


Mechanistic Insights into the Mechanism of Allosteric Inhibition of Ubiquitin-Specific Protease 7 (USP7)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. System Preparation
2.2. MD Simulations
2.3. DCCM Analysis
2.4. Community Network Analysis
3. Results and Discussion
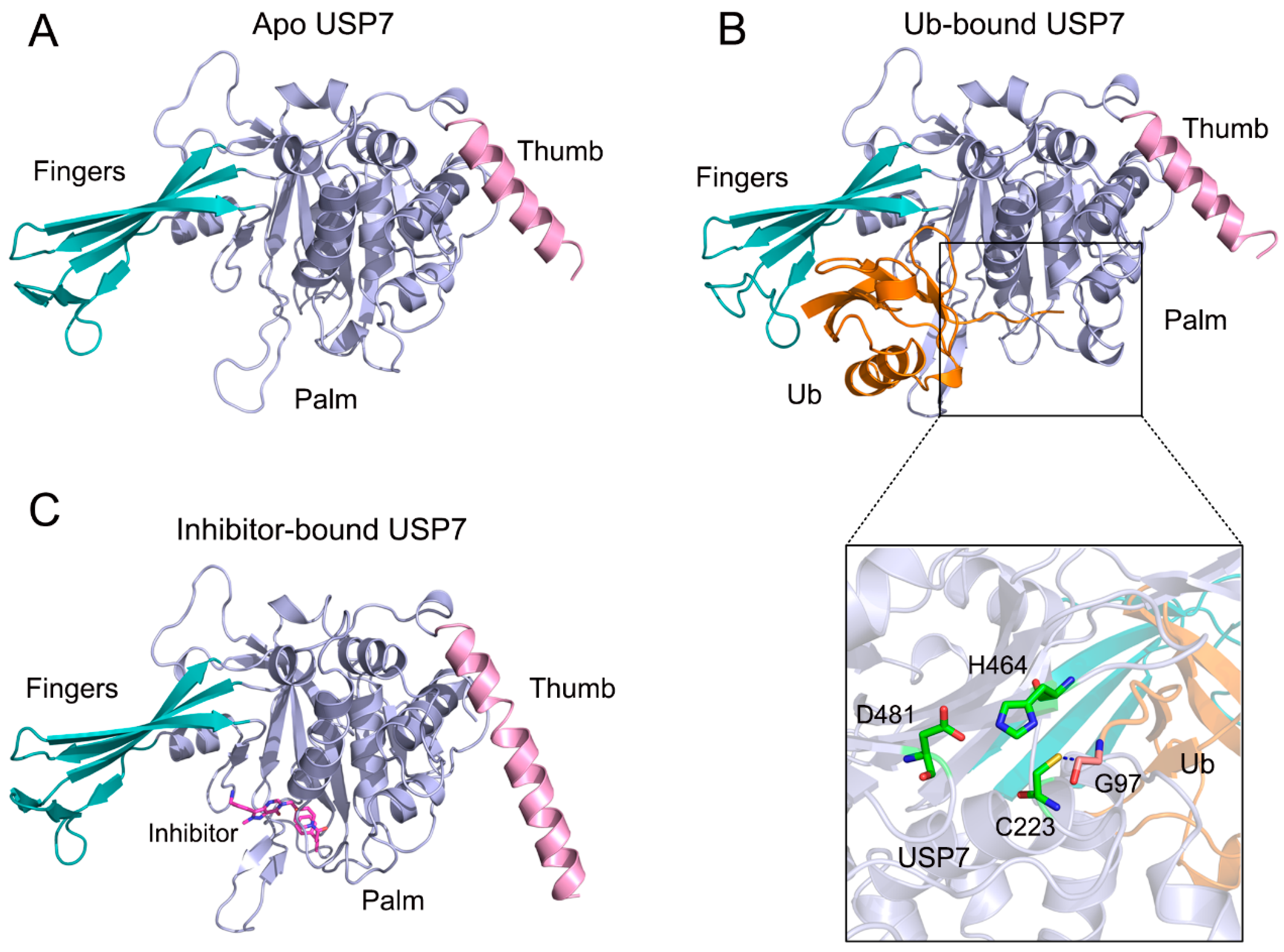
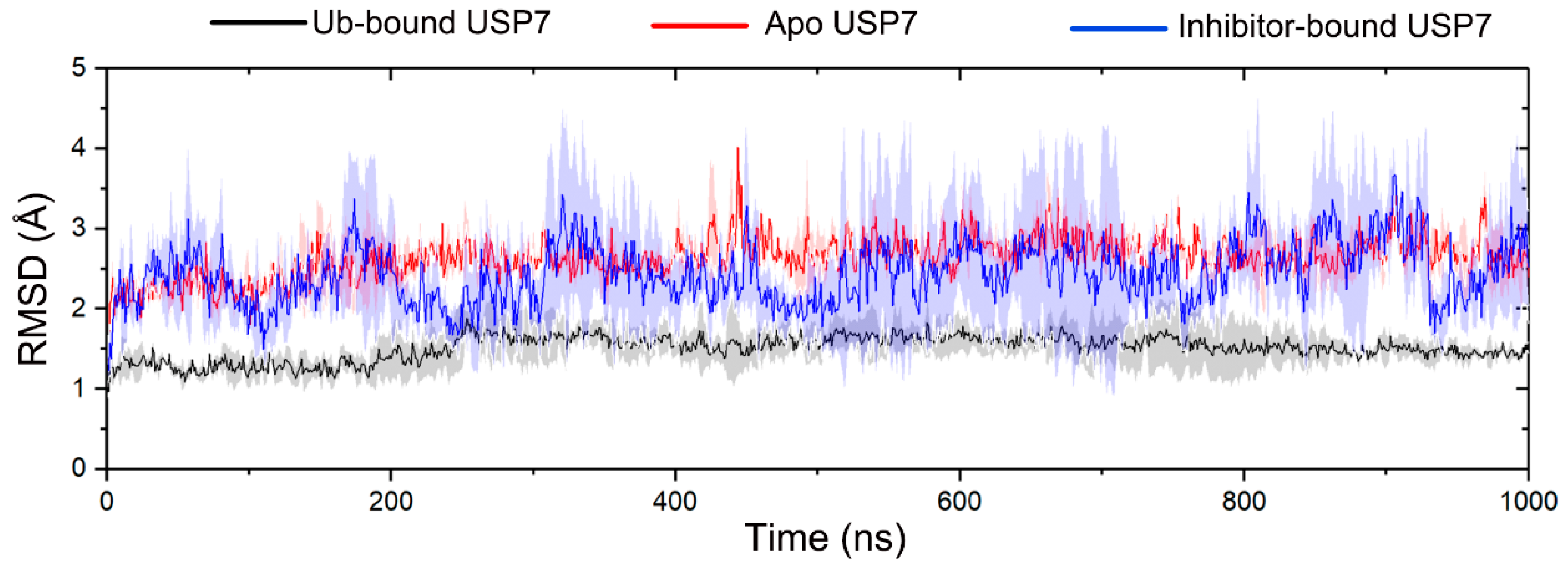
3.1. Allosteric Modulation Amplifies USP7 Structural Plasticity
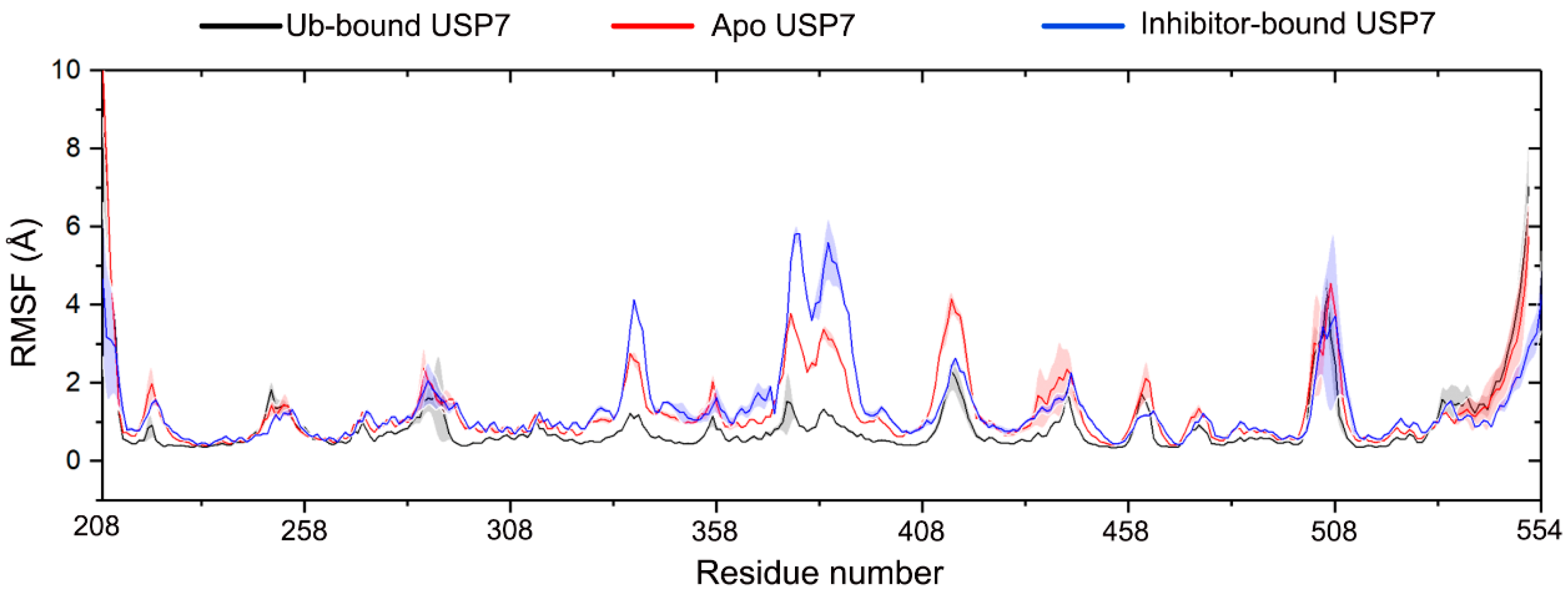
3.2. Allosteric Inhibitor Binding Enhances the Internal Motions of USP7
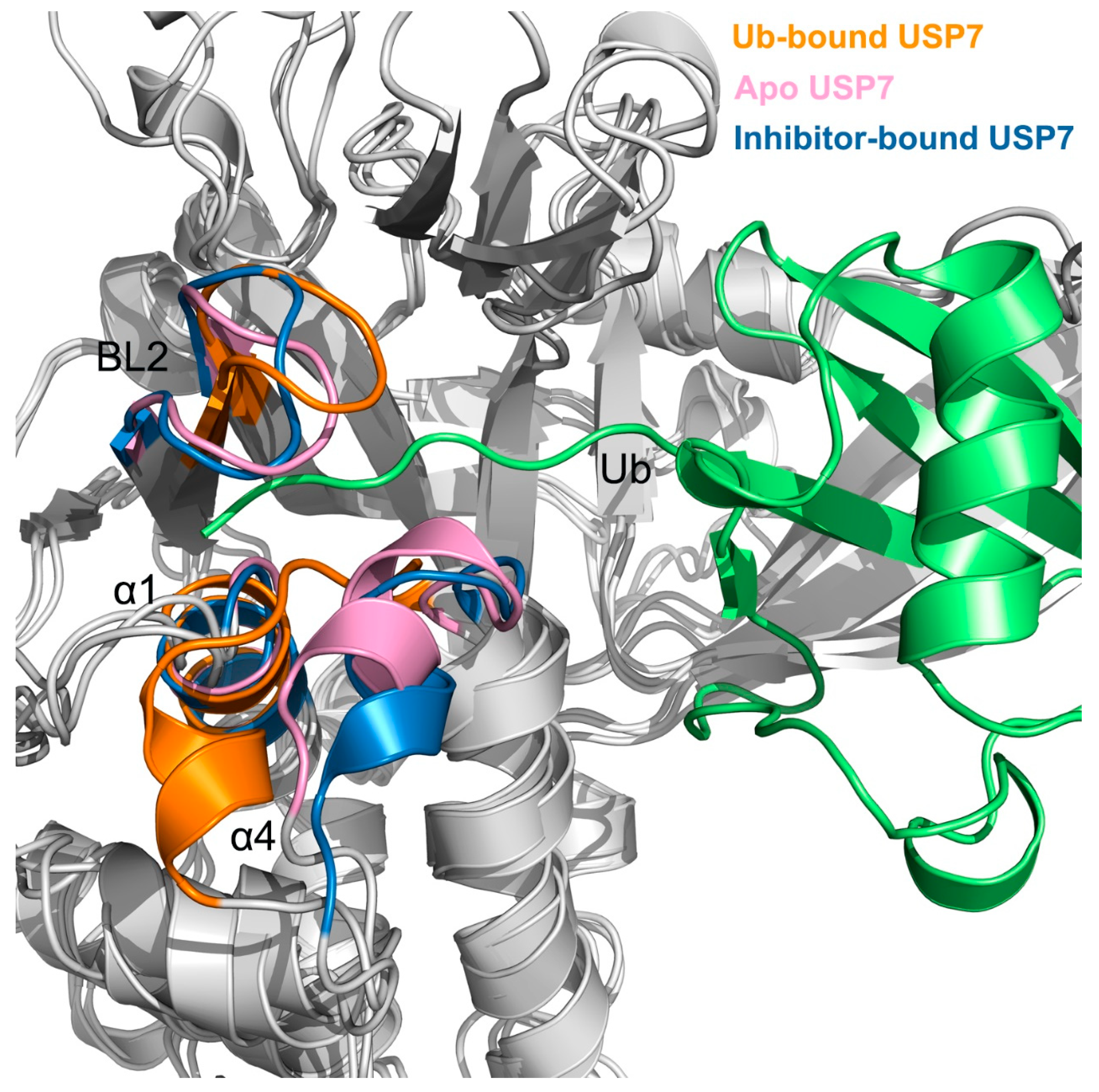
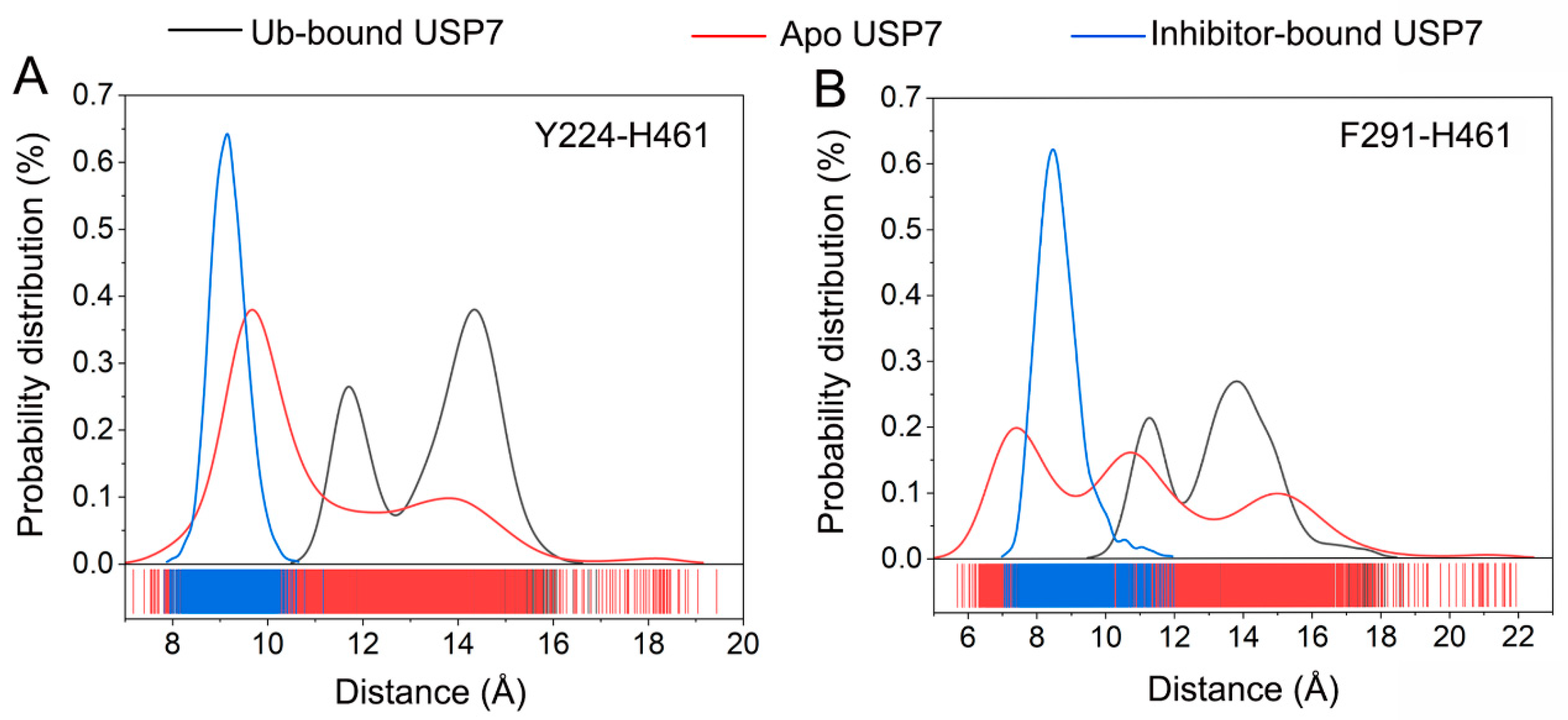
3.3. Allosteric Inhibitor Binding Restricts Ub Accessibility to USP7
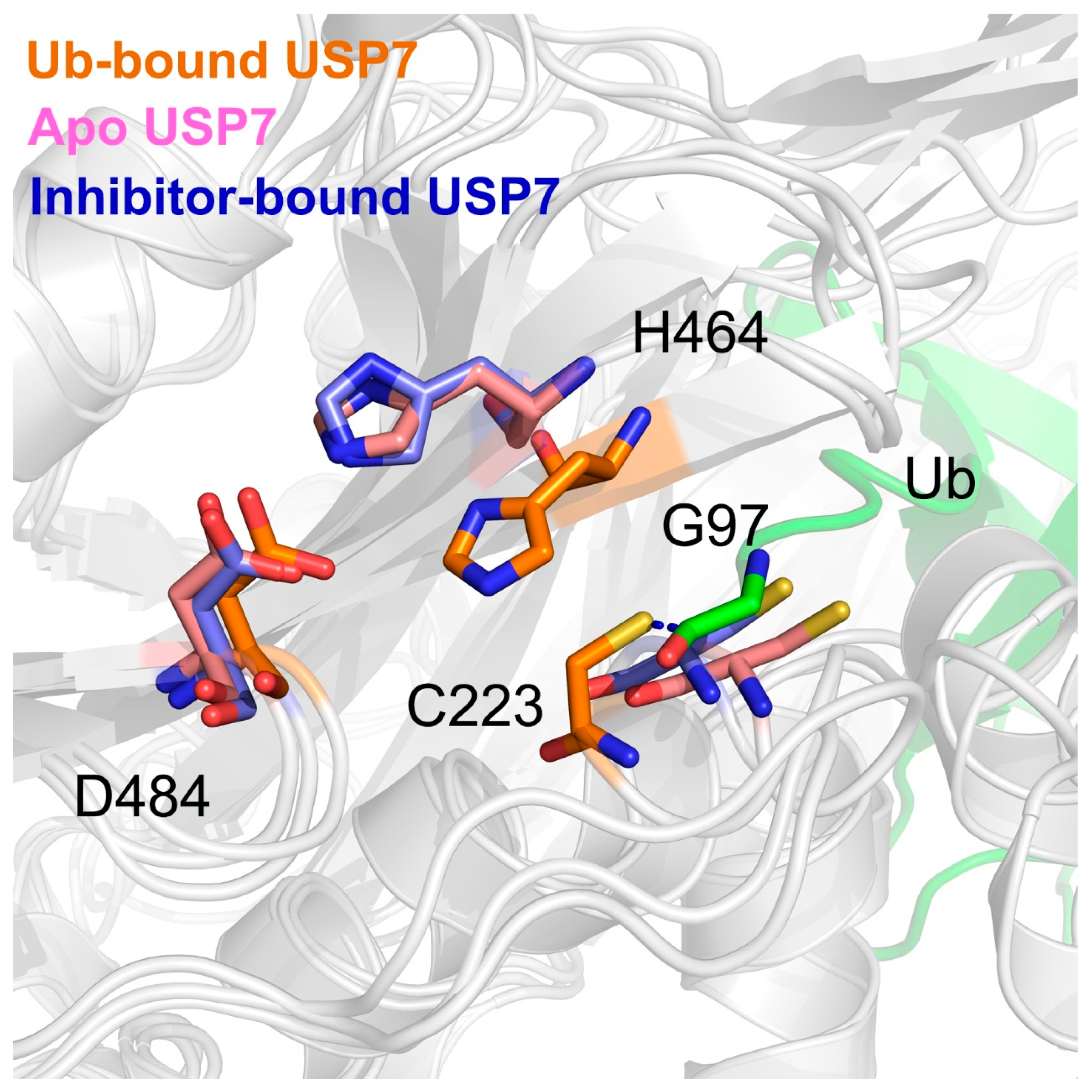
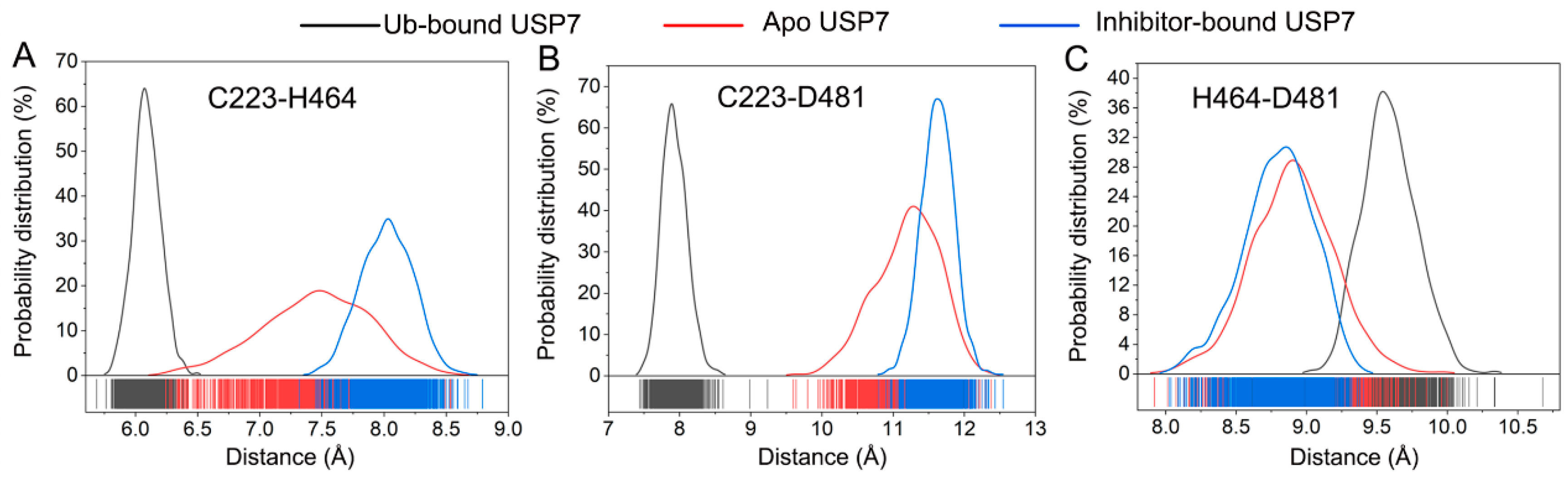
3.4. Allosteric Inhibitor Binding Disrupts the Catalytic Triad Arrangement
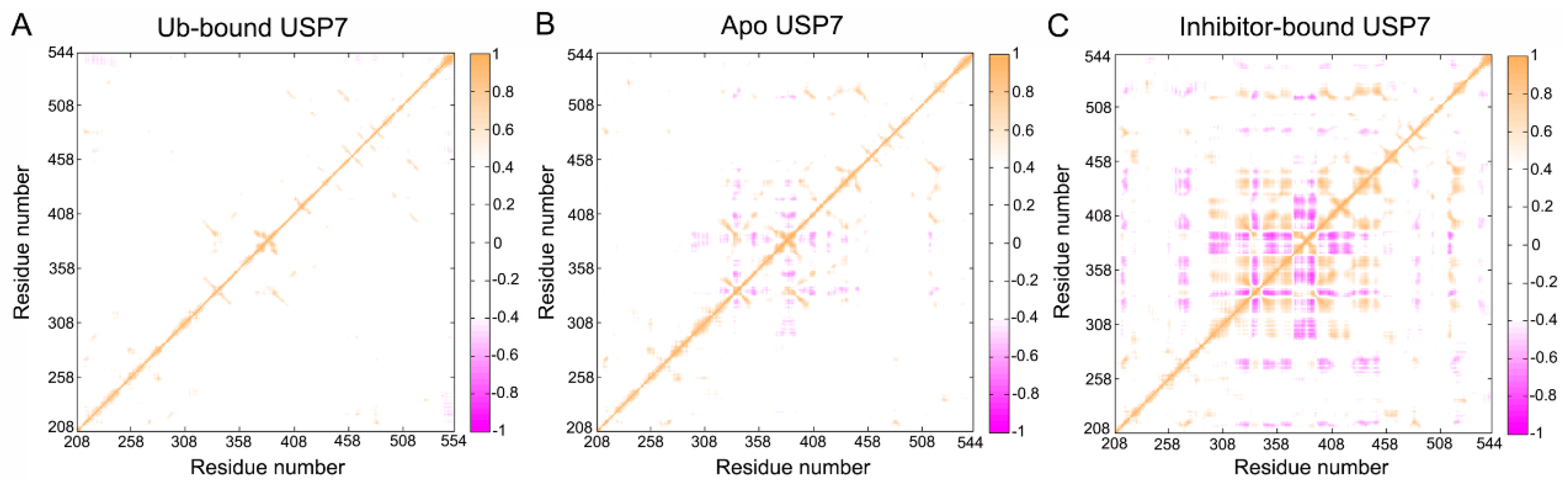
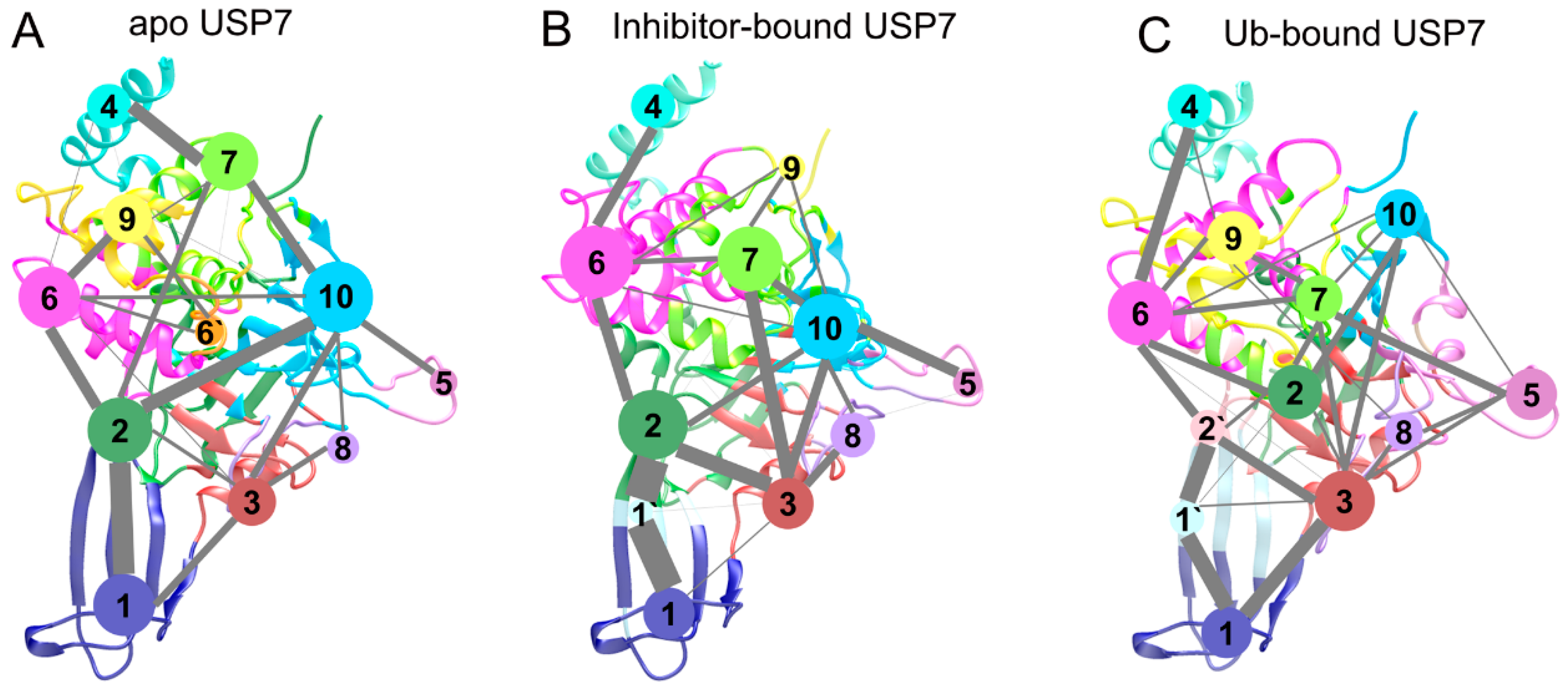
3.5. Topological Reorganization of Allosteric Networks
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Xue, H.; Jin, J. Applications of Protein Ubiquitylation and Deubiquitylation in Drug Discovery. J. Biol. Chem. 2024, 300, 107264. [Google Scholar] [CrossRef] [PubMed]
- Daviet, L.; Colland, F. Targeting Ubiquitin Specific Proteases for Drug Discovery. Biochimie 2008, 90, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Nininahazwe, L.; Liu, B.; He, C.; Zhang, H.; Chen, Z.S. The Emerging Nature of Ubiquitin-Specific Protease 7 (USP7): A New Target in Cancer Therapy. Drug Discov. Today 2021, 26, 490–502. [Google Scholar] [CrossRef]
- Rawat, R.; Starczynowski, D.T.; Ntziachristos, P. Nuclear Deubiquitination in the Spotlight: The Multifaceted Nature of USP7 Biology in Disease. Curr. Opin. Cell Biol. 2019, 58, 85–94. [Google Scholar] [CrossRef]
- Sun, L.-L.; Zhao, N.-L.; Sun, J.; Yuna, H.-F.; Wang, Y.-F.; Hou, C.-Y.; Lv, P.; Zhang, H.-H.; Yang, G.; Zhang, N.-N.; et al. Inhibition of USP7 enhances CD8+ T cell activity in liver cancer by suppressing PRDM1-mediated FGL1 upregulation. Acta Pharmacol. Sin. 2024, 45, 1686–1700. [Google Scholar] [CrossRef]
- Di Lello, P.; Pastor, R.; Murray, J.M.; Blake, R.A.; Cohen, F.; Crawford, T.D.; Drobnick, J.; Drummond, J.; Kategaya, L.; Kleinheinz, T.; et al. Discovery of Small-Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and in Silico Techniques. J. Med. Chem. 2017, 60, 10056–10070. [Google Scholar] [CrossRef]
- Leger, P.R.; Hu, D.X.; Biannic, B.; Bui, M.; Han, X.; Karbarz, E.; Maung, J.; Okano, A.; Osipov, M.; Shibuya, G.M.; et al. Discovery of Potent, Selective, and Orally Bioavailable Inhibitors of USP7 with in Vivo Antitumor Activity. J. Med. Chem. 2020, 63, 5398–5420. [Google Scholar] [CrossRef]
- Oliveira, R.I.; Guedes, R.A.; Salvador, J.A.R. Highlights in USP7 Inhibitors for Cancer Treatment. Front. Chem. 2022, 10, 1005727. [Google Scholar] [CrossRef]
- Rougé, L.; Bainbridge, T.W.; Kwok, M.; Tong, R.; Di Lello, P.; Wertz, I.E.; Maurer, T.; Ernst, J.A.; Murray, J. Molecular Understanding of USP7 Substrate Recognition and C-Terminal Activation. Structure 2016, 24, 1335–1345. [Google Scholar] [CrossRef]
- Cheng, J.; Li, Z.; Gong, R.; Fang, J.; Yang, Y.; Sun, C.; Yang, H.; Xu, Y. Molecular Mechanism for the Substrate Recognition of USP7. Protein Cell 2015, 6, 849–852. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, J.; Chen, C.; Yuan, H.; Wen, X.; Sun, H. USP7: Target Validation and Drug Discovery for Cancer Therapy. Med. Chem. 2018, 14, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Pozhidaeva, A.; Bezsonova, I. USP7: Structure, Substrate Specificity, and Inhibition. DNA Repair 2019, 76, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Özen, A.; Rougé, L.; Bashore, C.; Hearn, B.R.; Skelton, N.J.; Dueber, E.C. Selectively Modulating Conformational States of USP7 Catalytic Domain for Activation. Structure 2018, 26, 72–84.e7. [Google Scholar] [CrossRef]
- Gavory, G.; O’Dowd, C.R.; Helm, M.D.; Flasz, J.; Arkoudis, E.; Dossang, A.; Hughes, C.; Cassidy, E.; McClelland, K.; Odrzywol, E.; et al. Discovery and Characterization of Highly Potent and Selective Allosteric USP7 Inhibitors. Nat. Chem. Biol. 2018, 14, 118–125. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, X.; Li, M.; Zhao, W.; Zhou, S.; Cheng, K.; Xu, Q.; Chen, C.; Wen, X.; Sun, H.; et al. Discovery of Ubiquitin-Specific Protease 7 (USP7) Inhibitors with Novel Scaffold Structures by Virtual Screening, Molecular Dynamics Simulation, and Biological Evaluation. J. Chem. Inf. Model. 2020, 60, 3255–3264. [Google Scholar] [CrossRef]
- Zhao, L.-H.; Yuan, Q.-N.; Dai, A.-T.; He, X.-H.; Chen, C.-W.; Zhang, C.; Xu, Y.-W.; Zhou, Y.; Wang, M.W.; Yang, D.-H.; et al. Molecular Recognition of Two Endogenous Hormones by the Human Parathyroid Hormone Receptor-1. Acta Pharmacol. Sin. 2023, 44, 1227–1237. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Caballero, J.; Alzate-Morales, J.; Ruiz-Pernía, J.J.; Tuñón, I. Understanding the Interactions of Ubiquitin-Specific Protease 7 with Its Substrates through Molecular Dynamics Simulations: Insights into the Role of Its C-Terminal Domains in Substrate Recognition. J. Chem. Inf. Model. 2024, 64, 4134–4148. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of Cryptic Allosteric Sites Using Reversed Allosteric Communication by a Combined Computational and Experimental Strategy. Chem. Sci. 2021, 12, 464–476. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation Pathway of a G Protein-Coupled Receptor Uncovers Conformational Intermediates as Targets for Allosteric Drug Design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef]
- Chai, X.; Hu, X.-P.; Wang, X.-Y.; Wang, H.-T.; Pang, J.-P.; Zhou, W.-F.; Liao, J.-N.; Shan, L.-H.; Xu, X.-H.; Xu, L.; et al. Computational Guided Discovery of Novel Non-steroidal AR-GR dual antagonists demonstrating potency against antiandrogen resistance. Acta Pharmacol. Sin. 2023, 44, 1500–1518. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, Q.; Wang, W.; Sun, H.; Hu, G. Decoding the Identification Mechanism of an SAM-III Riboswitch on Ligands through Multiple Independent Gaussian-Accelerated Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 6118–6132. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-L.; Zhong, H.-Y.; Zhang, Y.-X.; Xue, H.-R.; Zhang, Z.-S.; Fu, K.-Q.; Cao, X.-D.; Xiong, X.-C.; Guo, D. Structural basis of tolvaptan binding to the vasopressin V2 receptor. Acta Pharmacol. Sin. 2024, 45, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, X.; Pang, L.; Zhang, J.Z.H.; Zhu, T. Effect of Mutations on Binding of Ligands to Guanine Riboswitch Probed by Free Energy Perturbation and Molecular Dynamics Simulations. Nucleic Acids Res. 2019, 47, 6618–6631. [Google Scholar] [CrossRef]
- Wei, J.; Chen, F.; Lu, X.; Fan, J.; Li, M.; Huang, J.; Liu, N.; Zhang, J.; Chai, Z.; Lu, S. In Silico Identification and Experimental Validation of Long-Range Allosteric Inhibition of Staphylococcus Aureus Cas9 Catalytic Activity by An Anti-CRISPR Protein AcrIIA14. Int. J. Biol. Macromol. 2025, 310, 143324. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Li, M.; Li, W.; Yao, T.; Wu, J.-W.; Gu, W.; Cohen, R.E.; Shi, Y. Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex with Ubiquitin Aldehyde. Cell 2002, 111, 1041–1054. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, L.; Schauperl, M.; Mobley, D.L.; Bayly, C.; Gilson, M.K. A Fast, Convenient, Polarizable Electrostatic Model for Molecular Dynamics. J. Chem. Theory Comput. 2024, 20, 1293–1305. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; SØndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N.Long(N)Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Wu, X.; Brooks, B.R. Self-Guided Langevin Dynamics Simulation Method. Chem. Phys. Lett. 2003, 381, 512–518. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, D.; Fan, J.; Li, M.; Zhang, J.; Hua, C.; Nussinov, R.; Lu, S. Markov State Models and Molecular Dynamics Simulations Reveal the Conformational Transition of the Intrinsically Disordered Hypervariable Region of K-Ras4B to the Ordered Conformation. J. Chem. Inf. Model. 2022, 62, 4222–4231. [Google Scholar] [CrossRef]
- Huang, J.-P.; Yun, S.-T.; Zhao, J.X.; Wang, X.T.; Wang, X.-C.; Guo, X.Y.; San, D.-M.; Zhou, Y.-X. The Two-step Strategy for Enhancing the Specific Activity and Thermostability of Alginate Lyase AlyG2 with Mechanism for Improved Thermostability. Int. J. Biol. Macromol. 2024, 273, 132685. [Google Scholar] [CrossRef]
- Zhang, Y.; Dou, W.; Zhao, Z.; Li, G.; Li, C.; Chen, X.; Mou, L. Stereo-Selectivity of Enantiomeric Inhibitors to Ubiquitin-Specific Protease 7 (USP7) Dissected by Molecular Docking, Molecular Dynamics Simulations, and Binding Free Energy Calculations. Mol. Divers. 2025, 29, 1725–1735. [Google Scholar] [CrossRef]
- Liu, J.; Jin, Q.; Xia, J.; Liu, L.; Liu, Y.; Jiang, L. Computational Insights into the Resistance of the Gatekeeper Mutation V564F of FGFR2 Against Pemigatinib. J. Comput. Biophys. Chem. 2024, 23, 891–900. [Google Scholar] [CrossRef]
- Eargle, J.; Luthey-Schulten, Z. Network View: 3D Display and Analysis of Protein·RNA Interaction Networks. Bioinformatics 2012, 28, 3000–3001. [Google Scholar] [CrossRef]
- Pan, F.; Wang, C.-N.; Yu, Z.-H.; Wu, Z.-R.; Wang, Z.; Lou, S.; Li, W.-H.; Liu, G.-X.; Li, T.; Zhao, Y.-Z.; et al. NADPHnet: A Novel Strategy to Predict Compounds for Regulation of NADPH Metabolism via Network-Based Methods. Acta Pharmacol. Sin. 2024, 45, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Caliman, A.D.; McCammon, J.A. Allosteric Effects of Sodium Ion Binding on Activation of the M3 Muscarinic G-Protein-Coupled Receptor. Biophys. J. 2015, 108, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational Stability and Dynamic of the Cancer-associated Isoform Δ133p53β Are Modulated by p53 Peptides and p53-specific DNA. FASEB J. 2019, 33, 4225–4235. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Xu, G.; Wu, B.; Liang, W.; Zhang, J.; Xu, S.; Du, T.; Li, M.; Zheng, G.; Zhou, B.; Wang, Z.; et al. Computational Revealing Infigratinib Resistance to the FGFR2 N549H and N549K Mutations. J. Exp. Nanosci. 2025, 20, 2442305. [Google Scholar] [CrossRef]
- Zheng, G.; Liu, W.; Kang, Y.; Xu, B.; Qiu, X.; Du, T.; Xu, S.; Chen, R.; Cheng, H.; Cai, C. Probing the Role of Zinc Ion in Metallo-β-Lactamase Inhibitor Binding by Using Multiple Molecular Dynamics Simulations. Results Chem. 2025, 15, 102171. [Google Scholar] [CrossRef]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Zhang, M.; Ao, J.; Liu, N.; Chen, T.; Lu, S. Exploring the Constitutive Activation Mechanism of the Class A Orphan GPR20. Acta Pharmacol. Sin. 2025, 46, 500–511. [Google Scholar] [CrossRef]
- Li, M.; Lan, X.; Shi, X.; Zhu, C.; Lu, X.; Pu, J.; Lu, S.; Zhang, J. Delineating the Stepwise Millisecond Allosteric Activation Mechanism of the Class C GPCR Dimer mGlu5. Nat. Commun. 2024, 15, 7519. [Google Scholar] [CrossRef]
- Basith, S.; Manavalan, B.; Lee, G. Unveiling Local and Global Conformational Changes and Allosteric Communications in SOD1 Systems Using Molecular Dynamics Simulation and Network Analyses. Comput. Biol. Med. 2024, 168, 107688. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, J.; Liu, Y.; Qu, Y.; Li, Y.Q.; Mu, Y.; Li, W. An Allosteric Mechanism for Potent Inhibition of SARS-CoV-2 Main Proteinase. Int. J. Biol. Macromol. 2024, 265, 130644. [Google Scholar] [CrossRef]
- Li, X.; Hou, C.; Yang, M.; Luo, B.; Mao, N.; Chen, K.; Chen, Z.; Bai, Y. The Effect of Phosphorylation on the Conformational Dynamics and Allostery of the Association of Death-associated Protein Kinase with Calmodulin. J. Biolmol. Struct. Dyn. 2024, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Qiao, X.; Zhu, R.; Zhou, L.; Zhang, Q.; Lu, S.; Chai, Z. Computational Elucidation of a Monobody Targeting the Phosphatase Domain of SHP2. Biomolecules 2025, 15, 217. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Liu, N.; Li, N.; Lu, S.; Chai, Z. Mechanistic Insights into the Mechanism of Allosteric Inhibition of Ubiquitin-Specific Protease 7 (USP7). Biomolecules 2025, 15, 749. https://doi.org/10.3390/biom15060749
Wang X, Liu N, Li N, Lu S, Chai Z. Mechanistic Insights into the Mechanism of Allosteric Inhibition of Ubiquitin-Specific Protease 7 (USP7). Biomolecules. 2025; 15(6):749. https://doi.org/10.3390/biom15060749
Chicago/Turabian StyleWang, Xuebin, Ning Liu, Nuan Li, Shaoyong Lu, and Zongtao Chai. 2025. "Mechanistic Insights into the Mechanism of Allosteric Inhibition of Ubiquitin-Specific Protease 7 (USP7)" Biomolecules 15, no. 6: 749. https://doi.org/10.3390/biom15060749
APA StyleWang, X., Liu, N., Li, N., Lu, S., & Chai, Z. (2025). Mechanistic Insights into the Mechanism of Allosteric Inhibition of Ubiquitin-Specific Protease 7 (USP7). Biomolecules, 15(6), 749. https://doi.org/10.3390/biom15060749