] [CrossRef]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Thomas, S.N.; Funk, K.E.; Wan, Y.; Liao, Z.; Davies, P.; Kuret, J.; Yang, A.J. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: A mass spectrometry approach. Acta Neuropathol. 2012, 123, 105–117. [Google Scholar] [CrossRef]
- Zhou, X.W.; Gustafsson, J.A.; Tanila, H.; Bjorkdahl, C.; Liu, R.; Winblad, B.; Pei, J.J. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol. Dis. 2008, 31, 386–394. [Google Scholar] [CrossRef]
- Reinecke, J.B.; DeVos, S.L.; McGrath, J.P.; Shepard, A.M.; Goncharoff, D.K.; Tait, D.N.; Fleming, S.R.; Vincent, M.P.; Steinhilb, M.L. Implicating calpain in tau-mediated toxicity in vivo. PLoS ONE 2011, 6, e23865. [Google Scholar] [CrossRef]
- Park, S.Y.; Ferreira, A. The generation of a 17 kDa neurotoxic fragment: An alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef]
- Carroll, E.C.; Marqusee, S. Site-specific ubiquitination: Deconstructing the degradation tag. Curr. Opin. Struct. Biol. 2022, 73, 102345. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Liu, J.; Wei, W.; Rezaeian, A.H. PROTACs technology for treatment of Alzheimer’s disease: Advances and perspectives. Acta Mater. Medica 2022, 1, 24–41. [Google Scholar] [CrossRef]
- Reits, E.A.; Benham, A.M.; Plougastel, B.; Neefjes, J.; Trowsdale, J. Dynamics of proteasome distribution in living cells. EMBO J. 1997, 16, 6087–6094. [Google Scholar] [CrossRef] [PubMed]
- von Mikecz, A.; Chen, M.; Rockel, T.; Scharf, A. The nuclear ubiquitin-proteasome system: Visualization of proteasomes, protein aggregates, and proteolysis in the cell nucleus. Methods Mol. Biol. 2008, 463, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.D.; Jasem, S.; Licchesi, J.D.F. The Ubiquitin System in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2020, 1233, 195–221. [Google Scholar] [CrossRef]
- Maruyama, S.; Hatakeyama, S.; Nakayama, K.; Ishida, N.; Kawakami, K.; Nakayama, K. Characterization of a mouse gene (Fbxw6) that encodes a homologue of Caenorhabditis elegans SEL-10. Genomics 2001, 78, 214–222. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, L.; Wu, R.; Wu, L.; Zhang, B.; Wang, J.Z.; Liu, R.; Liu, F.; Wang, J.; Wang, X. c-Src regulates δ-secretase activation and truncated Tau production by phosphorylating the E3 ligase Traf6. J. Biol. Chem. 2023, 299, 105462. [Google Scholar] [CrossRef]
- Oliveros, G.; Wallace, C.H.; Chaudry, O.; Liu, Q.; Qiu, Y.; Xie, L.; Rockwell, P.; Figueiredo-Pereira, M.E.; Serrano, P.A. Repurposing ibudilast to mitigate Alzheimer’s disease by targeting inflammation. Brain A J. Neurol. 2023, 146, 898–911. [Google Scholar] [CrossRef]
- Attali, I.; Tobelaim, W.S.; Persaud, A.; Motamedchaboki, K.; Simpson-Lavy, K.J.; Mashahreh, B.; Levin-Kravets, O.; Keren-Kaplan, T.; Pilzer, I.; Kupiec, M.; et al. Ubiquitylation-dependent oligomerization regulates activity of Nedd4 ligases. EMBO J. 2017, 36, 425–440. [Google Scholar] [CrossRef]
- Miller, L.V.C.; Papa, G.; Vaysburd, M.; Cheng, S.; Sweeney, P.W.; Smith, A.; Franco, C.; Katsinelos, T.; Huang, M.; Sanford, S.A.I.; et al. Co-opting templated aggregation to degrade pathogenic tau assemblies and improve motor function. Cell 2024, 187, 5967–5980.e5917. [Google Scholar] [CrossRef] [PubMed]
- Parolini, F.; Ataie Kachoie, E.; Leo, G.; Civiero, L.; Bubacco, L.; Arrigoni, G.; Munari, F.; Assfalg, M.; D’Onofrio, M.; Capaldi, S. Site-Specific Ubiquitination of Tau Amyloids Promoted by the E3 Ligase CHIP. Angew. Chem. 2023, 62, e202310230. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Fan, J.; Ji, C.; Wang, Z.; Ge, X.; Wang, J.; Ye, W.; Yin, G.; Cai, W.; Liu, W. USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ. 2022, 29, 1164–1175. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, X.; Chaput, D.; Shin, M.K.; Koh, Y.; Gan, L.; Pieper, A.A.; Woo, J.A.; Kang, D.E. X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 2022, 185, 3913–3930.E19. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.; Zhao, Y.; Zhang, Y.; Liu, Y.; Ma, F.; Wang, X.; Fu, J. Andrographolide ameliorates neuroinflammation in APP/PS1 transgenic mice. Int. Immunopharmacol. 2021, 96, 107808. [Google Scholar] [CrossRef]
- O’Connor, S.E.; Imperiali, B. Modulation of protein structure and function by asparagine-linked glycosylation. Chem. Biol. 1996, 3, 803–812. [Google Scholar] [CrossRef]
- Jayaprakash, N.G.; Surolia, A. Role of glycosylation in nucleating protein folding and stability. Biochem. J. 2017, 474, 2333–2347. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-glycans of pathological tau: Possible occurrence of aberrant processing of tau in Alzheimer’s disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Glycosylation of microtubule-associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat. Med. 1996, 2, 871–875. [Google Scholar] [CrossRef]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diagnostic and therapeutic approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef]
- Tapia-Monsalves, C.; Olesen, M.A.; Villavicencio-Tejo, F.; Quintanilla, R.A. Cyclosporine A (CsA) prevents synaptic impairment caused by truncated tau by caspase-3. Mol. Cell. Neurosci. 2023, 125, 103861. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, O.Y.; Glushakov, A.O.; Borlongan, C.V.; Valadka, A.B.; Hayes, R.L.; Glushakov, A.V. Role of Caspase-3-Mediated Apoptosis in Chronic Caspase-3-Cleaved Tau Accumulation and Blood-Brain Barrier Damage in the Corpus Callosum after Traumatic Brain Injury in Rats. J. Neurotrauma 2018, 35, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.A.; Villavicencio-Tejo, F.; Johnson, G.V.W.; Porter, G.A.; Quintanilla, R.A. Cyclophilin D (CypD) ablation prevents neurodegeneration and cognitive damage induced by caspase-3 cleaved tau. Free Radic. Biol. Med. 2025, 232, 128–141. [Google Scholar] [CrossRef]
- Zhang, B.; Wan, H.; Maierwufu, M.; Liu, Q.; Li, T.; He, Y.; Wang, X.; Liu, G.; Hong, X.; Feng, Q. STAT3 ameliorates truncated tau-induced cognitive deficits. Neural Regen. Res. 2024, 19, 915–922. [Google Scholar] [CrossRef]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.G.; Wang, Z.H.; Song, M.; Yu, S.P.; Kang, S.S.; Liu, X.; Zhang, Z.; Xie, M.; Liu, G.P.; et al. δ-Secretase-cleaved Tau stimulates Aβ production via upregulating STAT1-BACE1 signaling in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 586–603. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002, 530, 209–214. [Google Scholar] [CrossRef]
- Sarkar, P.; Banu, S.; Bhattacharya, S.; Bala, A.; Sur, D. Pathophysiology Associated with Diabetes-induced Tauopathy and Development of Alzheimer’s Disease. Curr. Diabetes Rev. 2023, 19, e130522204763. [Google Scholar] [CrossRef]
- Wei, Z.; Song, M.S.; MacTavish, D.; Jhamandas, J.H.; Kar, S. Role of calpain and caspase in beta-amyloid-induced cell death in rat primary septal cultured neurons. Neuropharmacology 2008, 54, 721–733. [Google Scholar] [CrossRef]
- Noël, A.; Foveau, B.; LeBlanc, A.C. Caspase-6-cleaved Tau fails to induce Tau hyperphosphorylation and aggregation, neurodegeneration, glial inflammation, and cognitive deficits. Cell Death Dis. 2021, 12, 227. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, Y.; Wu, G.; Mahaman, Y.A.R.; Ke, D.; Wang, Q.; Zhang, B.; Wang, J.Z.; Li, H.L.; Liu, R.; et al. Phosphorylation of Truncated Tau Promotes Abnormal Native Tau Pathology and Neurodegeneration. Mol. Neurobiol. 2022, 59, 6183–6199. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, S.; Sprenger, F.; Skachokova, Z.; Fraser, G.; Abramowski, D.; Clavaguera, F.; Probst, A.; Frank, S.; Müller, M.; Staufenbiel, M.; et al. Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol. Psychiatry 2016, 21, 1790–1798. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Bitto, A.; Giuliani, D.; Pallio, G.; Irrera, N.; Vandini, E.; Canalini, F.; Zaffe, D.; Ottani, A.; Minutoli, L.; Rinaldi, M.; et al. Effects of COX1-2/5-LOX blockade in Alzheimer transgenic 3xTg-AD mice. Inflamm. Res. 2017, 66, 389–398. [Google Scholar] [CrossRef]
- Kim, S.; Choi, Y.; Spencer, T.E.; Bazer, F.W. Effects of the estrous cycle, pregnancy and interferon tau on expression of cyclooxygenase two (COX-2) in ovine endometrium. Reprod. Biol. Endocrinol. RBE 2003, 1, 58. [Google Scholar] [CrossRef]
- Choi, S.H.; Aid, S.; Caracciolo, L.; Minami, S.S.; Niikura, T.; Matsuoka, Y.; Turner, R.S.; Mattson, M.P.; Bosetti, F. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J. Neurochem. 2013, 124, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Júnior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer’s disease: Current knowledge of signaling pathways and therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef]
- Wang, H.; Luo, W.; Chen, H.; Cai, Z.; Xu, G. Mitochondrial dynamics and mitochondrial autophagy: Molecular structure, orchestrating mechanism and related disorders. Mitochondrion 2024, 75, 101847. [Google Scholar] [CrossRef]
- Qu, C.; Li, Q.P.; Su, Z.R.; Ip, S.P.; Yuan, Q.J.; Xie, Y.L.; Xu, Q.Q.; Yang, W.; Huang, Y.F.; Xian, Y.F.; et al. Nano-Honokiol ameliorates the cognitive deficits in TgCRND8 mice of Alzheimer’s disease via inhibiting neuropathology and modulating gut microbiota. J. Adv. Res. 2022, 35, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.E.; He, B.; Nadeem, M.; Wuu, J.; Scheff, S.W.; Abrahamson, E.E.; Ikonomovic, M.D.; Mufson, E.J. Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer’s disease. Biol. Psychiatry 2015, 77, 693–703. [Google Scholar] [CrossRef]
- Olivera Santa-Catalina, M.; Caballero Bermejo, M.; Argent, R.; Alonso, J.C.; Centeno, F.; Lorenzo, M.J. JNK signaling pathway regulates sorbitol-induced Tau proteolysis and apoptosis in SH-SY5Y cells by targeting caspase-3. Arch. Biochem. Biophys. 2017, 636, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Atzori, C.; Ghetti, B.; Piva, R.; Srinivasan, A.N.; Zolo, P.; Delisle, M.B.; Mirra, S.S.; Migheli, A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Zheng, C.C.; Luo, Y.; Guo, K.W.; Gao, D.; Zhang, L.; Li, L.; Zhang, L. Cornel Iridoid Glycoside and Its Effective Component Regulate ATPase Vps4A/JNK to Alleviate Autophagy Deficit with Autophagosome Accumulation. Am. J. Chin. Med. 2022, 50, 1599–1615. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Hu, Y.; Zhao, J.N.; Huang, C.H.; Li, T.; Zhang, B.G.; He, Y.; Wu, Y.Q.; Zhang, Z.J.; et al. Acetylated tau exacerbates learning and memory impairment by disturbing with mitochondrial homeostasis. Redox Biol. 2023, 62, 102697. [Google Scholar] [CrossRef]
- Park, L.; Hochrainer, K.; Hattori, Y.; Ahn, S.J.; Anfray, A.; Wang, G.; Uekawa, K.; Seo, J.; Palfini, V.; Blanco, I.; et al. Tau induces PSD95-neuronal NOS uncoupling and neurovascular dysfunction independent of neurodegeneration. Nat. Neurosci. 2020, 23, 1079–1089. [Google Scholar] [CrossRef]
- Shen, Z.; Sun, D.; Savastano, A.; Varga, S.J.; Cima-Omori, M.S.; Becker, S.; Honigmann, A.; Zweckstetter, M. Multivalent Tau/PSD-95 interactions arrest in vitro condensates and clusters mimicking the postsynaptic density. Nat. Commun. 2023, 14, 6839. [Google Scholar] [CrossRef]
- Eitan, E.; Thornton-Wells, T.; Elgart, K.; Erden, E.; Gershun, E.; Levine, A.; Volpert, O.; Azadeh, M.; Smith, D.G.; Kapogiannis, D. Synaptic proteins in neuron-derived extracellular vesicles as biomarkers for Alzheimer’s disease: Novel methodology and clinical proof of concept. Extracell. Vesicles Circ. Nucleic Acids 2023, 4, 133–150. [Google Scholar] [CrossRef]
- Das, S.; Goossens, J.; Jacobs, D.; Dewit, N.; Pijnenburg, Y.A.L.; In ‘t Veld, S.; Teunissen, C.E.; Vanmechelen, E. Synaptic biomarkers in the cerebrospinal fluid associate differentially with classical neuronal biomarkers in patients with Alzheimer’s disease and frontotemporal dementia. Alzheimer’s Res. Ther. 2023, 15, 62. [Google Scholar] [CrossRef]
- Jiang, J.; Zou, Y.; Xie, C.; Yang, M.; Tong, Q.; Yuan, M.; Pei, X.; Deng, S.; Tian, M.; Xiao, L.; et al. Oxytocin alleviates cognitive and memory impairments by decreasing hippocampal microglial activation and synaptic defects via OXTR/ERK/STAT3 pathway in a mouse model of sepsis-associated encephalopathy. Brain Behav. Immun. 2023, 114, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Zheng, X.; Fang, T.; Yang, X.; Luo, X.; Guo, A.; Newell, K.A.; Huang, X.F.; Yu, Y. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J. Neuroinflammation 2018, 15, 112. [Google Scholar] [CrossRef]
- Puntambekar, S.S.; Moutinho, M.; Lin, P.B.; Jadhav, V.; Tumbleson-Brink, D.; Balaji, A.; Benito, M.A.; Xu, G.; Oblak, A.; Lasagna-Reeves, C.A.; et al. CX3CR1 deficiency aggravates amyloid driven neuronal pathology and cognitive decline in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.S.; Chung, H.J. Emerging Link between Alzheimer’s Disease and Homeostatic Synaptic Plasticity. Neural Plast. 2016, 2016, 7969272. [Google Scholar] [CrossRef]
- Griffiths, J.; Grant, S.G.N. Synapse pathology in Alzheimer’s disease. Semin. Cell Dev. Biol. 2023, 139, 13–23. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- Li, X.G.; Hong, X.Y.; Wang, Y.L.; Zhang, S.J.; Zhang, J.F.; Li, X.C.; Liu, Y.C.; Sun, D.S.; Feng, Q.; Ye, J.W.; et al. Tau accumulation triggers STAT1-dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep. 2019, 20, e47202. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.E7. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Sessa, G.; Podini, P.; Mariani, M.; Meroni, A.; Spreafico, R.; Sinigaglia, F.; Colonna, M.; Panina, P.; Meldolesi, J. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 2004, 20, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Liu, Y.; Hwang, S.; Archuleta, K.; Huang, H.; Campos, A.; Murad, R.; Piña-Crespo, J.; Xu, H.; Huang, T.Y. Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol. Neurodegener. 2022, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.C.; Wang, H.F.; Jiang, T.; Lu, H.; Tan, M.S.; Tan, C.C.; Tan, L.; Tan, L.; Yu, J.T. Effect of CR1 Genetic Variants on Cerebrospinal Fluid and Neuroimaging Biomarkers in Healthy, Mild Cognitive Impairment and Alzheimer’s Disease Cohorts. Mol. Neurobiol. 2017, 54, 551–562. [Google Scholar] [CrossRef]
- Wes, P.D.; Sayed, F.A.; Bard, F.; Gan, L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia 2016, 64, 1710–1732. [Google Scholar] [CrossRef]
- Zhao, Q.F.; Wan, Y.; Wang, H.F.; Sun, F.R.; Hao, X.K.; Tan, M.S.; Tan, C.C.; Zhang, D.Q.; Tan, L.; Yu, J.T. ABCA7 Genotypes Confer Alzheimer’s Disease Risk by Modulating Amyloid-β Pathology. J. Alzheimer’s Dis. JAD 2016, 52, 693–703. [Google Scholar] [CrossRef]
- Guo, F.; Tan, M.S.; Hu, H.; Ou, Y.N.; Zhang, M.Z.; Sheng, Z.H.; Chi, H.C.; Tan, L. sTREM2 Mediates the Correlation Between BIN1 Gene Polymorphism and Tau Pathology in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2024, 101, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef]
- Tang, J.J.; Huang, L.F.; Deng, J.L.; Wang, Y.M.; Guo, C.; Peng, X.N.; Liu, Z.; Gao, J.M. Cognitive enhancement and neuroprotective effects of OABL, a sesquiterpene lactone in 5xFAD Alzheimer’s disease mice model. Redox Biol. 2022, 50, 102229. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Park, J.; Griciuc, A.; Kim, E.; Choi, S.H.; Iwamoto, Y.; Kiss, M.G.; Christie, K.A.; Vinegoni, C.; Poller, W.C.; et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 2021, 595, 701–706. [Google Scholar] [CrossRef]
- He, J.; Tam, K.Y. Dual-target inhibitors of cholinesterase and GSK-3β to modulate Alzheimer’s disease. Drug Discov. Today 2024, 29, 103914. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M.; et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 2022, 185, 4153–4169.E19. [Google Scholar] [CrossRef]
- Shri, S.R.; Manandhar, S.; Nayak, Y.; Pai, K.S.R. Role of GSK-3β Inhibitors: New Promises and Opportunities for Alzheimer’s Disease. Adv. Pharm. Bull. 2023, 13, 688–700. [Google Scholar] [CrossRef]
- Baltissen, D.; Bold, C.S.; Rehra, L.; Banićević, M.; Fricke, J.; Just, J.; Ludewig, S.; Buchholz, C.J.; Korte, M.; Müller, U.C. APPsα rescues CDK5 and GSK3β dysregulation and restores normal spine density in Tau transgenic mice. Front. Cell. Neurosci. 2023, 17, 1106176. [Google Scholar] [CrossRef]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Alonso Adel, C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef]
- Carapeto, A.P.; Marcuello, C.; Faísca, P.F.N.; Rodrigues, M.S. Morphological and Biophysical Study of S100A9 Protein Fibrils by Atomic Force Microscopy Imaging and Nanomechanical Analysis. Biomolecules 2024, 14, 1091. [Google Scholar] [CrossRef]
- Sanders, E.; Csondor, R.; Šulskis, D.; Baronaitė, I.; Smirnovas, V.; Maheswaran, L.; Horrocks, J.; Munro, R.; Georgiadou, C.; Horvath, I.; et al. The Stabilization of S100A9 Structure by Calcium Inhibits the Formation of Amyloid Fibrils. Int. J. Mol. Sci. 2023, 24, 13200. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int. J. Mol. Sci. 2021, 22, 12382. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef]
- Fan, X.; Chen, H.; He, W.; Zhang, J. Emerging microglial biology highlights potential therapeutic targets for Alzheimer’s disease. Ageing Res. Rev. 2024, 101, 102471. [Google Scholar] [CrossRef] [PubMed]
- Gholami, A. Alzheimer’s disease: The role of proteins in formation, mechanisms, and new therapeutic approaches. Neurosci. Lett. 2023, 817, 137532. [Google Scholar] [CrossRef]
- Chen, J.; Li, S.; Zhang, F.; Chen, J.; Cai, C.; Guo, Y.; Lei, Z.; Zeng, L.H.; Zi, D.; Shen, Y.; et al. The pathogenic APP N-terminal Val225Ala mutation alters tau protein liquid-liquid phase separation and exacerbates synaptic damage. Mol. Psychiatry 2024, 30, 2316–2334. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Yang, Y.; Tapias, V.; Acosta, D.; Xu, H.; Chen, H.; Bhawal, R.; Anderson, E.T.; Ivanova, E.; Lin, H.; Sagdullaev, B.T.; et al. Altered succinylation of mitochondrial proteins, APP and tau in Alzheimer’s disease. Nat. Commun. 2022, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín, J. Alzheimer’s disease: Redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox Signal. 2012, 16, 974–1031. [Google Scholar] [CrossRef]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- Hosp, F.; Mann, M. A Primer on Concepts and Applications of Proteomics in Neuroscience. Neuron 2017, 96, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Fang, Z.T.; Zhao, J.N.; Liu, G.P.; Shen, X.; Jiang, G.F.; Liu, Q. Acetylated tau exacerbates apoptosis by disturbing mitochondrial dynamics in HEK293 cells. J. Neurochem. 2024, 168, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K.; et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 2020, 19, e13081. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Types of PTMs and Corresponding Biochemical Properties | Sites of PTMs | |||
|---|---|---|---|---|
| N-Terminus | Proline-Rich Domain | MT-Binding Domain | C-Terminus | |
| phosphorylation: microtubule stability hyperphosphorylation: tau aggregation | Tyr-29, Thr-30, Thr-39, Ser-46, Ser-56, Ser-68, Thr-69, Thr-71, Thr-102, Thr-111, Ser-113 | Thr-153, Thr-175, Thr-181, Ser-184, Ser-185, Ser-191, Ser-198, Ser-199, Ser-202, Thr-205, Ser-210, Thr-212, Ser-214, Thr-217, Thr-220, Thr-231, Ser-235, Ser-237, Ser-238, Ser241 | Ser-258, Ser-262, Thr-263, Ser-289, Ser-293, Ser-305, Tyr-310, Ser-316, Ser-352, Ser-356,Thr-361 | Thr-386, Tyr-393, Ser-396, Ser-400, Thr-403, Ser-404, Ser-409, Ser-412, Ser-413, Ser-414, Ser-422, Ser-433, Thr-435 |
| acetylation: causes dissociation from microtubules and prevents tau degradation | Lys-24, Lys-44 | Lys-240 | Lys-267, Lys-274, Lys-281, Lys-298, Lys-311, Lys-317, Lys-331, Lys-343, Lys-347, Lys-353 | Lys-369, Lys-370, Lys-375, Lys-385, Lys-395 |
| methylation: promote or prevent tau accumulation | Lys-67, Lys-87 | Lys-254, Lys-280, Lys-321, Lys-353 | Arg-406, Lys-438 | |
| ubiquitination: degradation and metabolism of tau | Lys-180, Lys-240 | Lys-254, Lys-257, Lys-259, Lys-267, Lys-274, Lys-290, Lys-298, Lys-311, Lys-317, Lys-321, Lys-343, Lys-353 | Lys-369, Lys-370, Lys-375, Lys-385, Lys-395 | |
| glycosylation: reduce solubility, stability, and increase tendency of tau to aggregate | Asn-167 | Asn-291, Asn-359, Asn-368 | Ser-409, Asn-410, Ser-412, Ser-413 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, X.; Huang, L.; Lei, F.; Li, T.; Luo, Y.; Zeng, M.; Wang, Z. The Role and Pathogenesis of Tau Protein in Alzheimer’s Disease. Biomolecules 2025, 15, 824. https://doi.org/10.3390/biom15060824
Hong X, Huang L, Lei F, Li T, Luo Y, Zeng M, Wang Z. The Role and Pathogenesis of Tau Protein in Alzheimer’s Disease. Biomolecules. 2025; 15(6):824. https://doi.org/10.3390/biom15060824
Chicago/Turabian StyleHong, Xiaoyue, Linshu Huang, Fang Lei, Tian Li, Yi Luo, Mengliu Zeng, and Zhuo Wang. 2025. "The Role and Pathogenesis of Tau Protein in Alzheimer’s Disease" Biomolecules 15, no. 6: 824. https://doi.org/10.3390/biom15060824
APA StyleHong, X., Huang, L., Lei, F., Li, T., Luo, Y., Zeng, M., & Wang, Z. (2025). The Role and Pathogenesis of Tau Protein in Alzheimer’s Disease. Biomolecules, 15(6), 824. https://doi.org/10.3390/biom15060824