

Molecular Pathogenesis of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension: A Narrative Review

Abstract
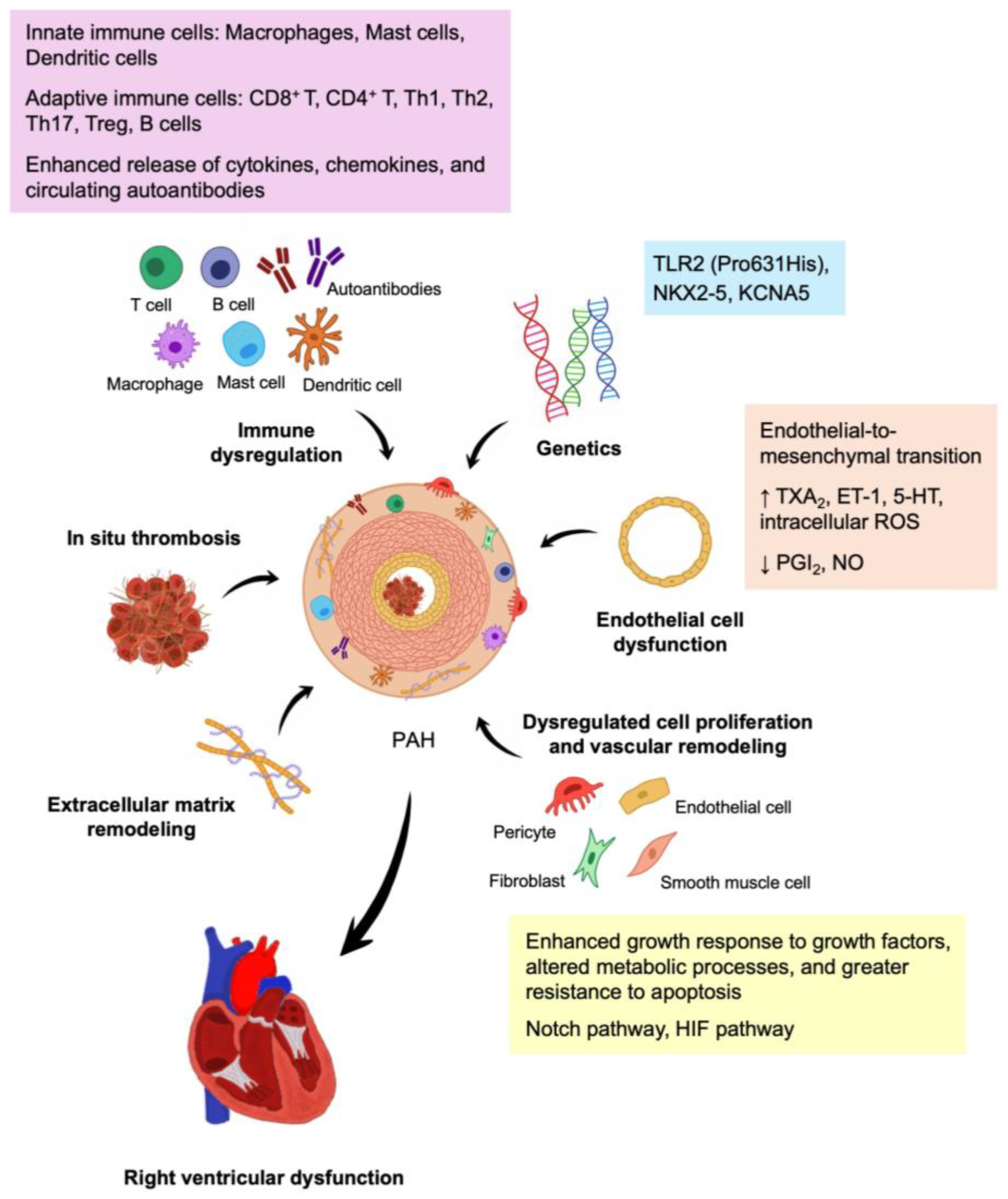
1. Introduction
2. Systemic Sclerosis-Associated PAH (SSc-PAH)
2.1. Pathology
2.2. Pathobiology
2.2.1. Endothelial Cell Dysfunction
- Prostacyclin
- Nitric oxide
- Endothelin 1
- 5-Hydroxytryptamine
- Reactive oxygen species
- Endothelial-to-mesenchymal transition
2.2.2. Dysregulated Cell Proliferation and Vascular Remodeling
- Pulmonary endothelium
- Pericytes
- Smooth muscle cells
- Fibroblasts
- Notch pathway
- Hypoxia-inducible factor pathway
2.2.3. In Situ Thrombosis
2.2.4. Extracellular Matrix Remodeling
2.2.5. Right Ventricular Dysfunction
2.3. Genetics
2.4. Potential Serum Biomarkers
- N-terminal pro-brain natriuretic peptide (NT-proBNP)
- Adipsin
- Lysyl oxidase (LOX)
- Endothelial microparticles (EMPs)
- Asymmetrical dimethylarginine (ADMA)
3. Non-SSc CTD-PAH
3.1. Systemic Lupus Erythematous-Associated PAH (SLE-PAH)
3.2. Primary Sjögren’s Disease-Associated PAH (pSS-PAH)
3.3. Mixed Connective Tissue Disease-Associated PAH (MCTD-PAH)
3.4. Rheumatoid Arthritis-Associated PAH (RA-PAH)
4. Immune Dysregulation in CTD-PAH
4.1. Innate Immune Cells
- Macrophages/monocytes
- Mast cells
- Dendritic cells
4.2. Adaptive Immune Cells
- T cells
- CD8+ Cytotoxic T cells
- CD4+ T cells
- Th1 and Th2
- Th17
- Treg
- B cells
- Circulating autoantibodies
- Cytokines and chemokines
5. CTD-PAH Animal Models
5.1. R-SU Rat
5.2. Pristane/Hypoxia Mice
5.3. Fra-2 Transgenic Mice
5.4. Fli1/Klf5 Mice
5.5. TNF Transgenic Mice (TNF-Tg Mice)

{kind=link}
| Animal Model | R-SU Rat | PriHx Mice | Fra-2 Tg Mice | Fli1/Klf5 Mice | TNF Tg Mice |
|---|---|---|---|---|---|
| Stimuli | VEGFR antagonist + TLR7/8 agonist | Pristane + chronic hypoxia | Fra-2 transgenic | Combined heterozygosity for Fli1 and Klf5 | TNF transgenic |
| Setup time | 5 weeks | 4 weeks | 16 weeks | 16 weeks | 12 weeks |
| Elevated mPAP/RVSP | Severe | Mild | n/a | n/a | Severe |
| RVH (RV/LV + S) | Severe | Moderate | Moderate | n/a | Severe |
| Main pulmonary vascular histological findings |
| Medial hypertrophy |
|
|
|
| Inflammatory cells in the lung |
|
| Perivascular inflammatory infiltrates (prominently T cell component) | Increased B cell accumulation and collagen deposition in the lung | Significant cellular interstitial infiltrate similar to NSIP pattern |
| PBMC | Treg decrease, Th17 increase, NK decrease, TIMP-1 increase | n/a | n/a | n/a | n/a |
| Features other than PH |
| Exacerbated lung fibrosis |
|
| Inflammatory arthritis |
| Advantages |
| Pulmonary vasculopathy, interstitial inflammation, and fibrosis model |
|
|
|
| PH classification | Group 1, CTD (SLE)-PAH | Group 1 + 3, CTD-PH | Group 1 + 3, SSc-PH | Group 1 + 3, SSc-PH | Group 1, CTD-PAH |
| Reference | [211] | [212] | [214] | [219] | [222] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Launay, D.; Sitbon, O.; Hachulla, E.; Mouthon, L.; Gressin, V.; Rottat, L.; Clerson, P.; Cordier, J.F.; Simonneau, G.; Humbert, M. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann. Rheum. Dis. 2013, 72, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Bazan, I.S.; Mensah, K.A.; Rudkovskaia, A.A.; Adonteng-Boateng, P.K.; Herzog, E.L.; Buckley, L. Fares WH: Pulmonary arterial hypertension in the setting of scleroderma is different than in the setting of lupus: A review. Respir. Med. 2018, 134, 42–46. [Google Scholar] [CrossRef]
- Huang, W.C.; Hsieh, S.C.; Wu, Y.W.; Hsieh, T.Y.; Wu, Y.J.; Li, K.J.; Charng, M.J.; Chen, W.S.; Sung, S.H.; Tsao, Y.P.; et al. 2023 Taiwan Society of Cardiology (TSOC) and Taiwan College of Rheumatology (TCR) Joint Consensus on Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Acta Cardiol. Sin. 2023, 39, 213–241. [Google Scholar]
- Lin, C.Y.; Ko, C.H.; Hsu, C.Y.; Chen, H.A. Epidemiology and mortality of connective tissue disease-associated pulmonary arterial hypertension: A national cohort study in taiwan. Semin. Arthritis Rheum. 2020, 50, 957–962. [Google Scholar] [CrossRef]
- Rhee, R.L.; Gabler, N.B.; Sangani, S.; Praestgaard, A.; Merkel, P.A.; Kawut, S.M. Comparison of Treatment Response in Idiopathic and Connective Tissue Disease-associated Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 1111–1117. [Google Scholar] [CrossRef]
- Khanna, D.; Zhao, C.; Saggar, R.; Mathai, S.C.; Chung, L.; Coghlan, J.G.; Shah, M.; Hartney, J.; McLaughlin, V. Long-Term Outcomes in Patients with Connective Tissue Disease-Associated Pulmonary Arterial Hypertension in the Modern Treatment Era: Meta-Analyses of Randomized, Controlled Trials and Observational Registries. Arthritis Rheumatol. 2021, 73, 837–847. [Google Scholar] [CrossRef]
- Thakkar, V.; Stevens, W.; Prior, D.; Youssef, P.; Liew, D.; Gabbay, E.; Roddy, J.; Walker, J.G.; Zochling, J.; Sahhar, J.; et al. The inclusion of N-terminal pro-brain natriuretic peptide in a sensitive screening strategy for systemic sclerosis-related pulmonary arterial hypertension: A cohort study. Arthritis Res. Ther. 2013, 15, R193. [Google Scholar] [CrossRef]
- Coghlan, J.G.; Denton, C.P.; Grunig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Muller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 73, 1340–1349. [Google Scholar] [CrossRef]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airo, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Muller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Ofner, C.; Huscher, D.; Jordan, S.; Ulrich, S.; Stahler, G.; Grunig, E.; Held, M.; Ghofrani, H.A.; Claussen, M.; et al. Treatment strategies and survival of patients with connective tissue disease and pulmonary arterial hypertension: A COMPERA analysis. Rheumatology 2024, 63, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Liu, J.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Badesch, D.B.; Miller, D.P.; Nicolls, M.R.; Zamanian, R.T. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: Identifying systemic sclerosis as a unique phenotype. Chest 2010, 138, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.L., Jr.; Gavilanes, F.; Jardim, C.; Fernandes, C.; Morinaga, L.T.K.; Dias, B.; Hoette, S.; Humbert, M.; Souza, R. Pulmonary arterial hypertension in the southern hemisphere: Results from a registry of incident Brazilian cases. Chest 2015, 147, 495–501. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Vonk, M.C.; Boonstra, A.; Voskuyl, A.E.; Vonk-Noordegraaf, A.; Smit, E.F.; Dijkmans, B.A.C.; Postmus, P.E.; Mooi, W.J.; Heijdra, Y.; et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: A distinctive vasculopathy. Eur. Respir. J. 2009, 34, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Dorfmuller, P.; Humbert, M.; Perros, F.; Sanchez, O.; Simonneau, G.; Muller, K.M.; Capron, F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum. Pathol. 2007, 38, 893–902. [Google Scholar] [CrossRef]
- Dorfmuller, P.; Montani, D.; Humbert, M. Beyond arterial remodelling: Pulmonary venous and cardiac involvement in patients with systemic sclerosis-associated pulmonary arterial hypertension. Eur. Respir. J. 2010, 35, 6–8. [Google Scholar] [CrossRef]
- Ghatnekar, A.; Chrobak, I.; Reese, C.; Stawski, L.; Seta, F.; Wirrig, E.; Paez-Cortez, J.; Markiewicz, M.; Asano, Y.; Harley, R.; et al. Endothelial GATA-6 deficiency promotes pulmonary arterial hypertension. Am. J. Pathol. 2013, 182, 2391–2406. [Google Scholar] [CrossRef]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef]
- Christman, B.W.; Mcpherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary-Hypertension. New Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J. Am. Coll. Cardiol. 2009, 53, 1573–1619. [Google Scholar] [PubMed]
- Dimitroulas, T.; Giannakoulas, G.; Sfetsios, T.; Karvounis, H.; Dimitroula, H.; Koliakos, G.; Settas, L. Asymmetrical dimethylarginine in systemic sclerosis-related pulmonary arterial hypertension. Rheumatology 2008, 47, 1682–1685. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Tochimoto, A.; Hara, M.; Kawamoto, M.; Sugiura, T.; Katsumata, Y.; Okada, J.; Kondo, H.; Okubo, M.; Kamatani, N. NOS2 polymorphisms associated with the susceptibility to pulmonary arterial hypertension with systemic sclerosis: Contribution to the transcriptional activity. Arthritis Res. Ther. 2006, 8, R104. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Wharton, J.; Grimminger, F.; Ghofrani, H.A. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur. Respir. J. 2008, 32, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Champion, H.C.; Bivalacqua, T.J.; Greenberg, S.S.; Giles, T.D.; Hyman, A.L.; Kadowitz, P.J. Adenoviral gene transfer of endothelial nitric-oxide synthase (eNOS) partially restores normal pulmonary arterial pressure in eNOS-deficient mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13248–13253. [Google Scholar] [CrossRef]
- Kozij, N.K.; Granton, J.T.; Silkoff, P.E.; Thenganatt, J.; Chakravorty, S.; Johnson, S.R. Exhaled Nitric Oxide in Systemic Sclerosis Lung Disease. Can. Respir. J. 2017, 2017, 6736239. [Google Scholar] [CrossRef]
- Girgis, R.E.; Champion, H.C.; Diette, G.B.; Johns, R.A.; Permutt, S.; Sylvester, J.T. Decreased exhaled nitric oxide in pulmonary arterial hypertension: Response to bosentan therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 352–357. [Google Scholar] [CrossRef]
- Giaid, A.Y.M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Bressollette, E.; Dupuis, J.; Bonan, R.; Doucet, S.; Cernacek, P.; Tardif, J.C. Intravascular ultrasound assessment of pulmonary vascular disease in patients with pulmonary hypertension. Chest 2001, 120, 809–815. [Google Scholar] [CrossRef]
- Hirata, Y.E.T.; Eguchi, S.; Kanno, K.; Imai, T.; Ohta, K.; Marumo, F. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J. Clin. Investig. 1993, 91, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kuhl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef]
- Tabeling, C.; Gonzalez Calera, C.R.; Lienau, J.; Hoppner, J.; Tschernig, T.; Kershaw, O.; Gutbier, B.; Naujoks, J.; Herbert, J.; Opitz, B.; et al. Endothelin B Receptor Immunodynamics in Pulmonary Arterial Hypertension. Front. Immunol. 2022, 13, 895501. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Shaul, P.W.; Borok, Z.; Willis, B.C. Endothelin-1 induces alveolar epithelial-mesenchymal transition through endothelin type A receptor-mediated production of TGF-beta1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 38–47. [Google Scholar] [CrossRef]
- Hajialilo, M.; Noorabadi, P.; Tahsini Tekantapeh, S.; Malek Mahdavi, A. Endothelin-1, alpha-Klotho, 25(OH) Vit D levels and severity of disease in scleroderma patients. Rheumatol. Int. 2017, 37, 1651–1657. [Google Scholar] [CrossRef]
- George, P.M.; Oliver, E.; Dorfmuller, P.; Dubois, O.D.; Reed, D.M.; Kirkby, N.S.; Mohamed, N.A.; Perros, F.; Antigny, F.; Fadel, E.; et al. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ. Res. 2014, 114, 677–688. [Google Scholar] [CrossRef]
- Coral-Alvarado, P.; Quintana, G.; Garces, M.F.; Cepeda, L.A.; Caminos, J.E.; Rondon, F.; Iglesias-Gamarra, A.; Restrepo, J.F. Potential biomarkers for detecting pulmonary arterial hypertension in patients with systemic sclerosis. Rheumatol. Int. 2009, 29, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Herve, P.; Launay, J.M.; Scrobohaci, M.L.; Brenot, F.; Simonneau, G.; Petitpretz, P.; Poubeau, P.; Cerrina, J.; Duroux, P.; Drouet, L. Increased plasma serotonin in primary pulmonary hypertension. Am. J. Med. 1995, 99, 249–254. [Google Scholar] [CrossRef]
- Gabrielli, A.; Svegliati, S.; Moroncini, G.; Amico, D. New insights into the role of oxidative stress in scleroderma fibrosis. Open Rheumatol. J. 2012, 6, 87–95. [Google Scholar] [CrossRef]
- Boin, F.E.G.; Posadino, A.M.; Cossu, A.; Giordo, R.; Spinetti, G.; Passiu, G.; Emanueli, C.; Pintus, G. Oxidative stress-dependent activation of collagen synthesis is induced in human pulmonary smooth muscle cells by sera from patients with scleroderma-associated pulmonary hypertension. Orphanet J Rare Dis 2014, 9, 123. [Google Scholar] [CrossRef]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Moonen, J.R.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.H.; Thum, T. Transforming Growth Factor-beta-Induced Endothelial-to-Mesenchymal Transition Is Partly Mediated by MicroRNA-21. Arterioscl Throm Vas. 2012, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra234. [Google Scholar] [CrossRef]
- International PPH Consortium; Lane, K.B.M.R.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Gilbane, A.J.; Derrett-Smith, E.; Trinder, S.L.; Good, R.B.; Pearce, A.; Denton, C.P.; Holmes, A.M. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-beta-dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am. J. Respir. Crit. Care Med. 2015, 191, 665–677. [Google Scholar] [CrossRef]
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmuller, P.; Guignabert, C.; Humbert, M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: A complex interplay. Circulation 2014, 129, 1332–1340. [Google Scholar] [CrossRef]
- Savai, R.; Al-Tamari, H.M.; Sedding, D.; Kojonazarov, B.; Muecke, C.; Teske, R.; Capecchi, M.R.; Weissmann, N.; Grimminger, F.; Seeger, W.; et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat. Med. 2014, 20, 1289–1300. [Google Scholar] [CrossRef]
- Le Hiress, M.; Tu, L.; Ricard, N.; Phan, C.; Thuillet, R.; Fadel, E.; Dorfmuller, P.; Montani, D.; de Man, F.; Humbert, M.; et al. Proinflammatory Signature of the Dysfunctional Endothelium in Pulmonary Hypertension. Role of the Macrophage Migration Inhibitory Factor/CD74 Complex. Am. J. Respir. Crit. Care Med. 2015, 192, 983–997. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Girerd, B.; Ricard, N.; Huertas, A.; Montani, D.; Humbert, M. New Molecular Targets of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension Importance of Endothelial Communication. Chest 2015, 147, 529–537. [Google Scholar] [CrossRef]
- Izikki, M.; Guignabert, C.; Fadel, E.; Humbert, M.; Tu, L.; Zadigue, P.; Dartevelle, P.; Simonneau, G.; Adnot, S.; Maitre, B.; et al. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J. Clin. Investig. 2009, 119, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Dewachter, L.; Gore, B.; Fadel, E.; Dartevelle, P.; Simonneau, G.; Humbert, M.; Eddahibi, S.; Guignabert, C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 311–322. [Google Scholar] [CrossRef]
- Tu, L.; De Man, F.S.; Girerd, B.; Huertas, A.; Chaumais, M.C.; Lecerf, F.; Francois, C.; Perros, F.; Dorfmuller, P.; Fadel, E.; et al. A critical role for p130Cas in the progression of pulmonary hypertension in humans and rodents. Am. J. Respir. Crit. Care Med. 2012, 186, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Ricard, N.; Tu, L.; Le Hiress, M.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; et al. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 2014, 129, 1586–1597. [Google Scholar] [CrossRef]
- de Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; Francois, C.; Schalij, I.; Dorfmuller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Dos Santos, M.C.; Guignabert, C.; Montani, D.; Phan, C.; Coste, F.; Tu, L.; Dubois, M.; Girerd, B.; Courtois, A.; et al. Role of Nerve Growth Factor in Development and Persistence of Experimental Pulmonary Hypertension. Am. J. Resp. Crit. Care 2015, 192, 342–355. [Google Scholar] [CrossRef]
- Huertas, A.; Tu, L.; Thuillet, R.; Le Hiress, M.; Phan, C.; Ricard, N.; Nadaud, S.; Fadel, E.; Humbert, M.; Guignabert, C. Leptin signalling system as a target for pulmonary arterial hypertension therapy. Eur. Respir. J. 2015, 45, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Phan, C.; Bordenave, J.; Tu, L.; Thuillet, R.; Le Hiress, M.; Avouac, J.; Tamura, Y.; Allanore, Y.; Jovan, R.; et al. Regulatory T Cell Dysfunction in Idiopathic, Heritable and Connective Tissue-Associated Pulmonary Arterial Hypertension. Chest 2016, 149, 1482–1493. [Google Scholar] [CrossRef]
- Patnaik, E.; Lyons, M.; Tran, K.; Pattanaik, D. Endothelial Dysfunction in Systemic Sclerosis. Int. J. Mol. Sci. 2023, 24, 14385. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef]
- Svegliati, S.; Amico, D.; Spadoni, T.; Fischetti, C.; Finke, D.; Moroncini, G.; Paolini, C.; Tonnini, C.; Grieco, A.; Rovinelli, M.; et al. Agonistic Anti-PDGF Receptor Autoantibodies from Patients with Systemic Sclerosis Impact Human Pulmonary Artery Smooth Muscle Cells Function In Vitro. Front. Immunol. 2017, 8, 75. [Google Scholar]
- Shi, T.Y.; Wen, X.H.; Meng, J.; Lu, Y.W. Effect of IL-17 on pulmonary artery smooth muscle cells and connective tissue disease-associated pulmonary arterial hypertension. Immun. Inflamm. Dis. 2024, 12, e1243. [Google Scholar] [CrossRef]
- Izikki, M.; Hoang, E.; Draskovic, I.; Mercier, O.; Lecerf, F.; Lamrani, L.; Liu, W.Y.; Guignabert, C.; Md, E.F.; Dorfmuller, P.; et al. Telomere Maintenance Is a Critical Determinant in the Physiopathology of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2015, 66, 1942–1943. [Google Scholar] [CrossRef]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; van Rooijen, N.; Stenmark, K.R. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.R.; Frid, M.G.; El Kasmi, K.C.; McKinsey, T.A.; Sokol, R.J.; Strassheim, D.; Meyrick, B.; Yeager, M.E.; Flockton, A.R.; et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J. Immunol. 2011, 187, 2711–2722. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef]
- Zmorzynski, S.; Styk, W.; Filip, A.A.; Krasowska, D. The Significance of NOTCH Pathway in the Development of Fibrosis in Systemic Sclerosis. Ann. Dermatol. 2019, 31, 365–371. [Google Scholar] [CrossRef]
- Bodas, M.; Subramaniyan, B.; Karmouty-Quintana, H.; Vitiello, P.F.; Walters, M.S. The emerging role of NOTCH3 receptor signalling in human lung diseases. Expert. Rev. Mol. Med. 2022, 24, e33. [Google Scholar] [CrossRef]
- Dees, C.; Tomcik, M.; Zerr, P.; Akhmetshina, A.; Horn, A.; Palumbo, K.; Beyer, C.; Zwerina, J.; Distler, O.; Schett, G.; et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1304–1310. [Google Scholar] [CrossRef]
- Kavian, N.; Servettaz, A.; Mongaret, C.; Wang, A.; Nicco, C.; Chereau, C.; Grange, P.; Vuiblet, V.; Birembaut, P.; Diebold, M.D.; et al. Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheum. 2010, 62, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Zerr, P.; Tomcik, M.; Beyer, C.; Horn, A.; Akhmetshina, A.; Palumbo, K.; Reich, N.; Zwerina, J.; Sticherling, M.; et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheum. 2011, 63, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Gong, D.; Wang, W. Soluble JAGGED1 inhibits pulmonary hypertension by attenuating notch signaling. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2733–2739. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef]
- Sahoo, S.; Li, Y.; de Jesus, D.; Sembrat, J.; Rojas, M.M.; Goncharova, E.; Cifuentes-Pagano, E.; Straub, A.C.; Pagano, P.J. Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H542–H557. [Google Scholar] [CrossRef]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Wang, L.; Chen, S.; Tian, B.; Huang, K.; Corrigan, C.J.; Ying, S.; Wang, W.; Wang, C. IL-33 Initiates Vascular Remodelling in Hypoxic Pulmonary Hypertension by up-Regulating HIF-1alpha and VEGF Expression in Vascular Endothelial Cells. EBioMedicine 2018, 33, 196–210. [Google Scholar] [CrossRef]
- Luo, Y.; Teng, X.; Zhang, L.; Chen, J.; Liu, Z.; Chen, X.; Zhao, S.; Yang, S.; Feng, J.; Yan, X. CD146-HIF-1alpha hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat. Commun. 2019, 10, 3551. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Shi, Y.; Zeng, Z.; Tang, B.; Xiao, X.; Yu, J.; Zou, P.; Liu, J.; Xiao, Y.; Luo, Y.; et al. Intimate intertwining of the pathogenesis of hypoxia and systemic sclerosis: A transcriptome integration analysis. Front. Immunol. 2022, 13, 929289. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, M.; Sikora, M.; Stec, A.; Zaremba, M.; Maciejewski, C.; Pawlik, K.; Rudnicka, L. Hypoxia-Inducible Factor-1alpha (HIF-1alpha) as a Biomarker for Changes in Microcirculation in Individuals with Systemic Sclerosis. Dermatol Ther 2023, 13, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Liu, J.; Zhou, M.; Wang, G.; Xiong, X.; Deng, Y. Hypoxia-induced interstitial transformation of microvascular endothelial cells by mediating HIF-1alpha/VEGF signaling in systemic sclerosis. PLoS ONE 2022, 17, e0263369. [Google Scholar] [CrossRef]
- Takagi, K.; Kawamoto, M.; Higuchi, T.; Tochimoto, A.; Harigai, M.; Kawaguchi, Y. Single nucleotide polymorphisms of the HIF1A gene are associated with susceptibility to pulmonary arterial hypertension in systemic sclerosis and contribute to SSc-PAH disease severity. Int. J. Rheum. Dis. 2020, 23, 674–680. [Google Scholar] [CrossRef]
- White, R.J.; Meoli, D.F.; Swarthout, R.F.; Kallop, D.Y.; Galaria, I.I.; Harvey, J.L.; Miller, C.M.; Blaxall, B.C.; Hall, C.M.; Pierce, R.A.; et al. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L583–L590. [Google Scholar] [CrossRef]
- van Hinsbergh, V.W. Endothelium--role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef]
- Moore, K.L.; Esmon, C.T.; Esmon, N.L. Tumor necrosis factor leads to the internalization and degradation of thrombomodulin from the surface of bovine aortic endothelial cells in culture. Blood 1989, 73, 159–165. [Google Scholar] [CrossRef]
- Elias, G.J.; Ioannis, M.; Theodora, P.; Dimitrios, P.P.; Despoina, P.; Kostantinos, V.; Charalampos, K.; Vassilios, V.; Petros, S.P. Circulating tissue inhibitor of matrix metalloproteinase-4 (TIMP-4) in systemic sclerosis patients with elevated pulmonary arterial pressure. Mediators Inflamm. 2008, 2008, 164134. [Google Scholar] [CrossRef]
- Chelladurai, P.; Seeger, W.; Pullamsetti, S.S. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur. Respir. J. 2012, 40, 766–782. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Hoffmann, J.; Marsh, L.M.; Pieper, M.; Stacher, E.; Ghanim, B.; Kovacs, G.; Konig, P.; Wilkens, H.; Haitchi, H.M.; Hoefler, G.; et al. Compartment-specific expression of collagens and their processing enzymes in intrapulmonary arteries of IPAH patients. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1002–L1013. [Google Scholar] [CrossRef]
- Lepetit, H.; Eddahibi, S.; Fadel, E.; Frisdal, E.; Munaut, C.; Noel, A.; Humbert, M.; Adnot, S.; D’Ortho, M.P.; Lafuma, C. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2005, 25, 834–842. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Ghataorhe, P.; Watson, G.; Girerd, B.; Howard, L.S.; Gibbs, J.S.R.; Condliffe, R.; Elliot, C.A.; Kiely, D.G.; et al. Plasma proteome analysis in patients with pulmonary arterial hypertension: An observational cohort study. Lancet Respir. Med. 2017, 5, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Chaouat, A.; Savale, L.; Chouaid, C.; Tu, L.; Sztrymf, B.; Canuet, M.; Maitre, B.; Housset, B.; Brandt, C.; Le Corvoisier, P.; et al. Role for interleukin-6 in COPD-related pulmonary hypertension. Chest 2009, 136, 678–687. [Google Scholar] [CrossRef] [PubMed]
- van Wolferen, S.A.; Marcus, J.T.; Boonstra, A.; Marques, K.M.; Bronzwaer, J.G.; Spreeuwenberg, M.D.; Postmus, P.E.; Vonk-Noordegraaf, A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur. Heart J. 2007, 28, 1250–1257. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef]
- Mukerji, B.; Alpert, M.A.; Mukerji, V. Right ventricular alterations in scuba divers: Findings on electrocardiography and echocardiography. South. Med. J. 2000, 93, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, M.J.; Lankhaar, J.W.; Westerhof, N.; Voskuyl, A.E.; Boonstra, A.; Bronzwaer, J.G.; Marques, K.M.; Smit, E.F.; Dijkmans, B.A.; Vonk-Noordegraaf, A. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2008, 31, 1160–1166. [Google Scholar] [CrossRef]
- Prins, K.W.; Duval, S.; Markowitz, J.; Pritzker, M.; Thenappan, T. Chronic use of PAH-specific therapy in World Health Organization Group III Pulmonary Hypertension: A systematic review and meta-analysis. Pulm. Circ. 2017, 7, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Tedford, R.J.; Mudd, J.O.; Girgis, R.E.; Mathai, S.C.; Zaiman, A.L.; Housten-Harris, T.; Boyce, D.; Kelemen, B.W.; Bacher, A.C.; Shah, A.A.; et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ. Heart Fail. 2013, 6, 953–963. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Mouchaers, K.T.; Niessen, H.M.; Hadi, A.M.; Kupreishvili, K.; Boonstra, A.; Voskuyl, A.E.; Belien, J.A.; Smit, E.F.; Dijkmans, B.C.; et al. Characteristics of interstitial fibrosis and inflammatory cell infiltration in right ventricles of systemic sclerosis-associated pulmonary arterial hypertension. Int. J. Rheumatol. 2010, 2010, 604615. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Kokkonen-Simon, K.M.; Kirk, J.A.; Kolb, T.M.; Damico, R.L.; Mathai, S.C.; Mukherjee, M.; Shah, A.A.; Wigley, F.M.; Margulies, K.B.; et al. Right Ventricular Myofilament Functional Differences in Humans With Systemic Sclerosis-Associated Versus Idiopathic Pulmonary Arterial Hypertension. Circulation 2018, 137, 2360–2370. [Google Scholar] [CrossRef]
- Zhu, N.; Swietlik, E.M.; Welch, C.L.; Pauciulo, M.W.; Hagen, J.J.; Zhou, X.; Guo, Y.; Karten, J.; Pandya, D.; Tilly, T.; et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med. 2021, 13, 80. [Google Scholar] [CrossRef]
- Wipff, J.; Kahan, A.; Hachulla, E.; Sibilia, J.; Cabane, J.; Meyer, O.; Mouthon, L.; Guillevin, L.; Junien, C.; Boileau, C.; et al. Association between an endoglin gene polymorphism and systemic sclerosis-related pulmonary arterial hypertension. Rheumatology 2006, 46, 622–625. [Google Scholar] [CrossRef]
- Morse, J.B.R.; Horn, E.; Cuervo, N.; Deng, Z.; Knowles, J. Pulmonary Hypertension in Scleroderma Spectrum of Disease- Lack of Bone Morphogenetic Protein Receptor 2 Mutations. J. Rheumatol. 2002, 29, 2379–2381. [Google Scholar]
- Unlu, B.; Tursen, U.; Rajabi, Z.; Jabalameli, N.; Rajabi, F. The Immunogenetics of Systemic Sclerosis. Adv. Exp. Med. Biol. 2022, 1367, 259–298. [Google Scholar]
- Broen, J.C.; Bossini-Castillo, L.; van Bon, L.; Vonk, M.C.; Knaapen, H.; Beretta, L.; Rueda, B.; Hesselstrand, R.; Herrick, A.; Worthington, J.; et al. A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. 2012, 64, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Dritsoula, A.; Kang, P.; Baliga, R.S.; Trinder, S.L.; Cook, E.; Shiwen, X.; Hobbs, A.J.; Denton, C.P.; Abraham, D.J.; et al. NKX2-5 regulates vessel remodeling in scleroderma-associated pulmonary arterial hypertension. JCI Insight 2024, 9, e164191. [Google Scholar] [CrossRef] [PubMed]
- Wipff, J.; Dieude, P.; Guedj, M.; Ruiz, B.; Riemekasten, G.; Cracowski, J.L.; Matucci-Cerinic, M.; Melchers, I.; Humbert, M.; Hachulla, E.; et al. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum. 2010, 62, 3093–3100. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gonzalez, I.; Tenorio-Castano, J.; Ochoa-Parra, N.; Gallego, N.; Perez-Olivares, C.; Lago-Docampo, M.; Palomino Doza, J.; Valverde, D.; Lapunzina, P.; Escribano-Subias, P. Novel Genetic and Molecular Pathways in Pulmonary Arterial Hypertension Associated with Connective Tissue Disease. Cells 2021, 10, 1488. [Google Scholar] [CrossRef] [PubMed]
- Mathai, S.C.; Bueso, M.; Hummers, L.K.; Boyce, D.; Lechtzin, N.; Le Pavec, J.; Campo, A.; Champion, H.C.; Housten, T.; Forfia, P.R.; et al. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur. Respir. J. 2010, 35, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Korman, B.D.; Marangoni, R.G.; Hinchcliff, M.; Shah, S.J.; Carns, M.; Hoffmann, A.; Ramsey-Goldman, R.; Varga, J. Elevated Adipsin Levels are Associated with Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis Rheumatol. 2017, 69, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, Z.; Balbir Gurman, A.; Meroni, P.; Farge, D.; Levi, Y.; Ingegnoli, F.; Braun-Moscovici, Y.; Rosner, I.; Slobodin, G.; Rozenbaum, M.; et al. Lysyl oxidase-a possible role in systemic sclerosis-associated pulmonary hypertension: A multicentre study. Rheumatology 2019, 58, 1547–1555. [Google Scholar] [CrossRef]
- Lammi, M.R.; Saketkoo, L.A.; Okpechi, S.C.; Ghonim, M.A.; Wyczechowska, D.; Bauer, N.; Pyakurel, K.; Saito, S.; deBoisblanc, B.P.; Boulares, A.H. Microparticles in systemic sclerosis: Potential pro-inflammatory mediators and pulmonary hypertension biomarkers. Respirology 2019, 24, 675–683. [Google Scholar] [CrossRef]
- Reiseter, S.; Molberg, O.; Gunnarsson, R.; Lund, M.B.; Aalokken, T.M.; Aukrust, P.; Ueland, T.; Garen, T.; Brunborg, C.; Michelsen, A.; et al. Associations between circulating endostatin levels and vascular organ damage in systemic sclerosis and mixed connective tissue disease: An observational study. Arthritis Res. Ther. 2015, 17, 231. [Google Scholar] [CrossRef]
- McMahan, Z.; Schoenhoff, F.; Van Eyk, J.E.; Wigley, F.M.; Hummers, L.K. Biomarkers of pulmonary hypertension in patients with scleroderma: A case-control study. Arthritis Res. Ther. 2015, 17, 201. [Google Scholar] [CrossRef]
- Bellan, M.; Dimagli, A.; Piccinino, C.; Giubertoni, A.; Ianniello, A.; Grimoldi, F.; Sguazzotti, M.; Nerviani, A.; Barini, M.; Carriero, A.; et al. Role of Gas6 and TAM Receptors in the Identification of Cardiopulmonary Involvement in Systemic Sclerosis and Scleroderma Spectrum Disorders. Dis. Markers 2020, 2020, 2696173. [Google Scholar] [CrossRef]
- Alotaibi, M.; Shao, J.; Pauciulo, M.W.; Nichols, W.C.; Hemnes, A.R.; Malhotra, A.; Kim, N.H.; Yuan, J.X.; Fernandes, T.; Kerr, K.M.; et al. Metabolomic Profiles Differentiate Scleroderma-PAH From Idiopathic PAH and Correspond With Worsened Functional Capacity. Chest 2023, 163, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Zaaroor Levy, M.; Rabinowicz, N.; Yamila Kohon, M.; Shalom, A.; Berl, A.; Hornik-Lurie, T.; Drucker, L.; Tartakover Matalon, S.; Levy, Y. MiRNAs in Systemic Sclerosis Patients with Pulmonary Arterial Hypertension: Markers and Effectors. Biomedicines 2022, 10, 629. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Meune, C.; Ruiz, B.; Couraud, P.O.; Uzan, G.; Boileau, C.; Kahan, A.; Chiocchia, G.; Allanore, Y. Angiogenic biomarkers predict the occurrence of digital ulcers in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 394–399. [Google Scholar] [CrossRef]
- Xing, Y.; Zhao, J.; Zhou, M.; Jing, S.; Zhao, X.; Mao, P.; Qian, J.; Huang, C.; Tian, Z.; Wang, Q.; et al. The LPS induced pyroptosis exacerbates BMPR2 signaling deficiency to potentiate SLE-PAH. FASEB J. 2021, 35, e22044. [Google Scholar] [CrossRef] [PubMed]
- Vegh, J.; Szodoray, P.; Kappelmayer, J.; Csipo, I.; Udvardy, M.; Lakos, G.; Aleksza, M.; Soltesz, P.; Szilagyi, A.; Zeher, M.; et al. Clinical and immunoserological characteristics of mixed connective tissue disease associated with pulmonary arterial hypertension. Scand. J. Immunol. 2006, 64, 69–76. [Google Scholar] [CrossRef]
- Tamura, Y.; Ono, T.; Kuwana, M.; Inoue, K.; Takei, M.; Yamamoto, T.; Kawakami, T.; Fujita, J.; Kataoka, M.; Kimura, K.; et al. Human pentraxin 3 (PTX3) as a novel biomarker for the diagnosis of pulmonary arterial hypertension. PLoS ONE 2012, 7, e45834. [Google Scholar] [CrossRef]
- Bellan, M.; Piccinino, C.; Tonello, S.; Minisini, R.; Giubertoni, A.; Sola, D.; Pedrazzoli, R.; Gagliardi, I.; Zecca, E.; Calzaducca, E.; et al. Role of Osteopontin as a Potential Biomarker of Pulmonary Arterial Hypertension in Patients with Systemic Sclerosis and Other Connective Tissue Diseases (CTDs). Pharmaceuticals 2021, 14, 394. [Google Scholar] [CrossRef]
- Adachi, S.; Kikuchi, R.; Shimokata, S.; Suzuki, A.; Yoshida, M.; Imai, R.; Nakano, Y.; Kondo, T.; Murohara, T. Endostatin and Vascular Endothelial Growth Factor-A(165)b May Contribute to Classification of Pulmonary Hypertension. Circ. Rep. 2021, 3, 161–169. [Google Scholar] [CrossRef]
- Lee, J.H.; Im Cho, K. Arterial stiffness, antiphospholipid antibodies, and pulmonary arterial hypertension in systemic lupus erythematosus. J. Cardiol. 2014, 64, 450–455. [Google Scholar] [CrossRef]
- Kim, K.J.; Baek, I.W.; Yoon, C.H.; Kim, W.U.; Cho, C.S. Association of Anemic Hypoxia and Increased Pulmonary Artery Systolic Pressure in Patients With Systemic Lupus Erythematosus. Arthritis Care Res. 2015, 67, 1702–1711. [Google Scholar] [CrossRef]
- Huang, H.; Chen, D.; Pu, J.; Yuan, A.; Fu, Q.; Li, J.; Leng, L.; Bucala, R.; Ye, S.; Lu, L. The small molecule macrophage migration inhibitory factor antagonist MIF098, inhibits pulmonary hypertension associated with murine SLE. Int. Immunopharmacol. 2019, 76, 105874. [Google Scholar] [CrossRef]
- Guo, L.; Li, M.; Chen, Y.; Wang, Q.; Tian, Z.; Pan, S.; Zeng, X.; Ye, S. Anti-Endothelin Receptor Type A Autoantibodies in Systemic Lupus Erythematosus-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2015, 67, 2394–2402. [Google Scholar] [CrossRef]
- Artim-Esen, B.; Cene, E.; Sahinkaya, Y.; Ertan, S.; Pehlivan, O.; Kamali, S.; Gul, A.; Ocal, L.; Aral, O.; Inanc, M. Cluster analysis of autoantibodies in 852 patients with systemic lupus erythematosus from a single center. J. Rheumatol. 2014, 41, 1304–1310. [Google Scholar] [CrossRef]
- Li, J.; Leng, X.; Li, Z.; Ye, Z.; Li, C.; Li, X.; Zhu, P.; Wang, Z.; Zheng, Y.; Li, X.; et al. Chinese SLE treatment and research group registry: III. association of autoantibodies with clinical manifestations in Chinese patients with systemic lupus erythematosus. J. Immunol. Res. 2014, 2014, 809389. [Google Scholar] [CrossRef]
- Zuily, S.; Domingues, V.; Suty-Selton, C.; Eschwege, V.; Bertoletti, L.; Chaouat, A.; Chabot, F.; Regnault, V.; Horn, E.M.; Erkan, D.; et al. Antiphospholipid antibodies can identify lupus patients at risk of pulmonary hypertension: A systematic review and meta-analysis. Autoimmun. Rev. 2017, 16, 576–586. [Google Scholar] [CrossRef]
- Li, M.; Shi, Y.; Zhao, J.; Wang, Q.; Li, M.; Zhao, X. Identification of potential susceptibility genes in patients with primary Sjogren’s syndrome-associated pulmonary arterial hypertension through whole exome sequencing. Arthritis Res. Ther. 2023, 25, 175. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Wang, Q.; Zhang, X.; Qian, J.; Zhao, J.; Xu, D.; Tian, Z.; Wei, W.; Zuo, X.; et al. Pulmonary arterial hypertension associated with primary Sjogren’s syndrome: A multicentre cohort study from China. Eur. Respir. J. 2020, 56, 1902157. [Google Scholar] [CrossRef]
- Shariff, N.; Kumar, A.; Narang, R.; Malhotra, A.; Mukhopadhyaya, S.; Sharma, S.K. A study of pulmonary arterial hypertension in patients with rheumatoid arthritis. Int. J. Cardiol. 2007, 115, 75–76. [Google Scholar] [CrossRef]
- Udayakumar, N.; Venkatesan, S.; Rajendiran, C. Pulmonary hypertension in rheumatoid arthritis--relation with the duration of the disease. Int. J. Cardiol. 2008, 127, 410–412. [Google Scholar] [CrossRef]
- Sadeghi, S.; Granton, J.T.; Akhavan, P.; Pasarikovski, C.R.; Roos, A.M.; Thenganatt, J.; Moric, J.; Johnson, S.R. Survival in rheumatoid arthritis-associated pulmonary arterial hypertension compared with idiopathic pulmonary arterial hypertension. Respirology 2015, 20, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, X.; Gu, B.; Su, D. Identifying the genetic association between rheumatoid arthritis and the risk of pulmonary arterial hypertension. Clin. Rheumatol. 2025, 44, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef]
- Cuttica, M.J.; Langenickel, T.; Noguchi, A.; Machado, R.F.; Gladwin, M.T.; Boehm, M. Perivascular T-cell infiltration leads to sustained pulmonary artery remodeling after endothelial cell damage. Am. J. Respir. Cell Mol. Biol. 2011, 45, 62–71. [Google Scholar] [CrossRef]
- Tamby, M.C.; Chanseaud, Y.; Humbert, M.; Fermanian, J.; Guilpain, P.; Garcia-de-la-Pena-Lefebvre, P.; Brunet, S.; Servettaz, A.; Weill, B.; Simonneau, G.; et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 2005, 60, 765–772. [Google Scholar] [CrossRef]
- Quismorio, F.P.; Jr Sharma, O.; Koss, M.; Boylen, T.; Edmiston, A.W.; Thornton, P.J.; Tatter, D. Immunopathologic and clinical studies in pulmonary hypertension associated with systemic lupus erythematosus. Semin. Arthritis Rheum. 1984, 13, 349–359. [Google Scholar] [CrossRef]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, S.A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Tamosiuniene, R.; Sung, Y.K.; Qian, J.; Dhillon, G.; Gera, L.; Farkas, L.; Rabinovitch, M.; Zamanian, R.T.; et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci. Transl. Med. 2013, 5, 200ra117. [Google Scholar] [CrossRef]
- Thenappan, T.; Goel, A.; Marsboom, G.; Fang, Y.H.; Toth, P.T.; Zhang, H.J.; Kajimoto, H.; Hong, Z.; Paul, J.; Wietholt, C.; et al. A central role for CD68(+) macrophages in hepatopulmonary syndrome. Reversal by macrophage depletion. Am. J. Respir. Crit. Care Med. 2011, 183, 1080–1091. [Google Scholar] [CrossRef]
- Heath, D.; Yacoub, M. Lung mast cells in plexogenic pulmonary arteriopathy. J. Clin. Pathol. 1991, 44, 1003–1006. [Google Scholar] [CrossRef]
- Miyata, M.; Sakuma, F.; Ito, M.; Ohira, H.; Sato, Y.; Kasukawa, R. Athymic nude rats develop severe pulmonary hypertension following monocrotaline administration. Int. Arch. Allergy Immunol. 2000, 121, 246–252. [Google Scholar] [CrossRef]
- Banasova, A.; Maxova, H.; Hampl, V.; Vizek, M.; Povysilova, V.; Novotna, J.; Vajnerova, O.; Hnilickova, O.; Herget, J. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration 2008, 76, 102–107. [Google Scholar] [CrossRef]
- Perros, F.; Dorfmuller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Herve, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef]
- Wang, W.; Yan, H.; Zhu, W.; Cui, Y.; Chen, J.; Wang, X.; Li, S.; Zhu, J. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J. Clin. Immunol. 2009, 29, 705–713. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Iannone, F.; Riccardi, M.T.; Guiducci, S.; Bizzoca, R.; Cinelli, M.; Matucci-Cerinic, M.; Lapadula, G. Bosentan regulates the expression of adhesion molecules on circulating T cells and serum soluble adhesion molecules in systemic sclerosis-associated pulmonary arterial hypertension. Ann. Rheum. Dis. 2008, 67, 1121–1126. [Google Scholar] [CrossRef]
- Ulrich, S.; Nicolls, M.R.; Taraseviciene, L.; Speich, R.; Voelkel, N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration 2008, 75, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Rock, M.T.; Mosse, C.A.; Vnencak-Jones, C.L.; Yoder, S.M.; Robbins, I.M.; Loyd, J.E.; Meyrick, B.O. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir. Med. 2010, 104, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Olewicz-Gawlik, A.; Danczak-Pazdrowska, A.; Kuznar-Kaminska, B.; Gornowicz-Porowska, J.; Katulska, K.; Trzybulska, D.; Batura-Gabryel, H.; Silny, W.; Poplawski, D.; Hrycaj, P. Interleukin-17 and interleukin-23: Importance in the pathogenesis of lung impairment in patients with systemic sclerosis. Int. J. Rheum. Dis. 2014, 17, 664–670. [Google Scholar] [CrossRef]
- Park, S.H.; Chen, W.C.; Esmaeil, N.; Lucas, B.; Marsh, L.M.; Reibman, J.; Grunig, G. Interleukin 13- and interleukin 17A-induced pulmonary hypertension phenotype due to inhalation of antigen and fine particles from air pollution. Pulm. Circ. 2014, 4, 654–668. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Cho, W.K.; Lee, C.M.; Kang, M.J.; Huang, Y.; Giordano, F.J.; Lee, P.J.; Trow, T.K.; Homer, R.J.; Sessa, W.C.; Elias, J.A.; et al. IL-13 receptor alpha2-arginase 2 pathway mediates IL-13-induced pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L112–L124. [Google Scholar] [CrossRef]
- Risbano, M.G.; Meadows, C.A.; Coldren, C.D.; Jenkins, T.J.; Edwards, M.G.; Collier, D.; Huber, W.; Mack, D.G.; Fontenot, A.P.; Geraci, M.W.; et al. Altered immune phenotype in peripheral blood cells of patients with scleroderma-associated pulmonary hypertension. Clin. Transl. Sci. 2010, 3, 210–218. [Google Scholar] [CrossRef]
- Swain, S.D.; Siemsen, D.W.; Pullen, R.R.; Han, S. CD4+ T cells and IFN-gamma are required for the development of Pneumocystis-associated pulmonary hypertension. Am. J. Pathol. 2014, 184, 483–493. [Google Scholar] [CrossRef]
- Daley, E.; Emson, C.; Guignabert, C.; de Waal Malefyt, R.; Louten, J.; Kurup, V.P.; Hogaboam, C.; Taraseviciene-Stewart, L.; Voelkel, N.F.; Rabinovitch, M.; et al. Pulmonary arterial remodeling induced by a Th2 immune response. J. Exp. Med. 2008, 205, 361–372. [Google Scholar] [CrossRef]
- Hautefort, A.; Girerd, B.; Montani, D.; Cohen-Kaminsky, S.; Price, L.; Lambrecht, B.N.; Humbert, M.; Perros, F. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest 2015, 147, 1610–1620. [Google Scholar] [CrossRef]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Gonzalez Bosc, L.V. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L609–L624. [Google Scholar] [CrossRef]
- Li, C.; Liu, P.P.; Tang, D.D.; Song, R.; Zhang, Y.Q.; Lei, S.; Wu, S.J. Targeting the RhoA-ROCK pathway to regulate T-cell homeostasis in hypoxia-induced pulmonary arterial hypertension. Pulm. Pharmacol. Ther. 2018, 50, 111–122. [Google Scholar] [CrossRef]
- Gaowa, S.; Zhou, W.; Yu, L.; Zhou, X.; Liao, K.; Yang, K.; Lu, Z.; Jiang, H.; Chen, X. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflamm. 2014, 2014, 247372. [Google Scholar] [CrossRef]
- Papadimitriou, T.I.; Lemmers, J.M.; van Caam, A.P.; Vos, J.L.; Vitters, E.L.; Stinissen, L.; van Leuven, S.I.; Koenders, M.I.; van der Kraan, P.M.; Koenen, H.J.P.M. Systemic sclerosis-associated pulmonary arterial hypertension is characterized by a distinct peripheral T helper cell profile. Rheumatology 2024, 63, 2525–2534. [Google Scholar] [CrossRef]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Chu, Y.; Xiangli, X.; Xiao, W. Regulatory T cells protect against hypoxia-induced pulmonary arterial hypertension in mice. Mol. Med. Rep. 2015, 11, 3181–3187. [Google Scholar] [CrossRef]
- Perros, F.; Dorfmuller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef]
- Colvin, K.L.; Cripe, P.J.; Ivy, D.D.; Stenmark, K.R.; Yeager, M.E. Bronchus-associated lymphoid tissue in pulmonary hypertension produces pathologic autoantibodies. Am. J. Respir. Crit. Care Med. 2013, 188, 1126–1136. [Google Scholar] [CrossRef]
- Sanges, S.; Guerrier, T.; Duhamel, A.; Guilbert, L.; Hauspie, C.; Largy, A.; Balden, M.; Podevin, C.; Lefevre, G.; Jendoubi, M.; et al. Soluble markers of B cell activation suggest a role of B cells in the pathogenesis of systemic sclerosis-associated pulmonary arterial hypertension. Front. Immunol. 2022, 13, 954007. [Google Scholar] [CrossRef]
- Nunes, J.P.L.; Cunha, A.C.; Meirinhos, T.; Nunes, A.; Araujo, P.M.; Godinho, A.R.; Vilela, E.M.; Vaz, C. Prevalence of auto-antibodies associated to pulmonary arterial hypertension in scleroderma—A review. Autoimmun. Rev. 2018, 17, 1186–1201. [Google Scholar] [CrossRef]
- Mehra, S.; Walker, J.; Patterson, K.; Fritzler, M.J. Autoantibodies in systemic sclerosis. Autoimmun. Rev. 2013, 12, 340–354. [Google Scholar] [CrossRef]
- Sobanski, V.; Giovannelli, J.; Lynch, B.M.; Schreiber, B.E.; Nihtyanova, S.I.; Harvey, J.; Handler, C.E.; Denton, C.P.; Coghlan, J.G. Characteristics and Survival of Anti-U1 RNP Antibody-Positive Patients With Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2016, 68, 484–493. [Google Scholar] [CrossRef]
- Merashli, M.; Alves, J.; Ames, P.R.J. Clinical relevance of antiphospholipid antibodies in systemic sclerosis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2017, 46, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Svegliati Baroni, S.; Santillo, M.; Bevilacqua, F.; Luchetti, M.; Spadoni, T.; Mancini, M.; Fraticelli, P.; Sambo, P.; Funaro, A.; Kazlauskas, A.; et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N. Engl. J. Med. 2006, 354, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Moroncini, G.; Grieco, A.; Nacci, G.; Paolini, C.; Tonnini, C.; Pozniak, K.N.; Cuccioloni, M.; Mozzicafreddo, M.; Svegliati, S.; Angeletti, M.; et al. Epitope Specificity Determines Pathogenicity and Detectability of Anti-Platelet-Derived Growth Factor Receptor alpha Autoantibodies in Systemic Sclerosis. Arthritis Rheumatol. 2015, 67, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, M.M.; Moroncini, G.; Jose Escamez, M.; Svegliati Baroni, S.; Spadoni, T.; Grieco, A.; Paolini, C.; Funaro, A.; Avvedimento, E.V.; Larcher, F.; et al. Induction of Scleroderma Fibrosis in Skin-Humanized Mice by Administration of Anti-Platelet-Derived Growth Factor Receptor Agonistic Autoantibodies. Arthritis Rheumatol. 2016, 68, 2263–2273. [Google Scholar] [CrossRef]
- Chizzolini, C.; Raschi, E.; Rezzonico, R.; Testoni, C.; Mallone, R.; Gabrielli, A.; Facchini, A.; Del Papa, N.; Borghi, M.O.; Dayer, J.M.; et al. Autoantibodies to fibroblasts induce a proadhesive and proinflammatory fibroblast phenotype in patients with systemic sclerosis. Arthritis Rheum. 2002, 46, 1602–1613. [Google Scholar] [CrossRef]
- Tamby, M.C.; Humbert, M.; Guilpain, P.; Servettaz, A.; Dupin, N.; Christner, J.J.; Simonneau, G.; Fermanian, J.; Weill, B.; Guillevin, L.; et al. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur. Respir. J. 2006, 28, 799–807. [Google Scholar] [CrossRef]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef]
- Gairhe, S.; Joshi, S.R.; Bastola, M.M.; McLendon, J.M.; Oka, M.; Fagan, K.A.; McMurtry, I.F. Sphingosine-1-phosphate is involved in the occlusive arteriopathy of pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 369–380. [Google Scholar] [CrossRef]
- Gluschke, H.; Siegert, E.; Minich, W.B.; Hackler, J.; Riemekasten, G.; Kuebler, W.M.; Simmons, S.; Schomburg, L. Autoimmunity to Sphingosine-1-Phosphate-Receptors in Systemic Sclerosis and Pulmonary Arterial Hypertension. Front. Immunol. 2022, 13, 935787. [Google Scholar] [CrossRef]
- Carvalho, D.; Savage, C.O.; Black, C.M.; Pearson, J.D. IgG antiendothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. Induction of adhesion molecule expression and involvement of endothelium-derived cytokines. J. Clin. Investig. 1996, 97, 111–119. [Google Scholar] [CrossRef]
- Liu, X.D.; Guo, S.Y.; Yang, L.L.; Zhang, X.L.; Fu, W.Y.; Wang, X.F. Anti-endothelial cell antibodies in connective tissue diseases associated with pulmonary arterial hypertension. J. Thorac. Dis. 2014, 6, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Khadilkar, P.; Chougule, D.; Tipnis, T.; Khopkar, U.; Nadkar, M.; Rajadhyaksha, A.; Kini, S.; Kharkar, V.; Athvale, A.; Athvale, T.; et al. A comparative study of modulatory interaction between cytokines and apoptotic proteins among Scleroderma patients with and without pulmonary involvement. Cytokine 2023, 166, 156183. [Google Scholar] [CrossRef] [PubMed]
- Christmann, R.B.; Hayes, E.; Pendergrass, S.; Padilla, C.; Farina, G.; Affandi, A.J.; Whitfield, M.L.; Farber, H.W.; Lafyatis, R. Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheum. 2011, 63, 1718–1728. [Google Scholar] [CrossRef]
- Pendergrass, S.A.; Hayes, E.; Farina, G.; Lemaire, R.; Farber, H.W.; Whitfield, M.L.; Lafyatis, R. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE 2010, 5, e12106. [Google Scholar] [CrossRef]
- Di Benedetto, P.; Guggino, G.; Manzi, G.; Ruscitti, P.; Berardicurti, O.; Panzera, N.; Grazia, N.; Badagliacca, R.; Riccieri, V.; Vizza, C.D.; et al. Interleukin-32 in systemic sclerosis, a potential new biomarker for pulmonary arterial hypertension. Arthritis Res. Ther. 2020, 22, 127. [Google Scholar] [CrossRef]
- Seki, N.; Tsujimoto, H.; Tanemura, S.; Ishigaki, S.; Takei, H.; Sugahara, K.; Yoshimoto, K.; Akiyama, M.; Kaneko, Y.; Chiba, K.; et al. Th17/IL-17A axis is critical for pulmonary arterial hypertension (PAH) in systemic sclerosis (SSc): SSc patients with high levels of serum IL-17A exhibit reduced lung functions and increased prevalence of PAH. Cytokine 2024, 176, 156534. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Asano, Y.; Taniguchi, T.; Minatsuki, S.; Inaba, T.; Maki, H.; Hatano, M.; Yamashita, T.; Saigusa, R.; Ichimura, Y.; et al. Serum levels of interleukin-18-binding protein isoform a: Clinical association with inflammation and pulmonary hypertension in systemic sclerosis. J. Dermatol. 2016, 43, 912–918. [Google Scholar] [CrossRef]
- Kolstad, K.D.; Khatri, A.; Donato, M.; Chang, S.E.; Li, S.; Steen, V.D.; Utz, P.J.; Khatri, P.; Chung, L. Cytokine signatures differentiate systemic sclerosis patients at high versus low risk for pulmonary arterial hypertension. Arthritis Res. Ther. 2022, 24, 39. [Google Scholar] [CrossRef]
- Meadows, C.A.; Risbano, M.G.; Zhang, L.; Geraci, M.W.; Tuder, R.M.; Collier, D.H.; Bull, T.M. Increased expression of growth differentiation factor-15 in systemic sclerosis-associated pulmonary arterial hypertension. Chest 2011, 139, 994–1002. [Google Scholar] [CrossRef]
- Vakkalanka, R.K.; Woo, C.; Kirou, K.A.; Koshy, M.; Berger, D.; Crow, M.K. Elevated levels and functional capacity of soluble CD40 ligand in systemic lupus erythematosus sera. Arthritis Rheum. 1999, 42, 871–881. [Google Scholar] [CrossRef]
- Jinnin, M.; Ihn, H.; Yazawa, N.; Asano, Y.; Yamane, K.; Tamaki, K. Elevated circulating soluble CD40 ligand in patients with mixed connective tissue disease. Clin. Rheumatol. 2003, 22, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Borderie, D.; Meune, C.; Lemarechal, H.; Weber, S.; Ekindjian, O.G.; Kahan, A. Increased plasma soluble CD40 ligand concentrations in systemic sclerosis and association with pulmonary arterial hypertension and digital ulcers. Ann. Rheum. Dis. 2004, 64, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, T.; Miyagawa, T.; Fukui, Y.; Minatsuki, S.; Maki, H.; Inaba, T.; Hatano, M.; Toyama, S.; Omatsu, J.; Awaji, K.; et al. Association of serum CCL20 levels with pulmonary vascular involvement and primary biliary cholangitis in patients with systemic sclerosis. Int. J. Rheum. Dis. 2021, 24, 711–718. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.M.; Hesselstrand, R.; Fretheim, H.; Ueland, T.; Andreassen, A.K.; Brunborg, C.; Palchevskiy, V.; Midtvedt, O.; Garen, T.; Aukrust, P.; et al. CCL21 as a Potential Serum Biomarker for Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis Rheumatol. 2018, 70, 1644–1653. [Google Scholar] [CrossRef]
- Didriksen, H.; Molberg, O.; Mehta, A.; Jordan, S.; Palchevskiy, V.; Fretheim, H.; Gude, E.; Ueland, T.; Brunborg, C.; Garen, T.; et al. Target organ expression and biomarker characterization of chemokine CCL21 in systemic sclerosis associated pulmonary arterial hypertension. Front. Immunol. 2022, 13, 991743. [Google Scholar] [CrossRef]
- Rabquer, B.J.; Tsou, P.S.; Hou, Y.; Thirunavukkarasu, E.; Haines, G.K., 3rd; Impens, A.J.; Phillips, K.; Kahaleh, B.; Seibold, J.R.; Koch, A.E. Dysregulated expression of MIG/CXCL9, IP-10/CXCL10 and CXCL16 and their receptors in systemic sclerosis. Arthritis Res. Ther. 2011, 13, R18. [Google Scholar] [CrossRef]
- van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef]
- Sakata, K.; Nakayamada, S.; Miyazaki, Y.; Kubo, S.; Ishii, A.; Nakano, K.; Tanaka, Y. Up-Regulation of TLR7-Mediated IFN-alpha Production by Plasmacytoid Dendritic Cells in Patients with Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1957. [Google Scholar] [CrossRef] [PubMed]
- Yokogawa, M.; Takaishi, M.; Nakajima, K.; Kamijima, R.; Fujimoto, C.; Kataoka, S.; Terada, Y.; Sano, S. Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: A new model of systemic Lupus erythematosus. Arthritis Rheumatol. 2014, 66, 694–706. [Google Scholar] [CrossRef]
- Taraseviciene-Stewart, L.; Nicolls, M.R.; Kraskauskas, D.; Scerbavicius, R.; Burns, N.; Cool, C.; Wood, K.; Parr, J.E.; Boackle, S.A.; Voelkel, N.F. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am. J. Respir. Crit. Care Med. 2007, 175, 1280–1289. [Google Scholar] [CrossRef]
- Yeh, F.C.; Chen, C.N.; Xie, C.Y.; Baxan, N.; Zhao, L.; Ashek, A.; Sabrin, F.; Lawrie, A.; Wilkins, M.; Zhao, L. TLR7/8 activation induces autoimmune vasculopathy and causes severe pulmonary arterial hypertension. Eur. Respir. J. 2023, 62, 2300204. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Ishibashi, T.; Inagaki, T.; Okazawa, M.; Masaki, T.; Asano, R.; Manabe, Y.; Ohta-Ogo, K.; Narazaki, M.; Ishibashi-Ueda, H.; et al. Pristane/Hypoxia (PriHx) Mouse as a Novel Model of Pulmonary Hypertension Reflecting Inflammation and Fibrosis. Circ. J. 2020, 84, 1163–1172. [Google Scholar] [CrossRef]
- Eferl, R.; Hasselblatt, P.; Rath, M.; Popper, H.; Zenz, R.; Komnenovic, V.; Idarraga, M.H.; Kenner, L.; Wagner, E.F. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc. Natl. Acad. Sci. USA 2008, 105, 10525–10530. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Reich, N.; Juengel, A.; Kriegsmann, J.; Gay, R.E.; Schett, G.; Michel, B.A.; Gay, S.; Distler, J.H.; Distler, O. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1382–1387. [Google Scholar] [CrossRef]
- Asano, Y.; Bujor, A.M.; Trojanowska, M. The impact of Fli1 deficiency on the pathogenesis of systemic sclerosis. J. Dermatol. Sci. 2010, 59, 153–162. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, P.S.; Kahaleh, B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006, 54, 2271–2279. [Google Scholar] [CrossRef]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Investig. 2010, 120, 254–265. [Google Scholar] [CrossRef]
- Fujiu, K.; Manabe, I.; Nagai, R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J. Clin. Investig. 2011, 121, 3425–3441. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Asano, Y.; Nishimura, S.; Taniguchi, T.; Fujiu, K.; Manabe, I.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Akamata, K.; et al. Simultaneous downregulation of KLF5 and Fli1 is a key feature underlying systemic sclerosis. Nat. Commun. 2014, 5, 5797. [Google Scholar] [CrossRef]
- Douni, E.; Akassoglou, K.; Alexopoulou, L.; Georgopoulos, S.; Haralambous, S.; Hill, S.; Kassiotis, G.; Kontoyiannis, D.; Pasparakis, M.; Plows, D.; et al. Transgenic and knockout analyses of the role of TNF in immune regulation and disease pathogenesis. J. Inflamm. 1995, 47, 27–38. [Google Scholar]
- Bawadekar, M.; Gendron-Fitzpatrick, A.; Rebernick, R.; Shim, D.; Warner, T.F.; Nicholas, A.P.; Lundblad, L.K.; Thompson, P.R.; Shelef, M.A. Tumor necrosis factor alpha, citrullination, and peptidylarginine deiminase 4 in lung and joint inflammation. Arthritis Res. Ther. 2016, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; White, R.J.; Garcia-Hernandez, M.L.; Wu, E.; Rahimi, H.; Marangoni, R.G.; Slattery, P.; Duemmel, S.; Nuzzo, M.; Huertas, N.; et al. Tumor Necrosis Factor Induces Obliterative Pulmonary Vascular Disease in a Novel Model of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2020, 72, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
| 1. SSc-PAH | |||
| Serum Levels | Biomarkers | Reference | |
| Higher in SSc-PAH patients than in SSc-nonPAH patients | ADMA, CD144+ EMP, endostatin, PlGF, soluble Mer, sFlt-1, fatty acyl esters of hydroxy fatty acid, 17β-estradiol, prostaglandin F2α, nervonic acid, a novel eicosanoid | [22,118,119,120,121,122] | |
| Lower in SSc-PAH patients than in SSc-nonPAH patients | miR-26, miR-let-7d | [123] | |
| Biomarkers | Function and Clinical Meanings | Reference | |
| ADMA | Negatively correlated with 6MWD in SSc-PAH patients | [22] | |
| Adipsin |
| [116] | |
| EMP | Modulated cellular signaling and was involved in the development of vascular disease | [118] | |
| Endostatin | Higher levels were associated with increased mortality in SSc patients | [119] | |
| NT-proBNP | Correlated with survival prediction in SSc-PAH patients | [27,115] | |
| LOX |
| [117] | |
| PlGF | Inversely correlated with DLCO in SSc patients | [120,124] | |
| sFlt-1 | Positively correlated with right ventricular systolic pressure but negatively correlated with DLCO in SSc patients | [120] | |
| 2. Non-SSc CTD-PAH | |||
| Serum Levels | Biomarkers | Reference | |
| Higher in SLE-PAH patients | Lipopolysaccharide | [125] | |
| Higher in MCTD-PAH patients | Thrombomodulin, von Willebrand factor antigen | [126] | |
| 3. CTD-PAH | |||
| Serum Levels | Biomarkers | Function and Clinical Meanings | Reference |
| Higher in CTD-PAH patients | PTX3 |
| [127] |
| Osteopontin | Associated with inflammation, fibrogenesis, and vascular remodeling process | [128] | |
| Endostatin, PlGF |
| [129] | |
| 1. SSc-PAH | |||
| Antibodies | Prevalence and Serum Levels | Function and Clinical Meanings | Reference |
| ANA | Positive in more than 80% of SSc-PAH patients | Hallmarks of CTDs | [178] |
| Anticentromere antibodies | Positive in almost half of SSc-PAH patients | One of the parameters in the DETECT study’s algorithm for detecting SSc-PAH | [10,178] |
| aPL | Positive in almost half of SSc-PAH patients | The potential role in thrombosis and other vascular manifestations in SSc patients | [178,181] |
| Anti-U1 RNP antibodies | Positive in 2–14% of SSc patients | SSc-PAH patients with anti-U1 RNP antibodies were younger at the time of PAH diagnosis and showed better predicted DLCO and WHO functional class | [179,180] |
| Stimulatory anti-PDGFR autoantibodies | High prevalence of anti-PDGFRα autoantibodies in SSc patients |
| [61,182,183,184] |
| Anti-fibroblast antibodies | Positive in 30% of SSc-PAH patients | Activated collagen synthesis by enhancing the expression of ICAM-1 | [185,186] |
| S1PR autoantibodies | Higher prevalence of S1PR2 and S1PR3 autoantibodies in SSc-PAH patients | Mediated cell proliferation, migration, activation, fibrosis, and vasoconstriction | [187,188,189] |
| Anti-AT1R antibodies Anti-ETAR antibodies | Higher serum levels in SSc-PAH and CTD-PAH patients than in other forms of PH | Predictive value for PAH development in SSc patients and prognosis in SSc-PAH patients | [32] |
| 2. Non-SSc CTD-PAH | |||
| SLE-PAH | |||
| Antibodies | Prevalence and Serum Levels | Function and Clinical Meanings | Reference |
| Anti-U1 RNP antibodies | Higher prevalence in SLE-PAH patients | Indicated the presence of vasculopathy | [133,134,135] |
| aPL |
|
| [130,136] |
| Anti-ETAR antibodies | Higher prevalence in SLE-PAH patients |
| [133] |
| pSS-PAH | |||
| Antibodies | Prevalence and Serum Levels | Function and Clinical Meanings | Reference |
| Anti-U1 RNP antibodies Anti-SSB antibodies | Higher prevalence in pSS-PAH patients | Associated with the risk of developing PAH in primary Sjögren’s disease patients | [138] |
| MCTD-PAH | |||
| Antibodies | Prevalence and Serum Levels | Reference | |
| AECA IgM aCL | Higher prevalence and serum levels in MCTD-PAH patients | [126] | |
| 3. CTD-PAH | |||
| Antibodies | Prevalence and Serum Levels | Function and Clinical Meanings | Reference |
| AECA |
|
| [190,191] |
| 1. SSc-PAH | |||
| Serum Levels | Cytokines and Chemokines | Reference | |
| Higher in SSc-PAH patients than in SSc-nonPAH patients | BDNF, CCL21, CXCL16, ET-1, EGF, GDF-15, IL-1β, IL-6, IL-13, IL-22, IL-32, IFN-β, IP10, Leptin, PAI-1, Resistin, sCD40L, TGF-α, TGF-β2, TNF-α, VEGF-D | [36,37,192,193,194,195,198,199,202,204,205,206] | |
| Lower in SSc-PAH patients than in SSc-nonPAH patients | IL-4, TGF-β1 | [192] | |
| Cytokines and Chemokines | Function and Clinical Meanings | Reference | |
| CCL 20 |
| [203] | |
| CCL 21 | Predictive value for the development of PAH in SSc patients | [204,205] | |
| CXCL 4 | Higher serum levels in SSc patients and correlated with PAH | [207] | |
| GDF-15 |
| [199] | |
| IL-13 | Stimulated the expression of MRC1, which served as a marker of alternative activation of macrophages, regulated the immune response, and correlated with PAP in SSc patients | [193] | |
| IL-17A | SSc patients with detected IL-17A had a higher prevalence of PAH | [196] | |
| IL-18BPa |
| [197] | |
| IL-32 | Regulated endothelial cell activities and positively correlated with mean PAP in SSc patients | [195] | |
| IP10 | Positively correlated with mean PAP, PVR, and serum BNP levels in SSc-PAH patients | [36] | |
| 2. Non-SSc CTD-PAH | |||
| Serum Levels | Cytokines and Chemokines | Function and Clinical Meanings | Reference |
| Higher in SLE-PAH patients | MIF | Mediated cell proliferation and survival | [132] |
| Higher in MCTD-PAH patients | IL-6 | Indicated active acute inflammation | [126] |
| 3. CTD-PAH | |||
| Serum Levels | Cytokines and Chemokines | Function and Clinical Meanings | Reference |
| Higher in CTD-PAH patients | IL-17 |
| [62,171] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, F.-C.; Tsai, I.-T.; Chyuan, I.-T. Molecular Pathogenesis of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension: A Narrative Review. Biomolecules 2025, 15, 772. https://doi.org/10.3390/biom15060772
Yeh F-C, Tsai I-T, Chyuan I-T. Molecular Pathogenesis of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension: A Narrative Review. Biomolecules. 2025; 15(6):772. https://doi.org/10.3390/biom15060772
Chicago/Turabian StyleYeh, Fu-Chiang, I-Ting Tsai, and I-Tsu Chyuan. 2025. "Molecular Pathogenesis of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension: A Narrative Review" Biomolecules 15, no. 6: 772. https://doi.org/10.3390/biom15060772
APA StyleYeh, F.-C., Tsai, I.-T., & Chyuan, I.-T. (2025). Molecular Pathogenesis of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension: A Narrative Review. Biomolecules, 15(6), 772. https://doi.org/10.3390/biom15060772