
Ensemble-Based Binding Free Energy Profiling and Network Analysis of the KRAS Interactions with DARPin Proteins Targeting Distinct Binding Sites: Revealing Molecular Determinants and Universal Architecture of Regulatory Hotspots and Allosteric Binding



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.2. Mutational Scanning and Sensitivity Analysis of the KRAS Residues: Quantifying Effects of Mutations on KRAS Binding and Protein Stability
2.3. MM-GBSA Binding Free Energy Computations of KRAS-DARPIN Complexes
2.4. Graph-Based Dynamic Network Analysis of KRAS-DARPIN Complexes
2.5. Network-Based Mutational Profiling of Allosteric Residue Centrality
3. Results
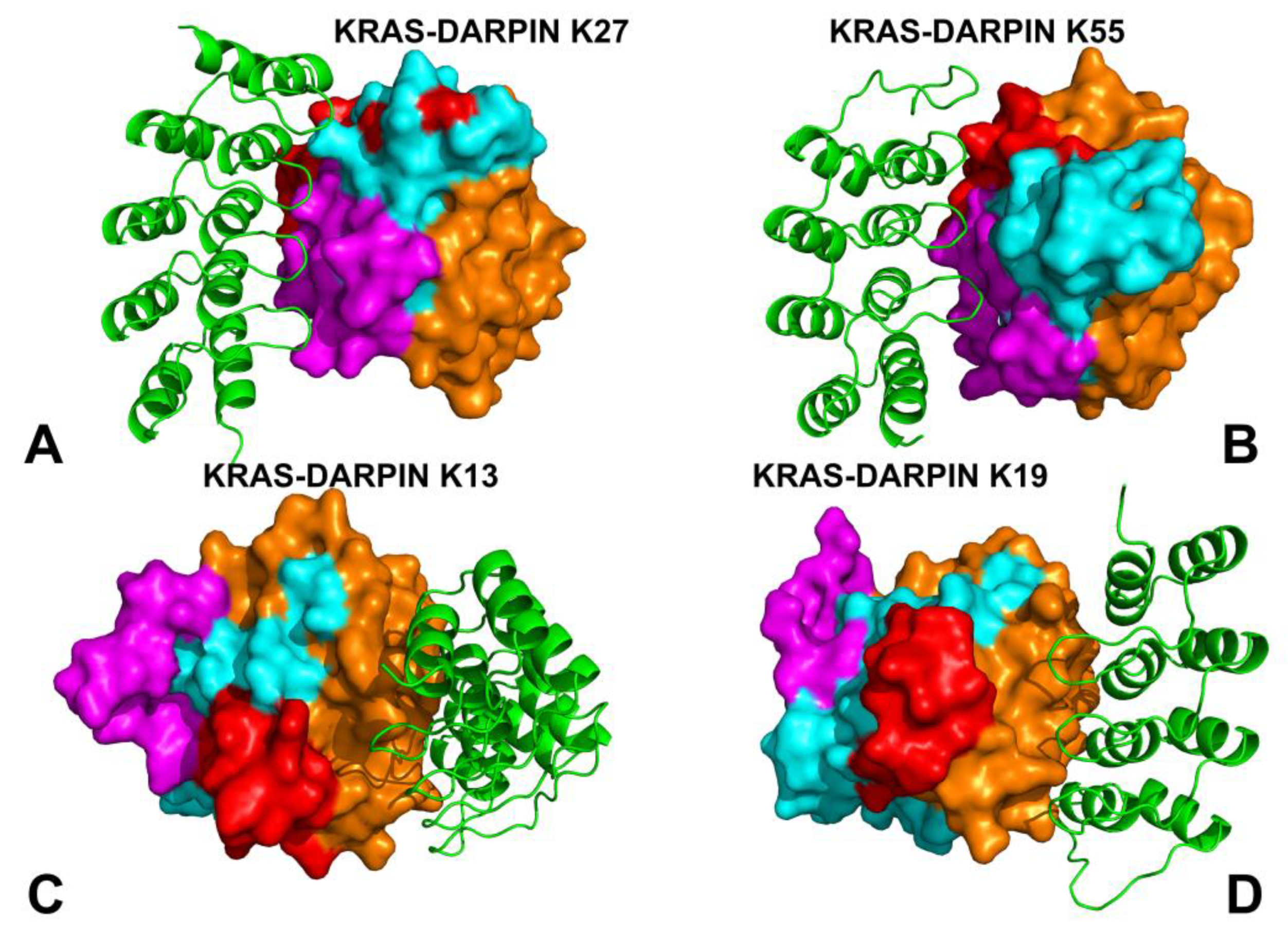
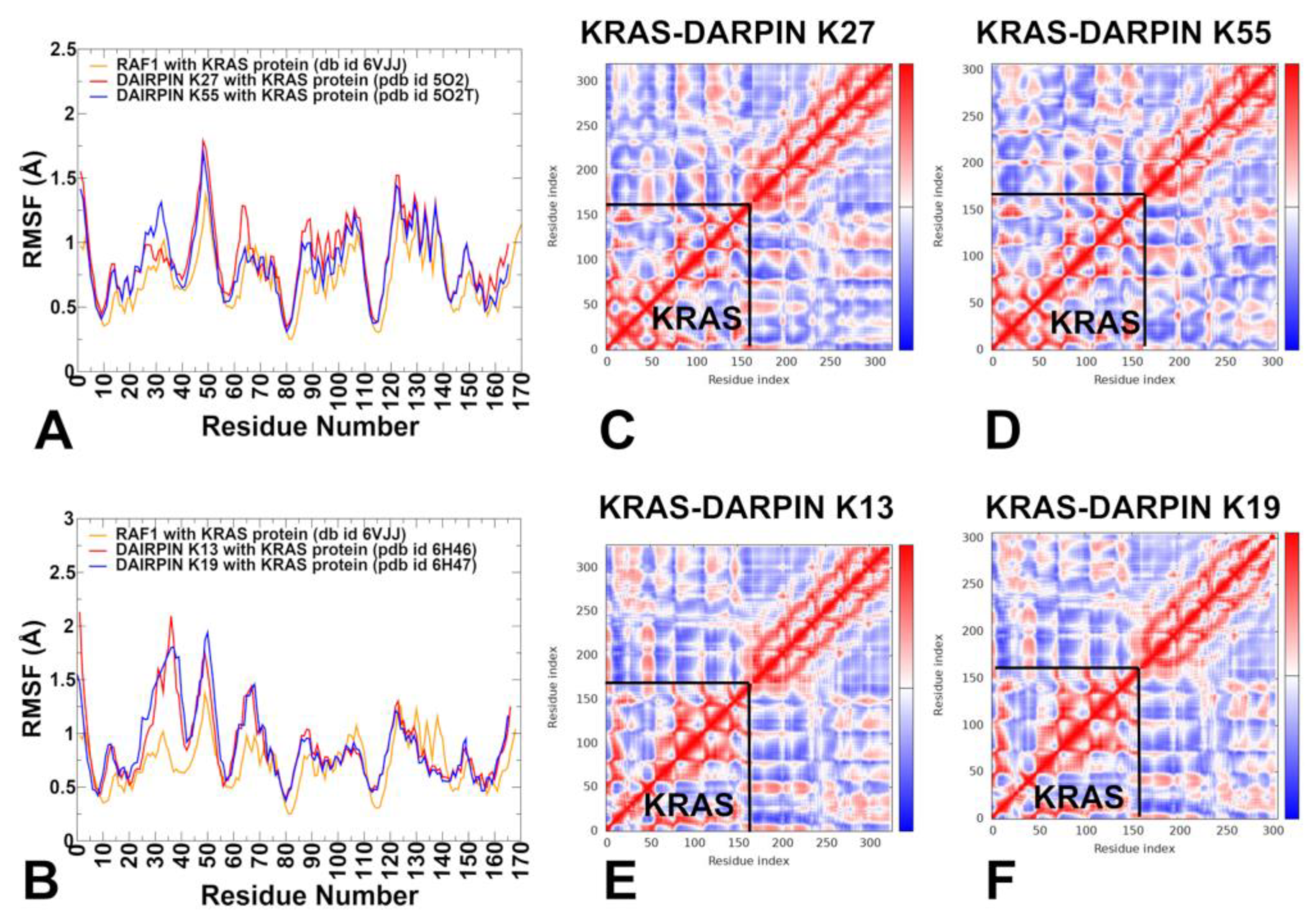
3.1. MD Simulations of the KRAS–Protein Complexes Reveal Distinct Dynamic Signatures
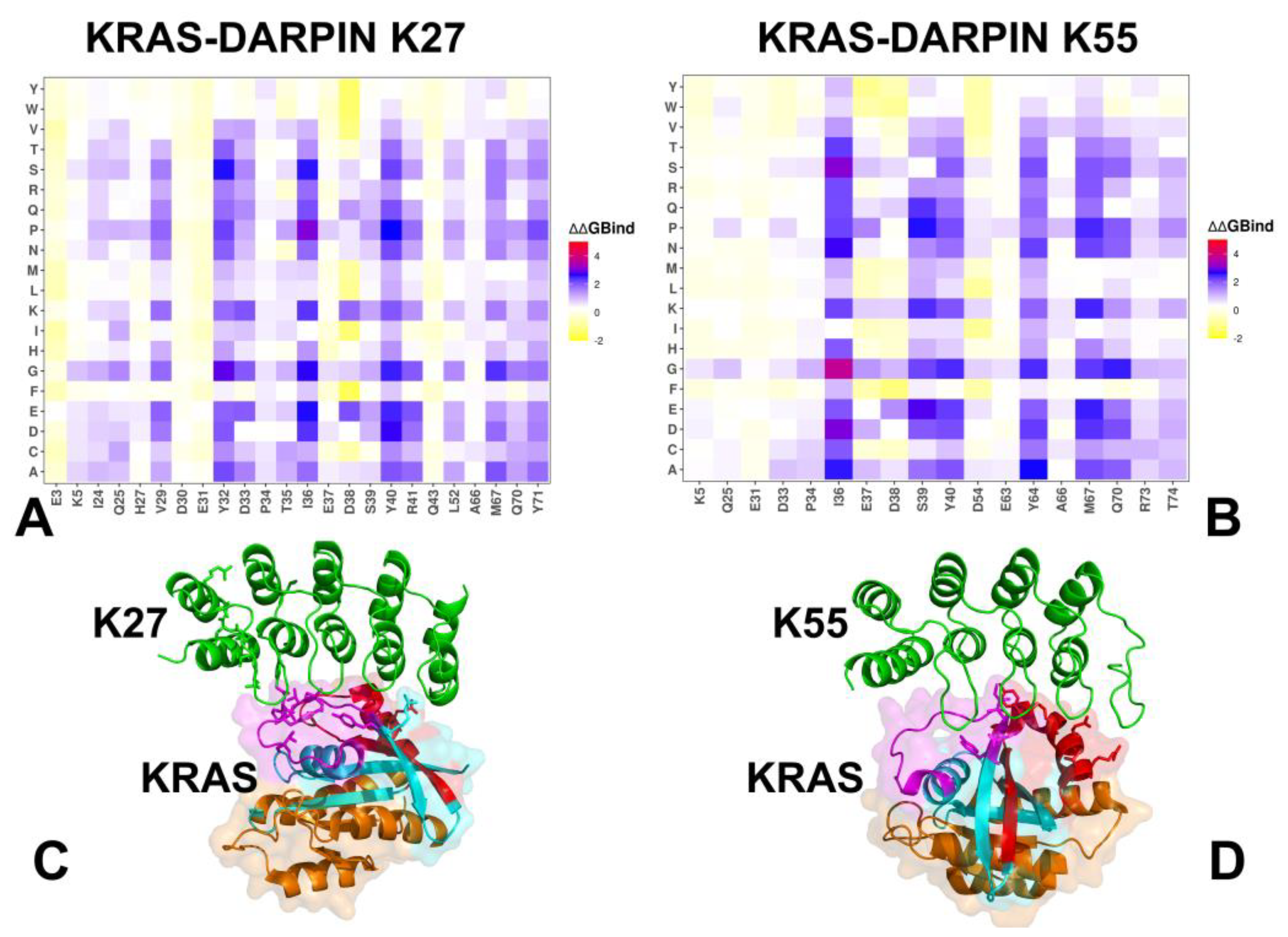
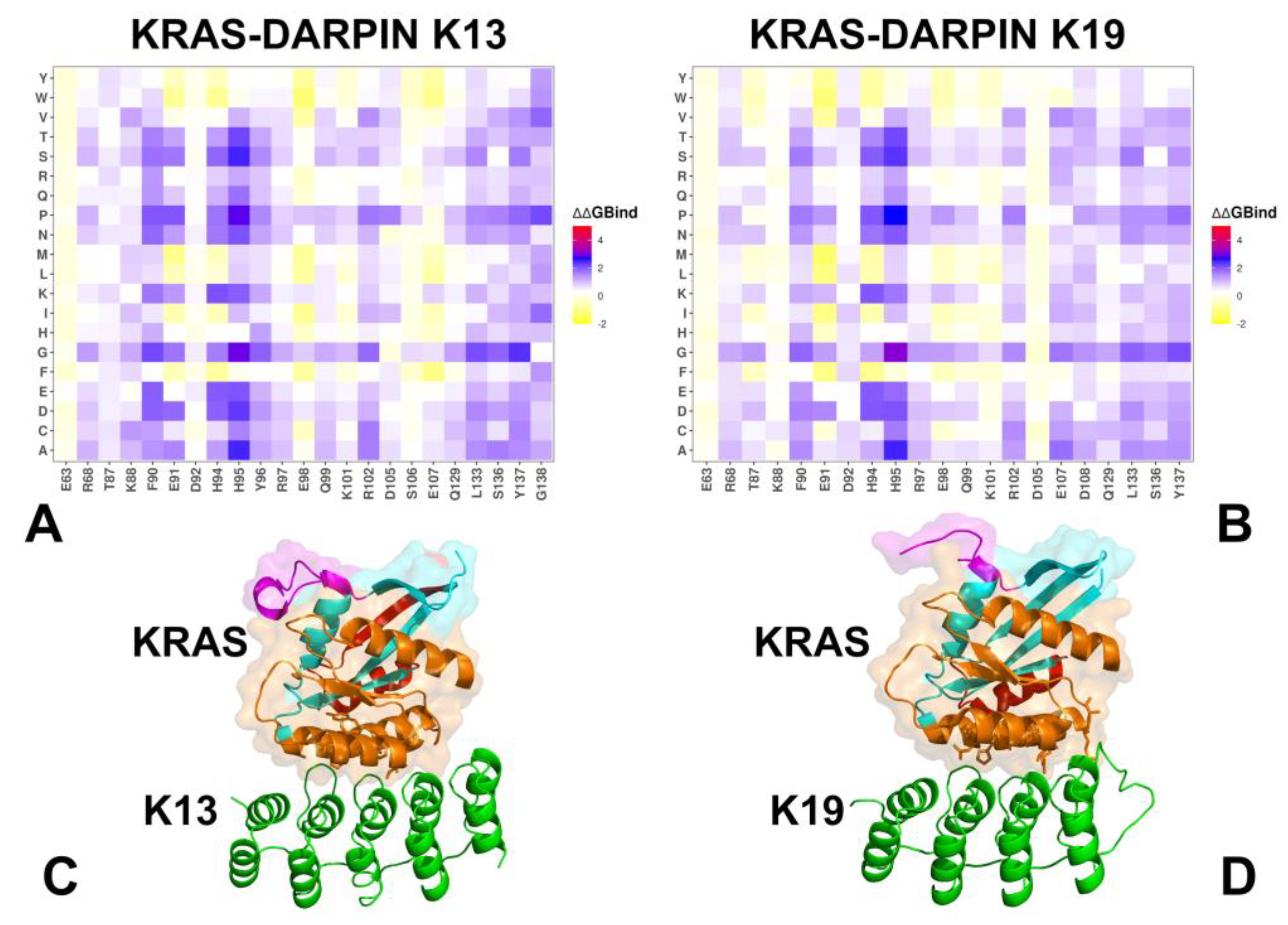
3.2. Mutational Scanning of KRAS-DARPin Complexes
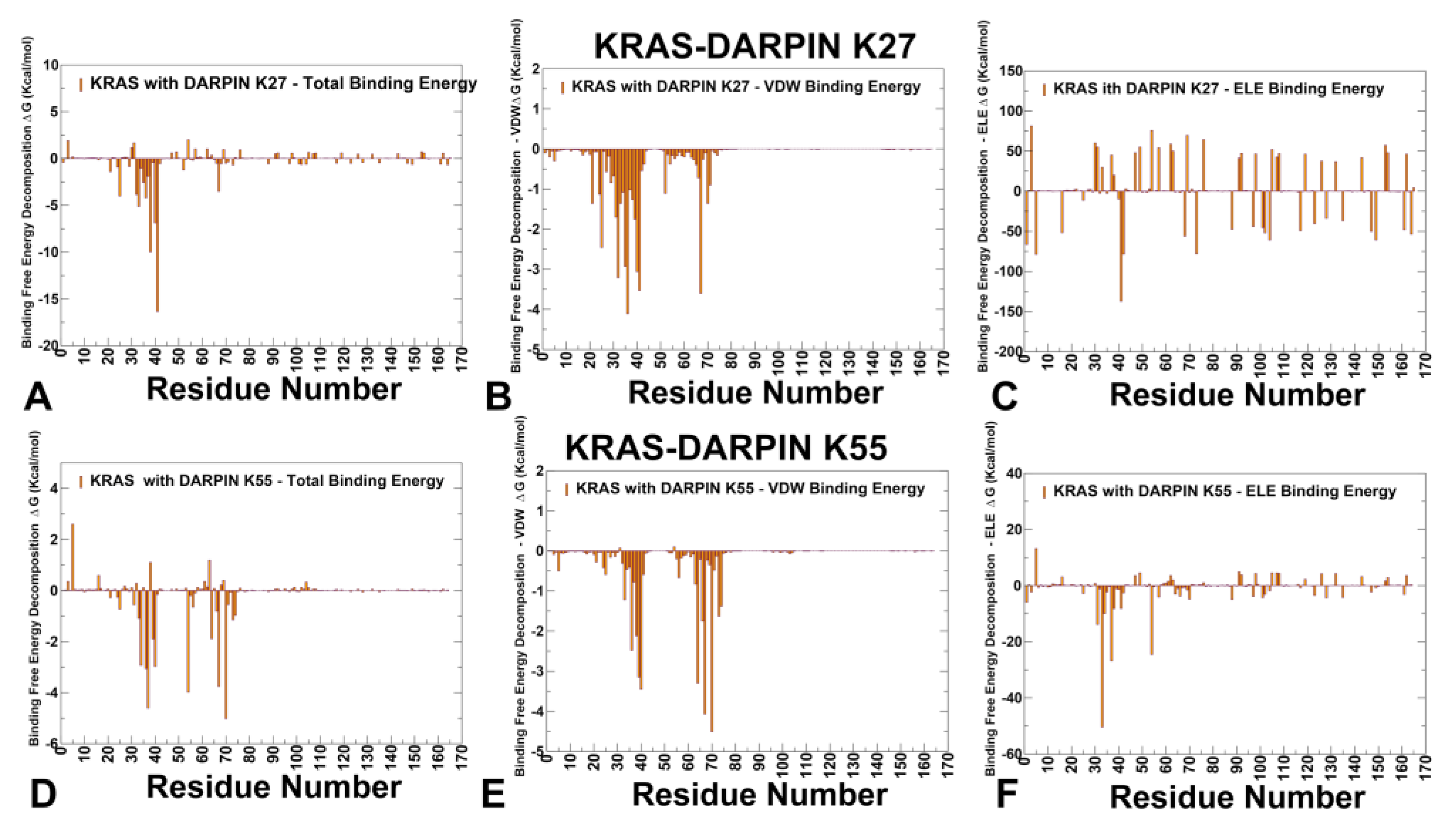
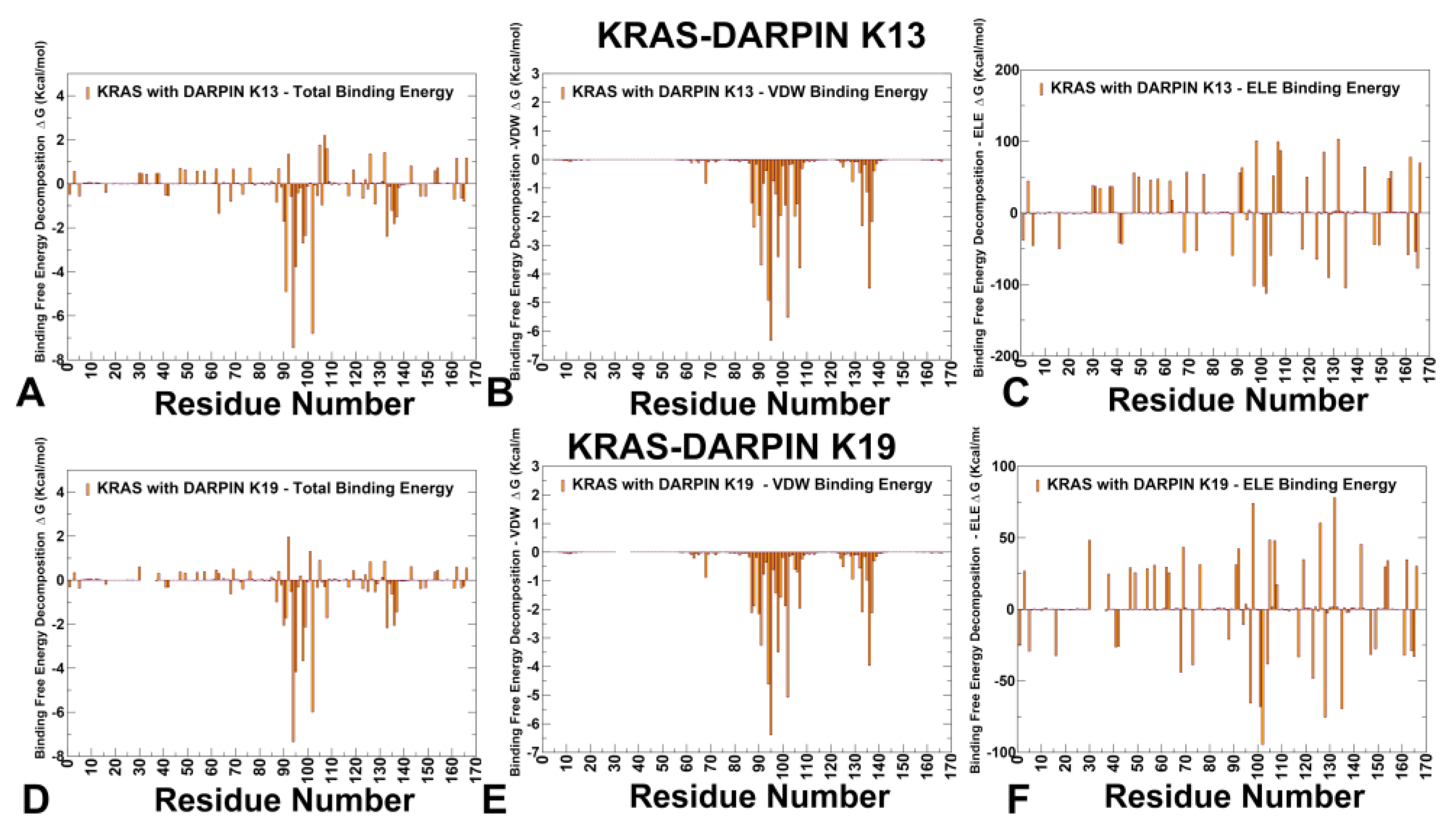
3.3. MM-GBSA Analysis of the Binding Energetics Provides Quantitative Characterization of Thermodynamic Drivers of DARPin Interactions
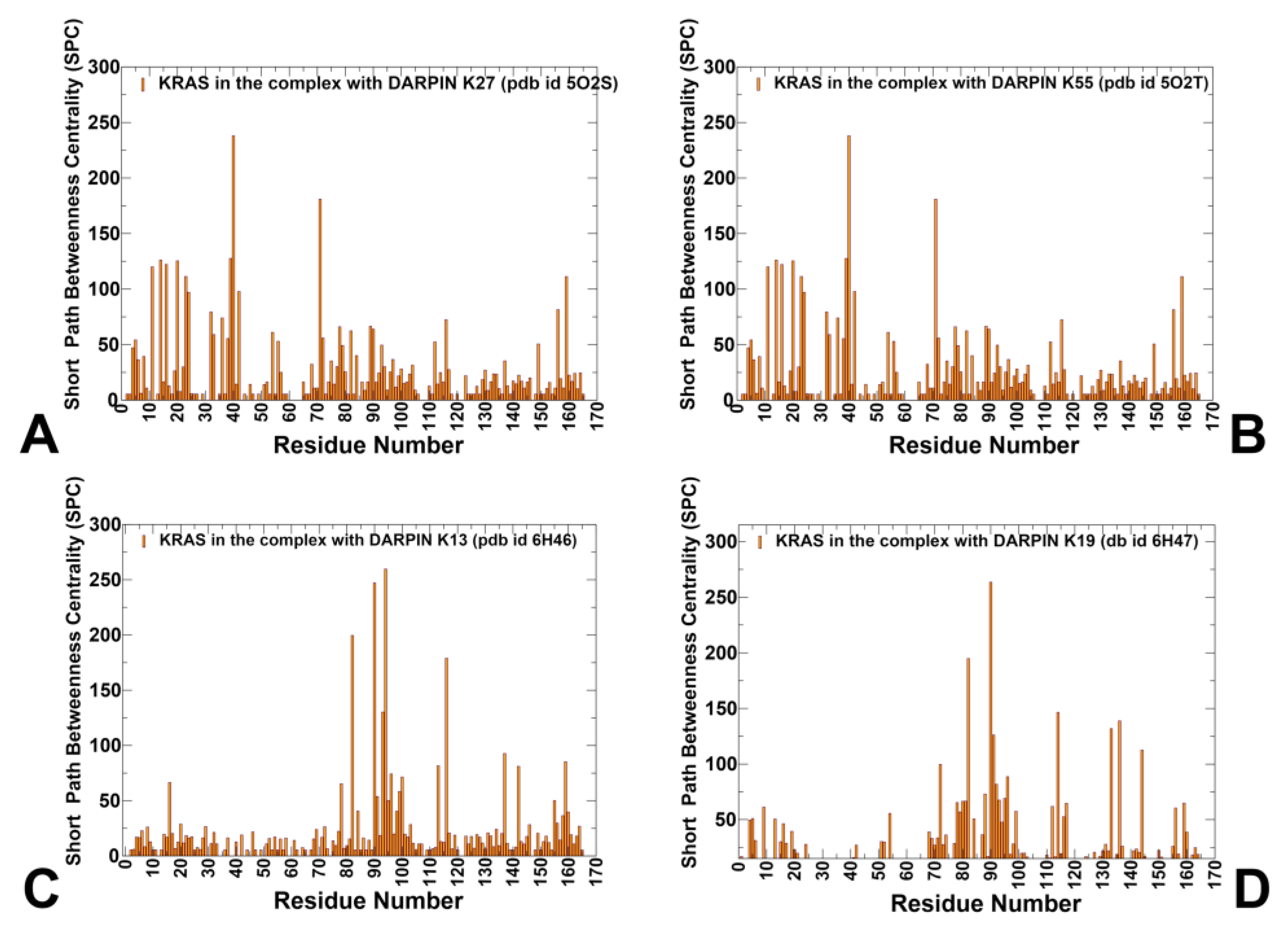
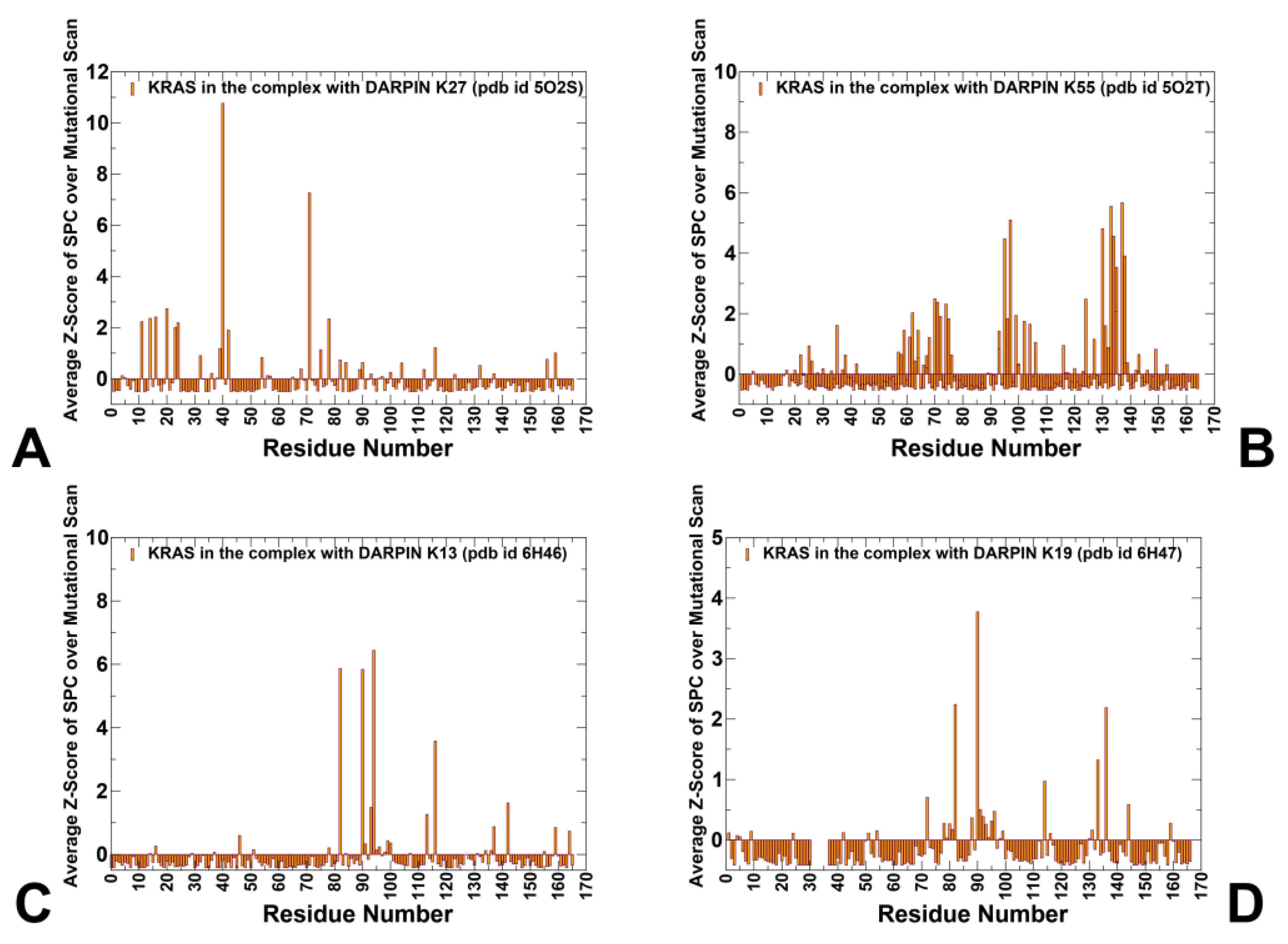
3.4. Network Analysis of Allosteric Communication in KRAS–DARPin Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saperstein, R.; Goel, S.; Maitra, R. Noncoding RNA Profile in Reovirus Treated KRAS-Mutated Colorectal Cancer Patients. Diseases 2023, 11, 142. [Google Scholar] [CrossRef]
- Spencer-Smith, R. The RAF Cysteine-Rich Domain: Structure, Function, and Role in Disease. Adv. Cancer Res. 2024, 164, 69–91. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Morrison, D.K. Regulation of RAF Family Kinases: New Insights from Recent Structural and Biochemical Studies. Biochem. Soc. Trans. 2024, 52, 1061–1069. [Google Scholar] [CrossRef]
- Jeon, H.; Tkacik, E.; Eck, M.J. Signaling from RAS to RAF: The Molecules and Their Mechanisms. Annu. Rev. Biochem. 2024, 93, 289–316. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Lee, K.-Y.; Huo, K.-G.; Gasmi-Seabrook, G.; Zheng, L.; Moghal, N.; Tsao, M.-S.; Ikura, M.; Marshall, C.B. Multivalent Assembly of KRAS with the RAS-Binding and Cysteine-Rich Domains of CRAF on the Membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 12101–12108. [Google Scholar] [CrossRef] [PubMed]
- Spencer-Smith, R.; Terrell, E.M.; Insinna, C.; Agamasu, C.; Wagner, M.E.; Ritt, D.A.; Stauffer, J.; Stephen, A.G.; Morrison, D.K. RASopathy Mutations Provide Functional Insight into the BRAF Cysteine-Rich Domain and Reveal the Importance of Autoinhibition in BRAF Regulation. Mol. Cell 2022, 82, 4262–4276.e5. [Google Scholar] [CrossRef]
- Fernandez-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Ghimessy, A.; Radeczky, P.; Laszlo, V.; Hegedus, B.; Renyi-Vamos, F.; Fillinger, J.; Klepetko, W.; Lang, C.; Dome, B.; Megyesfalvi, Z. Current Therapy of KRAS-Mutant Lung Cancer. Cancer Metastasis Rev. 2020, 39, 1159–1177. [Google Scholar] [CrossRef]
- Vigil, D.; Martin, T.D.; Williams, F.; Yeh, J.J.; Campbell, S.L.; Der, C.J. Aberrant Overexpression of the Rgl2 Ral Small GTPase-Specific Guanine Nucleotide Exchange Factor Promotes Pancreatic Cancer Growth through Ral-Dependent and Ral-Independent Mechanisms. J. Biol. Chem. 2010, 285, 34729–34740. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Gu, S.; Zhang, J.; Nussinov, R. Drugging Ras GTPase: A Comprehensive Mechanistic and Signaling Structural View. Chem. Soc. Rev. 2016, 45, 4929–4952. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D Mutation Alters Local Conformations and Dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef] [PubMed]
- Baussand, J.; Kleinjung, J. Specific Conformational States of Ras GTPase upon Effector Binding. J. Chem. Theory Comput. 2013, 9, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Spoerner, M.; Herrmann, C.; Vetter, I.R.; Kalbitzer, H.R.; Wittinghofer, A. Dynamic Properties of the Ras Switch I Region and Its Importance for Binding to Effectors. Proc. Natl. Acad. Sci. USA 2001, 98, 4944–4949. [Google Scholar] [CrossRef]
- Spoerner, M.; Nuehs, A.; Ganser, P.; Herrmann, C.; Wittinghofer, A.; Kalbitzer, H.R. Conformational States of Ras Complexed with the GTP Analogue GppNHp or GppCH2p: Implications for the Interaction with Effector Proteins. Biochemistry 2005, 44, 2225–2236. [Google Scholar] [CrossRef]
- Spoerner, M.; Hozsa, C.; Poetzl, J.A.; Reiss, K.; Ganser, P.; Geyer, M.; Kalbitzer, H.R. Conformational States of Human Rat Sarcoma (Ras) Protein Complexed with Its Natural Ligand GTP and Their Role for Effector Interaction and GTP Hydrolysis. J. Biol. Chem. 2010, 285, 39768–39778. [Google Scholar] [CrossRef]
- Chao, F.-A.; Dharmaiah, S.; Taylor, T.; Messing, S.; Gillette, W.; Esposito, D.; Nissley, D.V.; McCormick, F.; Byrd, R.A.; Simanshu, D.K.; et al. Insights into the Cross Talk between Effector and Allosteric Lobes of KRAS from Methyl Conformational Dynamics. J. Am. Chem. Soc. 2022, 144, 4196–4205. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Murugan, A.K.; Grieco, M.; Tsuchida, N. RAS Mutations in Human Cancers: Roles in Precision Medicine. Semin. Cancer Biol. 2019, 59, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Fetics, S.K.; Guterres, H.; Kearney, B.M.; Buhrman, G.; Ma, B.; Nussinov, R.; Mattos, C. Allosteric Effects of the Oncogenic RasQ61L Mutant on Raf-RBD. Structure 2015, 23, 505–516. [Google Scholar] [CrossRef]
- Tran, T.H.; Chan, A.H.; Young, L.C.; Bindu, L.; Neale, C.; Messing, S.; Dharmaiah, S.; Taylor, T.; Denson, J.-P.; Esposito, D.; et al. KRAS Interaction with RAF1 RAS-Binding Domain and Cysteine-Rich Domain Provides Insights into RAS-Mediated RAF Activation. Nat. Commun. 2021, 12, 1176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jang, H.; Li, Z.; Sacks, D.B.; Nussinov, R. B-Raf Autoinhibition in the Presence and Absence of 14-3-3. Structure 2021, 29, 768–777.e2. [Google Scholar] [CrossRef]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of Autoinhibited and Active BRAF–MEK1–14-3-3 Complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Martinez Fiesco, J.A.; Durrant, D.E.; Morrison, D.K.; Zhang, P. Structural Insights into the BRAF Monomer-to-Dimer Transition Mediated by RAS Binding. Nat. Commun. 2022, 13, 486. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rawson, S.; Schmoker, A.; Kim, B.W.; Oh, S.; Song, K.; Jeon, H.; Eck, M.J. Cryo-EM Structure of an RAS/RAF Recruitment Complex. Nat. Commun. 2023, 14, 4580. [Google Scholar] [CrossRef]
- Lee, S.Y.; Eun, H.J.; Lee, K.Y. Effector Binding Sequentially Alters KRAS Dimerization on the Membrane: New Insights into RAS-Mediated RAF Activation. Adv. Sci. 2024, 11, e2401530. [Google Scholar] [CrossRef]
- Zhou, Z.W.; Ambrogio, C.; Bera, A.K.; Li, Q.; Li, X.X.; Li, L.; Son, J.; Gondi, S.; Li, J.; Campbell, E.; et al. KRASQ61H Preferentially Signals through MAPK in a RAF Dimer-Dependent Manner in Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, 3719–3731. [Google Scholar] [CrossRef]
- Kwon, J.J.; Dilly, J.; Liu, S.; Kim, E.; Bian, Y.; Dharmaiah, S.; Tran, T.H.; Kapner, K.S.; Ly, S.H.; Yang, X.; et al. Comprehensive Structure-Function Analysis Reveals Gain- and Loss-of-Function Mechanisms Impacting Oncogenic KRAS Activity. bioRxiv 2024, 2024.10.22.618529. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guillard, S.; Kolasinska-Zwierz, P.; Debreczeni, J.; Breed, J.; Zhang, J.; Bery, N.; Marwood, R.; Tart, J.; Overman, R.; Stocki, P.; et al. Structural and Functional Characterization of a DARPin Which Inhibits Ras Nucleotide Exchange. Nat. Commun. 2017, 8, 16111. [Google Scholar] [CrossRef]
- Bery, N.; Legg, S.; Debreczeni, J.; Breed, J.; Embrey, K.; Stubbs, C.; Kolasinska-Zwierz, P.; Barrett, N.; Marwood, R.; Watson, J.; et al. KRAS-Specific Inhibition Using a DARPin Binding to a Site in the Allosteric Lobe. Nat. Commun. 2019, 10, 2607. [Google Scholar] [CrossRef]
- Weng, C.; Faure, A.J.; Escobedo, A.; Lehner, B. The Energetic and Allosteric Landscape for KRAS Inhibition. Nature 2024, 626, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Er, T.K.; Liu, Y.Y.; Hwang, J.K.; Barrio, M.J.; Rodrigo, M.; Garcia-Toro, E.; Herreros-Villanueva, M. Computational Analysis of KRAS Mutations: Implications for Different Effects on the KRAS p.G12D and p.G13D Mutations. PLoS ONE 2013, 8, e55793. [Google Scholar] [CrossRef] [PubMed]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Comparative Effects of Oncogenic Mutations G12C, G12V, G13D, and Q61H on Local Conformations and Dynamics of K-Ras. Comput. Struct. Biotechnol. J. 2020, 18, 1000–1011. [Google Scholar] [CrossRef]
- Nguyen, K.; López, C.A.; Neale, C.; Van, Q.N.; Carpenter, T.S.; Di Natale, F.; Travers, T.; Tran, T.H.; Chan, A.H.; Bhatia, H.; et al. Exploring CRD Mobility during RAS/RAF Engagement at the Membrane. Biophys. J. 2022, 121, 3630–3650. [Google Scholar] [CrossRef]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.J.; Gorfe, A.A.; Hancock, J.F. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell 2017, 168, 239–251.e16. [Google Scholar] [CrossRef]
- Sayyed-Ahmad, A.; Prakash, P.; Gorfe, A.A. Distinct Dynamics and Interaction Patterns in H- and K-Ras Oncogenic P-Loop Mutants. Proteins 2017, 85, 1618–1632. [Google Scholar] [CrossRef]
- Li, Z.L.; Prakash, P.; Buck, M. A “Tug of War” Maintains a Dynamic Protein-Membrane Complex: Molecular Dynamics Simulations of C-Raf RBD-CRD Bound to K-Ras4B at an Anionic Membrane. ACS Cent. Sci. 2018, 4, 298–305. [Google Scholar] [CrossRef]
- Ancy, I.; Penislusshiyan, S.; Ameen, F.; Chitra, L. Microsecond Molecular Dynamics Simulation to Gain Insight into the Binding of MRTX1133 and Trametinib with KRASG12D Mutant Protein for Drug Repurposing. J. Mol. Recognit. 2024, 37, e3103. [Google Scholar] [CrossRef] [PubMed]
- Tu, G.; Gong, Y.; Yao, X.; Liu, Q.; Xue, W.; Zhang, R. Pathways and Mechanism of MRTX1133 Binding to KRAS G12D Elucidated by Molecular Dynamics Simulations and Markov State Models. Int. J. Biol. Macromol. 2024, 274, 133374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Bohidar, N.; Bourne, P.E. Analysis of KRAS-Ligand Interaction Modes and Flexibilities Reveals the Binding Characteristics. J. Chem. Inf. Model. 2023, 63, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, L.; Zhang, Z. Understanding the Influence of AMG 510 on the Structure of KRASG12C Empowered by Molecular Dynamics Simulation. Comput. Struct. Biotechnol. J. 2022, 20, 1056–1067. [Google Scholar] [CrossRef]
- Mir, S.A.; Nayak, B.; Aljarba, N.H.; Kumarasamy, V.; Subramaniyan, V.; Dhara, B. Exploring KRas Protein Dynamics: An Integrated Molecular Dynamics Analysis of KRas Wild and Mutant Variants. ACS Omega 2024, 9, 30665–30674. [Google Scholar] [CrossRef]
- Pandey, D.; Roy, K.K. Decoding KRAS Dynamics: Exploring the Impact of Mutations and Inhibitor Binding. Arch. Biochem. Biophys. 2025, 764, 110279. [Google Scholar] [CrossRef]
- Pantsar, T.; Rissanen, S.; Dauch, D.; Laitinen, T.; Vattulainen, I.; Poso, A. Assessment of Mutation Probabilities of KRAS G12 Missense Mutants and Their Long-Timescale Dynamics by Atomistic Molecular Simulations and Markov State Modeling. PLoS Comput. Biol. 2018, 14, e1006458. [Google Scholar] [CrossRef]
- Shi, S.; Zheng, L.; Ren, Y.; Wang, Z. Impacts of Mutations in the P-Loop on Conformational Alterations of KRAS Investigated with Gaussian Accelerated Molecular Dynamics Simulations. Molecules 2023, 28, 2886. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Yang, W.; Zhao, L.; Hu, G. Conformations of KRAS4B Affected by Its Partner Binding and G12C Mutation: Insights from GaMD Trajectory-Image Transformation-Based Deep Learning. J. Chem. Inf. Model. 2024, 64, 6880–6898. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Yang, W.; Zhao, L.; Zhao, J.; Hu, G. Molecular Mechanism of Phosphorylation-Mediated Impacts on the Conformation Dynamics of GTP-Bound KRAS Probed by GaMD Trajectory-Based Deep Learning. Molecules 2024, 29, 2317. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A Template Based Loop Structure Prediction Server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved Prediction of Protein Side-Chain Conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. VMD Store—A VMD Plugin to Browse, Discover, and Install VMD Extensions. J. Chem. Inf. Model. 2019, 59, 4519–4523. [Google Scholar] [CrossRef]
- Grant, B.J.; Skjaerven, L.; Yao, X.Q. The Bio3D Packages for Structural Bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of Changes in Protein-Protein Binding Affinity on Mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef]
- Dehouck, Y.; Gilis, D.; Rooman, M. A New Generation of Statistical Potentials for Proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and Accurate Predictions of Protein Stability Changes upon Mutations Using Statistical Potentials and Neural Networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A Web Server to Predict and Analyze the Protein–Protein Complex Based on Computational Docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 7. Entropy Effects on the Performance of End-Point Binding Free Energy Calculation Approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Chen, F.; Sun, H.; Wang, Z.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 9. Prediction Reliability of Binding Affinities and Binding Poses for Protein-Peptide Complexes. Phys. Chem. Chem. Phys. 2019, 21, 10135–10145. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Brinda, K.V.; Vishveshwara, S. A Network Representation of Protein Structures: Implications for Protein Stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction Energy Based Protein Structure Networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical Networks in tRNA:Protein Complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLoS Comput. Biol. 2017, 13, e1005299. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.J.; Vidotto, M.; Boscariol, F.; Di Domenico, T.; Walsh, I.; Tosatto, S.C. RING: Networking Interacting Residues, Evolutionary Information and Energetics in Protein Structures. Bioinformatics 2011, 27, 2003–2005. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Minervini, G.; Tosatto, S.C. The RING 2.0 Web Server for High Quality Residue Interaction Networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C.E. RING 3.0: Fast Generation of Probabilistic Residue Interaction Networks from Structural Ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef]
- Del Conte, A.; Camagni, G.F.; Clementel, D.; Minervini, G.; Monzon, A.M.; Ferrari, C.; Piovesan, D.; Tosatto, S.C.E. RING 4.0: Faster Residue Interaction Networks with Novel Interaction Types across over 35,000 Different Chemical Structures. Nucleic Acids Res. 2024, 52, W306–W312. [Google Scholar] [CrossRef]
- Floyd, R.W. Algorithm 97: Shortest Path. Commun. ACM 1962, 5, 345. [Google Scholar] [CrossRef]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring Network Structure, Dynamics, and Function Using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; pp. 11–15. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshahrani, M.; Parikh, V.; Foley, B.; Verkhivker, G. Ensemble-Based Binding Free Energy Profiling and Network Analysis of the KRAS Interactions with DARPin Proteins Targeting Distinct Binding Sites: Revealing Molecular Determinants and Universal Architecture of Regulatory Hotspots and Allosteric Binding. Biomolecules 2025, 15, 819. https://doi.org/10.3390/biom15060819
Alshahrani M, Parikh V, Foley B, Verkhivker G. Ensemble-Based Binding Free Energy Profiling and Network Analysis of the KRAS Interactions with DARPin Proteins Targeting Distinct Binding Sites: Revealing Molecular Determinants and Universal Architecture of Regulatory Hotspots and Allosteric Binding. Biomolecules. 2025; 15(6):819. https://doi.org/10.3390/biom15060819
Chicago/Turabian StyleAlshahrani, Mohammed, Vedant Parikh, Brandon Foley, and Gennady Verkhivker. 2025. "Ensemble-Based Binding Free Energy Profiling and Network Analysis of the KRAS Interactions with DARPin Proteins Targeting Distinct Binding Sites: Revealing Molecular Determinants and Universal Architecture of Regulatory Hotspots and Allosteric Binding" Biomolecules 15, no. 6: 819. https://doi.org/10.3390/biom15060819
APA StyleAlshahrani, M., Parikh, V., Foley, B., & Verkhivker, G. (2025). Ensemble-Based Binding Free Energy Profiling and Network Analysis of the KRAS Interactions with DARPin Proteins Targeting Distinct Binding Sites: Revealing Molecular Determinants and Universal Architecture of Regulatory Hotspots and Allosteric Binding. Biomolecules, 15(6), 819. https://doi.org/10.3390/biom15060819