
Molecular Mechanisms of Epithelial–Mesenchymal Transition in Retinal Pigment Epithelial Cells: Implications for Age-Related Macular Degeneration (AMD) Progression

Abstract
1. Introduction
2. Molecular Mechanisms Underlying EMT
3. Molecular Drivers and Regulatory Mechanisms of EMT in AMD
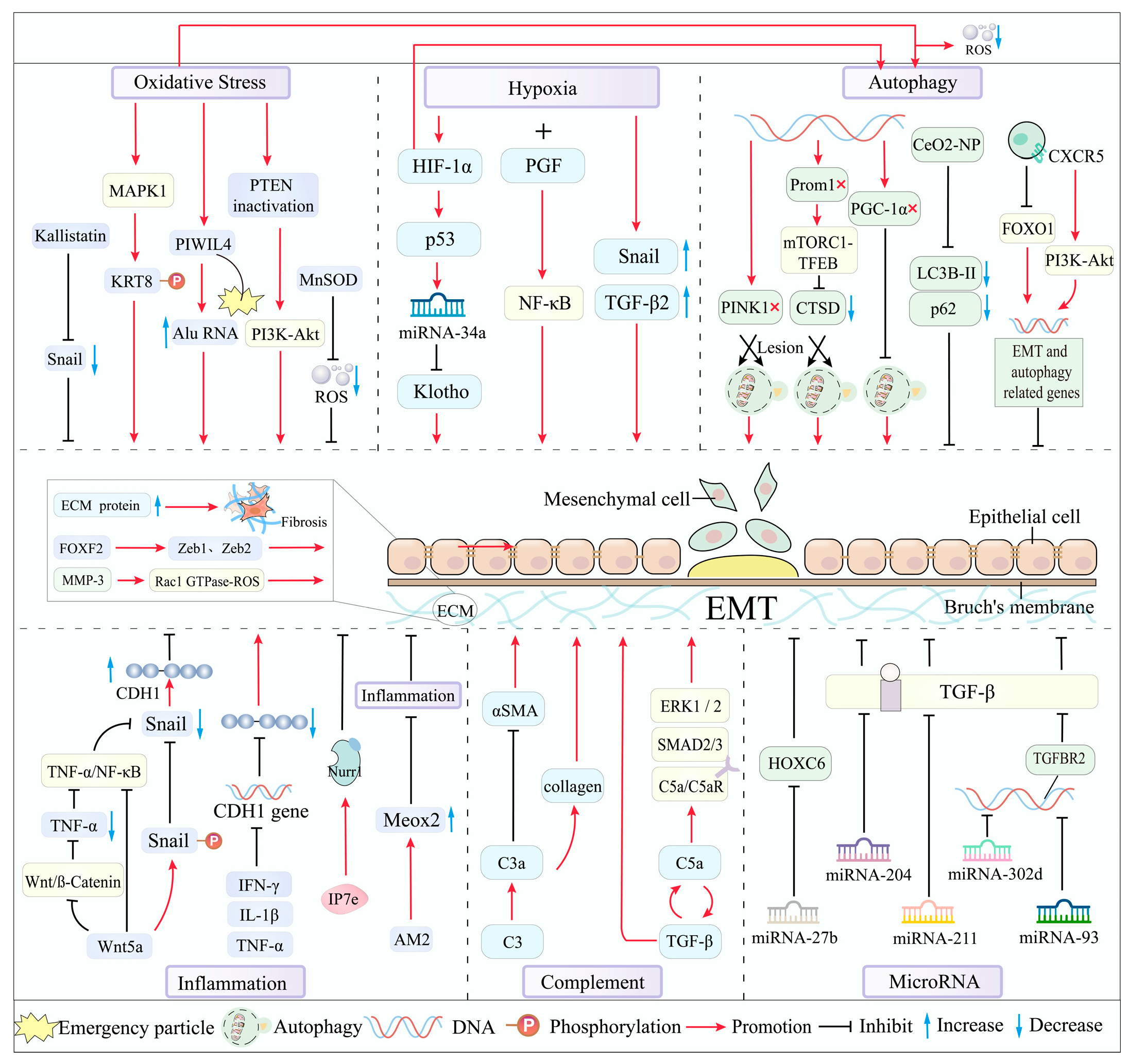
3.1. Oxidative Stress: A Potent Driver of the EMT/AMD Cascade
3.2. Hypoxia: Inducing the EMT Process in RPE Cells
3.3. Autophagy: Orchestrating the EMT Process in RPE Cells
3.4. Inflammation: Inhibiting EMT by Protecting RPE Cells
3.5. Complement Activation: Promoting Fibrosis Through EMT
3.6. MicroRNA: A Regulator of EMT
3.7. Extracellular Matrix (ECM): Balancing Dynamics in EMT of AMD
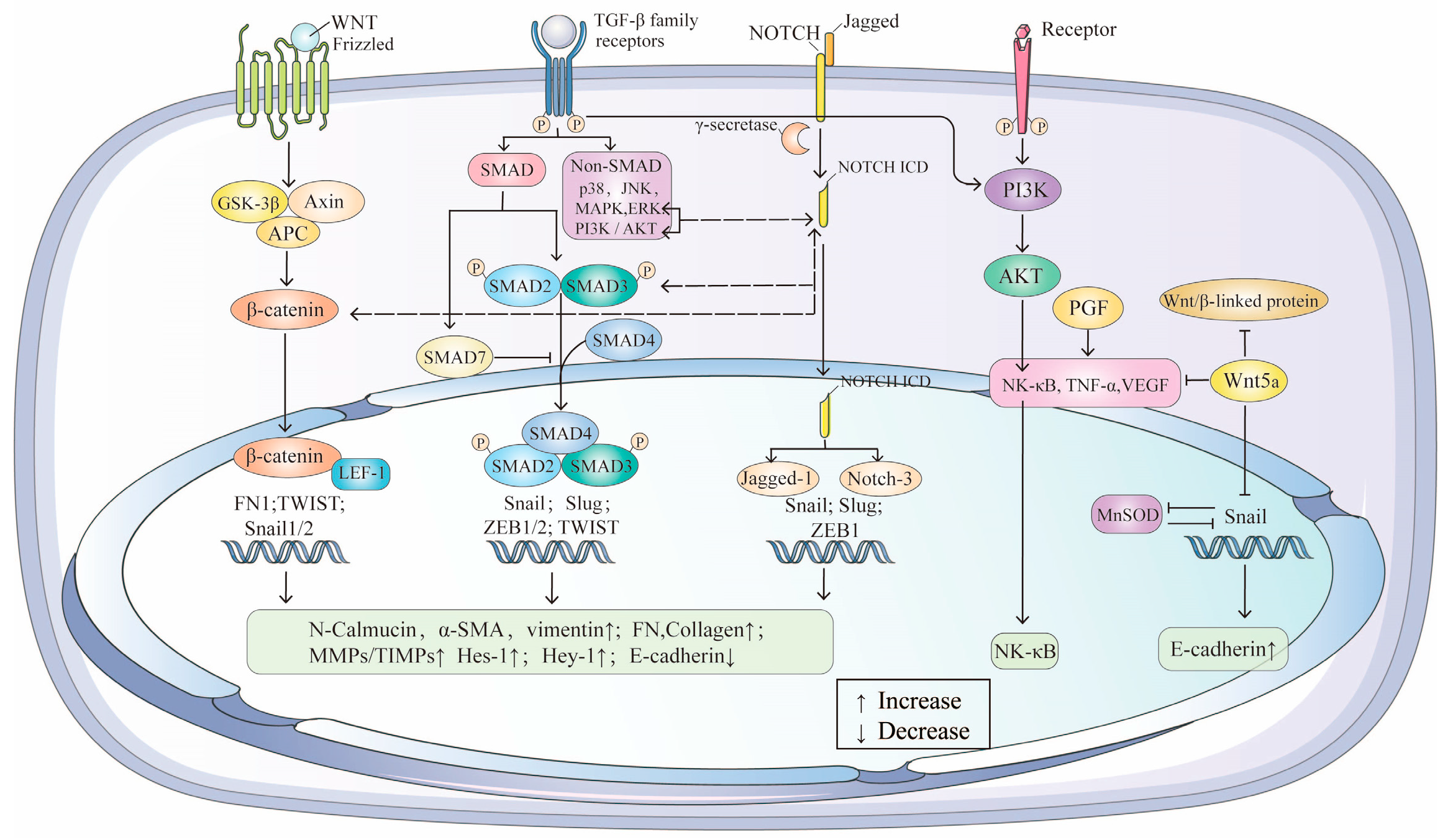
4. Roles of Key Cytokine-Mediated Signaling Pathways in EMT/AMD
4.1. TGF-β/SMAD: The Central Signaling Pathway in EMT/AMD
4.2. Wnt/β-Catenin: A Collaborative Pathway in EMT/AMD
4.3. Jagged/Notch: A Fibrosis-Related Pathway in EMT/AMD
5. Therapeutic Strategies Targeting EMT in AMD
5.1. Inhibiting EMT in AMD Through TGF-β Pathway Modulation
5.2. Targeting Oxidative Stress to Inhibit EMT in AMD
5.3. Reversing EMT in AMD to Restore Epithelial Function
5.4. Addressing Risk Factors and Cellular Senescence
5.5. Cell Replacement Strategies to Target RPE-EMT in AMD
5.6. Omics Landscapes of RPE-EMT
5.7. Epigenetic Regulation of RPE-EMT: Implications for AMD Therapy
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| AMD | Age-related Macular Degeneration |
| RPE | Retinal Pigment Epithelial |
| EMT | Epithelial–mesenchymal Transition |
| ECM | Extracellular Matrix |
| VEGF | Vascular Endothelial Growth Factor |
| CNV | Choroidal Neovascularization |
| TGF-β | Transforming Growth Factor Beta |
| MET | Mesenchymal–epithelial Transition |
| ROS | Reactive Oxygen Species |
| KRT8 | Keratin 8 |
| MnSOD | Manganese Superoxide Dismutase |
| CSE | Cigarette Smoke Extract |
| HIF | Hypoxia-inducible Factor |
| PGF | Placental Growth Factor |
| PINK1 | PTEN Induced Kinase 1 |
| RMNS | Retrograde Mitochondrial Nuclear Signaling |
| PGC-1α | Peroxisome proliferator-activated Receptor γ Coactivator-1α |
| CXCR5 | C-X-C Motif Chemokine Receptor 5 |
| CeO2-NP | Cerium Oxide Nanoparticles |
| MiRNA | MicroRNA |
| HucMSC-Exo | Human Umbilical Cord Mesenchymal Stem Cell Exosomes |
| MMPs | Matrix Metalloproteinases |
| TIMPs | MMP Tissue Inhibitors |
| CTGF | Connective Tissue Growth Factor |
| FN | Fibronectin |
| POSTN | Periostin |
| FAK | Focal Adhesion Kinase |
| TIMPs | MMP Tissue Inhibitors |
| MAPK | Mitogen-activated Protein Kinase |
| PI3K | Phosphatidylinositol-3-kinase |
| NICD | Extracellular and Intracellular Structural Domains |
| RAR | Retinoic Acid Receptor |
| ERK | Extracellular Signal-regulated Kinase |
| CeO2-NPs | Cerium Oxide Nanoparticles |
| hRPESC-RPE | Human RPE Stem Cells |
References
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The e-cadherin and n-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.Q.; Saint-Geniez, M.; Kim, L.A.; Shu, D.Y. Divergent metabolomic signatures of tgfβ2 and tnfα in the induction of retinal epithelial-mesenchymal transition. Metabolites 2023, 13, 213. [Google Scholar] [CrossRef]
- Yonekawa, Y.; Miller, J.W.; Kim, I.K. Age-related macular degeneration: Advances in management and diagnosis. J. Clin. Med. 2015, 4, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.; Sheridan, C. Choroidal neovascularization: A wound healing perspective. Mol. Vis. 2003, 9, 747–755. [Google Scholar]
- Gao, F.; Wang, L.; Wu, B.; Ou, Q.; Tian, H.; Xu, J.; Jin, C.; Zhang, J.; Wang, J.; Lu, L.; et al. Elimination of senescent cells inhibits epithelial-mesenchymal transition of retinal pigment epithelial cells. Exp. Eye Res. 2022, 223, 109207. [Google Scholar] [CrossRef]
- Ishikawa, K.; Kannan, R.; Hinton, D.R. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 2016, 142, 19–25. [Google Scholar] [CrossRef]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor. Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wu, K.J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef]
- Ghosh, S.; Shang, P.; Terasaki, H.; Stepicheva, N.; Hose, S.; Yazdankhah, M.; Weiss, J.; Sakamoto, T.; Bhutto, I.A.; Xia, S.; et al. A role for βa3/a1-crystallin in type 2 emt of rpe cells occurring in dry age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 59, Amd104–Amd113. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, C.; Zhang, J.; Xu, G.T.; Zhang, J. Molecular pathogenesis of subretinal fibrosis in neovascular AMD focusing on epithelial-mesenchymal transformation of retinal pigment epithelium. Neurobiol. Dis. 2023, 185, 106250. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The role of inflammation in age-related macular degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Finnegan, L.K.; Post, I.J.M.; Carrigan, M.; Gardiner, T.; Peixoto, E.; Maloney, D.; Humphries, M.M.; Stitt, A.; et al. AAV-mediated gene therapy improving mitochondrial function provides benefit in age-related macular degeneration models. Clin. Transl. Med. 2022, 12, e952. [Google Scholar] [CrossRef]
- Xu, M.; Gao, Y.; Yin, W.; Liu, Q.; Yuan, S. RNA-sequencing expression profile and functional analysis of retinal pigment epithelium in atrophic age-related macular degeneration. J. Biomed. Res. 2024, 38, 500. [Google Scholar] [CrossRef]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Hobbs, S.D.; Tripathy, K.; Pierce, K. Wet age-related macular degeneration (amd). In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Holz, F.G.; Schmitz-Valckenberg, S.; Fleckenstein, M. Recent developments in the treatment of age-related macular degeneration. J. Clin. Investig. 2014, 124, 1430–1438. [Google Scholar] [CrossRef]
- Daniel, E.; Toth, C.A.; Grunwald, J.E.; Jaffe, G.J.; Martin, D.F.; Fine, S.L.; Huang, J.; Ying, G.S.; Hagstrom, S.A.; Winter, K.; et al. Risk of scar in the comparison of age-related macular degeneration treatments trials. Ophthalmology 2014, 121, 656–666. [Google Scholar] [CrossRef]
- Im, S.; Song, M.H.; Elangovan, M.; Woo, K.M.; Park, W.J. The matricellular protein CCN5 prevents anti-VEGF drug-induced epithelial-mesenchymal transition of retinal pigment epithelium. Sci. Rep. 2024, 14, 13920. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D.; Kim, Y.S.; Kim, J.W.; Sun, H.; Roh, K.H.; Yang, J.W.; Hur, D.Y. Regulation of adam10 and adam17 by sorafenib inhibits epithelial-to-mesenchymal transition in epstein-barr virus-infected retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5162–5173. [Google Scholar] [CrossRef] [PubMed]
- McCabe, E.M.; Rasmussen, T.P. lncRNA involvement in cancer stem cell function and epithelial-mesenchymal transitions. Semin. Cancer Biol. 2021, 75, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging roles in age-related macular degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef]
- Lovisa, S.; Genovese, G.; Danese, S. Role of epithelial-to-mesenchymal transition in inflammatory bowel disease. J. Crohn’s Colitis 2019, 13, 659–668. [Google Scholar] [CrossRef]
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of epithelial-mesenchymal transition in retinal pigment epithelium dysfunction. Front. Cell Dev. Biol. 2020, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Di, Y.; Li, Q.; Zhan, Q.; He, X.; Liu, S.; Zou, H.; Corpe, C.; Chen, L.; Wang, J. LncRNAs as epigenetic regulators of epithelial to mesenchymal transition in pancreatic cancer. Discov. Oncol. 2022, 13, 61. [Google Scholar] [CrossRef]
- Kimura, K.; Orita, T.; Liu, Y.; Yang, Y.; Tokuda, K.; Kurakazu, T.; Noda, T.; Yanai, R.; Morishige, N.; Takeda, A.; et al. Attenuation of EMT in RPE cells and subretinal fibrosis by an RAR-γ agonist. J. Mol. Med. 2015, 93, 749–758. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef]
- Ge, A.; Ma, Y.; Liu, Y.N.; Li, Y.S.; Gu, H.; Zhang, J.X.; Wang, Q.X.; Zeng, X.N.; Huang, M. Diosmetin prevents TGF-β1-induced epithelial-mesenchymal transition via ROS/MAPK signaling pathways. Life Sci. 2016, 153, 1–8. [Google Scholar] [CrossRef]
- Baek, A.; Yoon, S.; Kim, J.; Baek, Y.M.; Park, H.; Lim, D.; Chung, H.; Kim, D.E. Autophagy and KRT8/keratin 8 protect degeneration of retinal pigment epithelium under oxidative stress. Autophagy 2017, 13, 248–263. [Google Scholar] [CrossRef]
- Hwang, Y.E.; Baek, Y.M.; Baek, A.; Kim, D.E. Oxidative stress causes Alu RNA accumulation via PIWIL4 sequestration into stress granules. BMB Rep. 2019, 52, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef]
- Shen, G.; Li, Y.; Hong, F.; Zhang, J.; Fang, Z.; Xiang, W.; Qi, W.; Yang, X.; Gao, G.; Zhou, T. A role for Snail-MnSOD axis in regulating epithelial-to-mesenchymal transition markers expression in RPE cells. Biochem. Biophys. Res. Commun. 2021, 585, 146–154. [Google Scholar] [CrossRef]
- Li, H.; Li, M.; Xu, D.; Zhao, C.; Liu, G.; Wang, F. Overexpression of Snail in retinal pigment epithelial triggered epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2014, 446, 347–351. [Google Scholar] [CrossRef]
- Li, D.; Wei, T.T.; Cai, J.; Xie, T.H.; Yao, Y.; Zhu, L. Smurf1: A possible therapeutic target in dry age-related macular degeneration. Exp. Eye Res. 2023, 233, 109549. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kang, K.H.; Burrola, P.; Mak, T.W.; Lemke, G. Retinal degeneration triggered by inactivation of PTEN in the retinal pigment epithelium. Genes Dev. 2008, 22, 3147–3157. [Google Scholar] [CrossRef]
- Shen, G.; Li, Y.; Zeng, Y.; Hong, F.; Zhang, J.; Wang, Y.; Zhang, C.; Xiang, W.; Wang, J.; Fang, Z.; et al. Kallistatin deficiency induces the oxidative stress-related epithelial-mesenchymal transition of retinal pigment epithelial cells: A novel protagonist in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2023, 64, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhu, Y.; Zhou, J.; Wu, R.; Yang, N.; Bao, Q.; Xu, X. Luteolin alleviates epithelial-mesenchymal transformation induced by oxidative injury in arpe-19 cell via nrf2 and akt/gsk-3β pathway. Oxid. Med. Cell Longev. 2022, 2022, 2265725. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, H.; Huang, L.; Zhu, X.; Sha, J.; Li, G.; Ma, G.; Zhang, W.; Gu, M.; Guo, Y. Nrf2 signaling attenuates epithelial-to-mesenchymal transition and renal interstitial fibrosis via PI3K/Akt signaling pathways. Exp. Mol. Pathol. 2019, 111, 104296. [Google Scholar] [CrossRef]
- Blasiak, J.; Koskela, A.; Pawlowska, E.; Liukkonen, M.; Ruuth, J.; Toropainen, E.; Hyttinen, J.M.T.; Viiri, J.; Eriksson, J.E.; Xu, H.; et al. Epithelial-mesenchymal transition and senescence in the retinal pigment epithelium of nfe2l2/pgc-1α double knock-out mice. Int. J. Mol. Sci. 2021, 22, 1684. [Google Scholar] [CrossRef]
- Blasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef] [PubMed]
- Shoda, C.; Lee, D.; Miwa, Y.; Yamagami, S.; Nakashizuka, H.; Nimura, K.; Okamoto, K.; Kawagishi, H.; Negishi, K.; Kurihara, T. Inhibition of hypoxia-inducible factors suppresses subretinal fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2024, 38, e23792. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Wang, Y.; Li, Q.; Ji, X.; Tu, Y.; Du, S.; Lou, H.; Zeng, X.; Zhu, L.; Zhang, J.; et al. The HIF-1α/p53/miRNA-34a/Klotho axis in retinal pigment epithelial cells promotes subretinal fibrosis and exacerbates choroidal neovascularization. J. Cell Mol. Med. 2021, 25, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Reichmuth, K.; Prager, P.; Wiedemann, P.; Bringmann, A.; Kohen, L. Osmotic induction of placental growth factor in retinal pigment epithelial cells in vitro: Contribution of NFAT5 activity. Mol. Biol. Rep. 2016, 43, 803–814. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, L.; Wang, L.; Yang, X.; Zhou, A.; Wang, J. Placental growth factor promotes epithelial-mesenchymal transition-like changes in ARPE-19 cells under hypoxia. Mol. Vis. 2018, 24, 340–352. [Google Scholar]
- Feng, Z.; Li, R.; Shi, H.; Bi, W.; Hou, W.; Zhang, X. Combined silencing of TGF-β2 and Snail genes inhibit epithelial-mesenchymal transition of retinal pigment epithelial cells under hypoxia. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 875–884. [Google Scholar] [CrossRef]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Yin, J.; Huo, W.; Chaum, E. Loss of Prom1 impairs autophagy and promotes epithelial-mesenchymal transition in mouse retinal pigment epithelial cells. J. Cell Physiol. 2023, 238, 2373–2389. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Satyanarayana, G.; Liu, T.; Wang, L.; Wang, J.; Cheng, J.; Itoh, K.; Sharma, A.; Bhutto, I.; et al. Mitophagy initiates retrograde mitochondrial-nuclear signaling to guide retinal pigment cell heterogeneity. Autophagy 2022, 19, 966–983. [Google Scholar] [CrossRef]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2, e201900436. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Durando, M.; Cooke, M. CXCL13 and its receptor CXCR5 in cancer: Inflammation, immune response, and beyond. Front. Endocrinol. (Lausanne) 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Saddala, M.S.; Lennikov, A.; Mukwaya, A.; Huang, H. Transcriptome-wide analysis of CXCR5 deficient retinal pigment epithelial (RPE) cells reveals molecular signatures of rpe homeostasis. Biomedicines 2020, 8, 147. [Google Scholar] [CrossRef]
- Lennikov, A.; Mukwaya, A.; Saddala, M.S.; Huang, H. Deficiency of C-X-C chemokine receptor type 5 (CXCR5) gene causes dysfunction of retinal pigment epithelium cells. Lab. Investig. 2021, 101, 228–244. [Google Scholar] [CrossRef]
- Tisi, A.; Flati, V.; Delle Monache, S.; Lozzi, L.; Passacantando, M.; Maccarone, R. Nanoceria particles are an eligible candidate to prevent age-related macular degeneration by inhibiting retinal pigment epithelium cell death and autophagy alterations. Cells 2020, 9, 1617. [Google Scholar] [CrossRef]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Kutty, R.K.; Samuel, W.; Boyce, K.; Cherukuri, A.; Duncan, T.; Jaworski, C.; Nagineni, C.N.; Redmond, T.M. Proinflammatory cytokines decrease the expression of genes critical for RPE function. Mol. Vis. 2016, 22, 1156–1168. [Google Scholar]
- Kinoshita, K.; Matsumoto, K.; Kurauchi, Y.; Hisatsune, A.; Seki, T.; Katsuki, H. A Nurr1 agonist amodiaquine attenuates inflammatory events and neurological deficits in a mouse model of intracerebral hemorrhage. J. Neuroimmunol. 2019, 330, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.L.; Parmar, V.M.; Choudhary, M.; Malek, G. NURR1 expression regulates retinal pigment epithelial-mesenchymal transition and age-related macular degeneration phenotypes. Proc. Natl. Acad. Sci. USA 2022, 119, e2202256119. [Google Scholar] [CrossRef]
- Chen, H.C.; Zhu, Y.T.; Chen, S.Y.; Tseng, S.C. Wnt signaling induces epithelial-mesenchymal transition with proliferation in ARPE-19 cells upon loss of contact inhibition. Lab. Investig. 2012, 92, 676–687. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, S.; Chung, H.; Oh, S. Wnt5a attenuates the pathogenic effects of the Wnt/β-catenin pathway in human retinal pigment epithelial cells via down-regulating β-catenin and Snail. BMB Rep. 2015, 48, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Xu, M.J.; Wang, X. Adrenomedullin 2/intermedin: A putative drug candidate for treatment of cardiometabolic diseases. Br. J. Pharmacol. 2018, 175, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Kakihara, S.; Matsuda, Y.; Hirabayashi, K.; Imai, A.; Iesato, Y.; Sakurai, T.; Kamiyoshi, A.; Tanaka, M.; Ichikawa-Shindo, Y.; Kawate, H.; et al. Role of adrenomedullin 2/intermedin in the pathogenesis of neovascular age-related macular degeneration. Lab. Investig. 2023, 103, 100038. [Google Scholar] [CrossRef]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725. [Google Scholar] [CrossRef]
- Chinchilla, B.; Fernandez-Godino, R. AMD-like substrate causes epithelial mesenchymal transition in ipsc-derived retinal pigment epithelial cells wild type but not c3-knockout. Int. J. Mol. Sci. 2021, 22, 8183. [Google Scholar] [CrossRef] [PubMed]
- Llorián-Salvador, M.; Byrne, E.M.; Szczepan, M.; Little, K.; Chen, M.; Xu, H. Complement activation contributes to subretinal fibrosis through the induction of epithelial-to-mesenchymal transition (EMT) in retinal pigment epithelial cells. J. Neuroinflammation 2022, 19, 182. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Wang, F.E.; Zhang, C.; Maminishkis, A.; Dong, L.; Zhi, C.; Li, R.; Zhao, J.; Majerciak, V.; Gaur, A.B.; Chen, S.; et al. MicroRNA-204/211 alters epithelial physiology. Faseb J. 2010, 24, 1552–1571. [Google Scholar] [CrossRef]
- Fuchs, H.R.; Meister, R.; Lotke, R.; Framme, C. The microRNAs miR-302d and miR-93 inhibit TGFB-mediated EMT and VEGFA secretion from ARPE-19 cells. Exp. Eye Res. 2020, 201, 108258. [Google Scholar] [CrossRef]
- Li, D.; Zhang, J.; Liu, Z.; Gong, Y.; Zheng, Z. Human umbilical cord mesenchymal stem cell-derived exosomal miR-27b attenuates subretinal fibrosis via suppressing epithelial-mesenchymal transition by targeting HOXC6. Stem Cell Res. Ther. 2021, 12, 24. [Google Scholar] [CrossRef]
- Ishikawa, M.; Sawada, Y.; Yoshitomi, T. Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp. Eye Res. 2015, 133, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Cai, H.; Tezel, T.H.; Paik, D.; Gaillard, E.R.; Del Priore, L.V. Bruch’s membrane aging decreases phagocytosis of outer segments by retinal pigment epithelium. Mol. Vis. 2007, 13, 2310–2319. [Google Scholar] [PubMed]
- Saito, A. EMT and EndMT: Regulated in similar ways? J. Biochem. 2013, 153, 493–495. [Google Scholar] [CrossRef]
- Das, A.; Puklin, J.E.; Frank, R.N.; Zhang, N.L. Ultrastructural immunocytochemistry of subretinal neovascular membranes in age-related macular degeneration. Ophthalmology 1992, 99, 1368–1376. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J. Yes-associated protein is essential for proliferative vitreoretinopathy development via the epithelial-mesenchymal transition in retinal pigment epithelial fibrosis. J. Cell. Mol. Med. 2021, 25, 10213–10223. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; He, S.; Wörpel, V.; Ryan, S.J.; Hinton, D.R. Promotion of adhesion and migration of RPE cells to provisional extracellular matrices by TNF-alpha. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4324–4332. [Google Scholar]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763. [Google Scholar] [CrossRef]
- Meyer-Schaller, N.; Heck, C.; Tiede, S.; Yilmaz, M.; Christofori, G. Foxf2 plays a dual role during transforming growth factor beta-induced epithelial to mesenchymal transition by promoting apoptosis yet enabling cell junction dissolution and migration. Breast Cancer Res. 2018, 20, 118. [Google Scholar] [CrossRef]
- Liu, Y.; Xin, Y.; Ye, F.; Wang, W.; Lu, Q.; Kaplan, H.J.; Dean, D.C. Taz-tead1 links cell-cell contact to zeb1 expression, proliferation, and dedifferentiation in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3372–3378. [Google Scholar] [CrossRef]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate functions of matrix metalloproteinases in physiological and pathological conditions. J. Cell Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef]
- Nita, M.; Strzałka-Mrozik, B.; Grzybowski, A.; Mazurek, U.; Romaniuk, W. Age-related macular degeneration and changes in the extracellular matrix. Med. Sci. Monit. 2014, 20, 1003–1016. [Google Scholar] [CrossRef]
- Steen, B.; Sejersen, S.; Berglin, L.; Seregard, S.; Kvanta, A. Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2194–2200. [Google Scholar]
- Chau, K.Y.; Sivaprasad, S.; Patel, N.; Donaldson, T.A.; Luthert, P.J.; Chong, N.V. Plasma levels of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9) in age-related macular degeneration. Eye (Lond) 2007, 21, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef]
- Schlingemann, R.O. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 91–101. [Google Scholar] [CrossRef]
- Chen, X.; Xiao, W.; Liu, X.; Zeng, M.; Luo, L.; Wu, M.; Ye, S.; Liu, Y. Blockade of Jagged/Notch pathway abrogates transforming growth factor β2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Curr. Mol. Med. 2014, 14, 523–534. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of tgf-β and tgf-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Goumans, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Tanaka, T.; Yamanaka, O.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Yoo, J.; Flanders, K.C.; et al. Smad3 is required for dedifferentiation of retinal pigment epithelium following retinal detachment in mice. Lab. Investig. 2004, 84, 1245–1258. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, S.J.; Jo, D.H.; Park, K.S.; Kim, J.H. Blockade of mTORC1-NOX signaling pathway inhibits TGF-β1-mediated senescence-like structural alterations of the retinal pigment epithelium. Faseb J. 2021, 35, e21403. [Google Scholar] [CrossRef]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, K.; Sponseller, T.L.; Danielpour, D. Novel function of androgen receptor-associated protein 55/Hic-5 as a negative regulator of Smad3 signaling. J. Biol. Chem. 2005, 280, 5154–5162. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Attisano, L.; Wrana, J.L. Signal transduction by the TGF-beta superfamily. Science 2002, 296, 1646–1647. [Google Scholar] [CrossRef]
- Medici, D.; Hay, E.D.; Goodenough, D.A. Cooperation between snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol. Biol. Cell 2006, 17, 1871–1879. [Google Scholar] [CrossRef]
- Hannigan, G.; Troussard, A.A.; Dedhar, S. Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 2005, 5, 51–63. [Google Scholar] [CrossRef]
- Han, J.W.; Lyu, J.; Park, Y.J.; Jang, S.Y.; Park, T.K. Wnt/β-catenin signaling mediates regeneration of retinal pigment epithelium after laser photocoagulation in mouse eye. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8314–8324. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y.; Lin, M.; Lee, K.; Mott, R.A.; Ma, J.X. Pathogenic role of the Wnt signaling pathway activation in laser-induced choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2013, 54, 141–154. [Google Scholar] [CrossRef]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Verras, M.; Sun, Z. Roles and regulation of Wnt signaling and beta-catenin in prostate cancer. Cancer Lett. 2006, 237, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Yoon, S.O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial-mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef]
- Zhou, B.P.; Hung, M.C. Wnt, hedgehog and snail: Sister pathways that control by GSK-3beta and beta-Trcp in the regulation of metastasis. Cell Cycle 2005, 4, 772–776. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef]
- Chen, X.; Xiao, W.; Wang, W.; Luo, L.; Ye, S.; Liu, Y. The complex interplay between ERK1/2, TGFβ/Smad, and Jagged/Notch signaling pathways in the regulation of epithelial-mesenchymal transition in retinal pigment epithelium cells. PLoS ONE 2014, 9, e96365. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Tokuda, K.; Yamashiro, C.; Higashijima, F.; Yoshimoto, T.; Ota, M.; Ogata, T.; Ashimori, A.; Hatano, M.; Kobayashi, M.; et al. Inhibition of epithelial-mesenchymal transition in retinal pigment epithelial cells by a retinoic acid receptor-α agonist. Sci. Rep. 2021, 11, 11842. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Shao, M.; Ren, M.; Zhang, W.; Zhou, J.; Wang, J. Pirfenidone attenuates the emt process and the secretion of vegf in tgf-β2-induced arpe-19 cells via inhibiting the activation of the nf-κb/snail signaling pathway. J. Ophthalmol. 2023, 2023, 4798071. [Google Scholar] [CrossRef]
- Sripathi, S.R.; Hu, M.W.; Turaga, R.C.; Mikeasky, R.; Satyanarayana, G.; Cheng, J.; Duan, Y.; Maruotti, J.; Wahlin, K.J.; Berlinicke, C.A.; et al. IKKβ inhibition attenuates epithelial mesenchymal transition of human stem cell-derived retinal pigment epithelium. Cells 2023, 12, 1155. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tokuda, K.; Kobayashi, Y.; Yamashiro, C.; Uchi, S.H.; Hatano, M.; Kimura, K. Suppression of epithelial-mesenchymal transition in retinal pigment epithelial cells by an mrtf-a inhibitor. Investig. Ophthalmol. Vis. Sci. 2019, 60, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Ramos de Carvalho, J.E.; Verwoert, M.T.; Vogels, I.M.C.; Reits, E.A.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Involvement of the ubiquitin-proteasome system in the expression of extracellular matrix genes in retinal pigment epithelial cells. Biochem. Biophys. Rep. 2018, 13, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Park, G.B.; Kim, D. Cigarette smoke-induced EGFR activation promotes epithelial mesenchymal migration of human retinal pigment epithelial cells through regulation of the FAK-mediated Syk/Src pathway. Mol. Med. Rep. 2018, 17, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Y.; Hu, X.; Zhao, Z.; Chen, Z.; Wang, X.; Zhang, Z.; Jin, H.; Zhang, J. Luteolin inhibits subretinal fibrosis and epithelial-mesenchymal transition in laser-induced mouse model via suppression of Smad2/3 and YAP signaling. Phytomedicine 2023, 116, 154865. [Google Scholar] [CrossRef]
- Josifovska, N.; Albert, R.; Nagymihály, R.; Lytvynchuk, L.; Moe, M.C.; Kaarniranta, K.; Veréb, Z.J.; Petrovski, G. Resveratrol as inducer of autophagy, pro-survival, and anti-inflammatory stimuli in cultured human RPE cells. Int. J. Mol. Sci. 2020, 21, 813. [Google Scholar] [CrossRef]
- Shu, D.Y.; Chaudhary, S.; Cho, K.S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of oxidative stress in ocular diseases: A balancing act. Metabolites 2023, 13, 187. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Ng, T.K.; Brelén, M.E.; Wu, D.; Wang, J.X.; Chan, K.P.; Yung, J.S.Y.; Cao, D.; Wang, Y.; Zhang, S.; et al. Continuous exposure to non-lethal doses of sodium iodate induces retinal pigment epithelial cell dysfunction. Sci. Rep. 2016, 6, 37279. [Google Scholar] [CrossRef]
- Yang, Y.C.; Chien, Y.; Yarmishyn, A.A.; Lim, L.Y.; Tsai, H.Y.; Kuo, W.C.; Tsai, P.H.; Yang, S.H.; Hong, S.I.; Chen, S.J.; et al. Inhibition of oxidative stress-induced epithelial-mesenchymal transition in retinal pigment epithelial cells of age-related macular degeneration model by suppressing ERK activation. J. Adv. Res. 2023, 60, 141–157. [Google Scholar] [CrossRef]
- Kanlaya, R.; Kapincharanon, C.; Fong-Ngern, K.; Thongboonkerd, V. Induction of mesenchymal-epithelial transition (MET) by epigallocatechin-3-gallate to reverse epithelial-mesenchymal transition (EMT) in SNAI1-overexpressed renal cells: A potential anti-fibrotic strategy. J. Nutr. Biochem. 2022, 107, 109066. [Google Scholar] [CrossRef]
- Sethi, S.; Li, Y.; Sarkar, F.H. Regulating miRNA by natural agents as a new strategy for cancer treatment. Curr. Drug Targets 2013, 14, 1167–1174. [Google Scholar] [CrossRef]
- Ishikawa, K.; Sreekumar, P.G.; Spee, C.; Nazari, H.; Zhu, D.; Kannan, R.; Hinton, D.R. αB-crystallin regulates subretinal fibrosis by modulation of epithelial-mesenchymal transition. Am. J. Pathol. 2016, 186, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Wang, L.; Wan, J.; Barnett, B.P.; Ebrahimi, K.; Qian, J.; Handa, J.T. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Radic. Biol. Med. 2014, 69, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.Y.; Kim, S.J.; Park, K.S.; Kim, J.H. Klotho prevents transforming growth factor-β2-induced senescent-like morphological changes in the retinal pigment epithelium. Cell Death Dis. 2023, 14, 334. [Google Scholar] [CrossRef]
- Jiang, Y.; Wen, X.; Jian, X.; Chen, Q.; Li, Y. Klotho attenuates epithelial-mesenchymal transition of retinal pigment epithelial cells in subretinal fibrosis by suppressing the ERK1/2 and Wnt/β-catenin signaling pathways. Int. J. Mol. Med. 2025, 55, 45. [Google Scholar] [CrossRef]
- Nadeem, U.; Xie, B.; Xie, E.F.; D’Souza, M.; Dao, D.; Sulakhe, D.; Skondra, D. Using advanced bioinformatics tools to identify novel therapeutic candidates for age-related macular degeneration. Transl. Vis. Sci. Technol. 2022, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Zhu, Q.; Yang, J.; Zheng, Y.; Yang, W.; Li, D.; Cui, Y.; Shen, L.; Rao, L.; Zhang, X.; et al. Metformin inhibits subretinal fibrosis by activating Klotho by miR-126-5p. Cytotechnology 2025, 77, 84. [Google Scholar] [CrossRef]
- Shimizu, H.; Yamada, K.; Suzumura, A.; Kataoka, K.; Takayama, K.; Sugimoto, M.; Terasaki, H.; Kaneko, H. Caveolin-1 promotes cellular senescence in exchange for blocking subretinal fibrosis in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 21. [Google Scholar] [CrossRef]
- da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.M.; Mitra, D.; Zhu, D.; Thomas, B.B.; et al. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Transl. Med. 2018, 10, eaao4097. [Google Scholar] [CrossRef]
- Liu, Z.; Parikh, B.H.; Tan, Q.S.W.; Wong, D.S.L.; Ong, K.H.; Yu, W.; Seah, I.; Holder, G.E.; Hunziker, W.; Tan, G.S.W.; et al. Surgical transplantation of human rpe stem cell-derived rpe monolayers into non-human primates with immunosuppression. Stem Cell Rep. 2021, 16, 237–251. [Google Scholar] [CrossRef]
- Tian, H.; Chen, Z.; Zhu, X.; Ou, Q.; Wang, Z.; Wu, B.; Xu, J.Y.; Jin, C.; Gao, F.; Wang, J.; et al. Induced retinal pigment epithelial cells with anti-epithelial-to-mesenchymal transition ability delay retinal degeneration. iScience 2022, 25, 105050. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Z.; Wang, L.; Ou, Q.; Feng, Z.; Xiao, H.; Shen, Q.; Li, Y.; Jin, C.; Xu, J.Y.; et al. Direct conversion of human umbilical cord mesenchymal stem cells into retinal pigment epithelial cells for treatment of retinal degeneration. Cell Death Dis. 2022, 13, 785. [Google Scholar] [CrossRef]
- Shen, H.; Ding, C.; Yuan, S.; Pan, T.; Li, D.; Li, H.; Huang, B.; Liu, Q. Vitamin C- and valproic acid-induced fetal rpe stem-like cells recover retinal degeneration via regulating sox2. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1645–1657. [Google Scholar] [CrossRef]
- Ma, X.; Wu, W.; Hara, M.; Zhou, J.; Panzarin, C.; Schafer, C.M.; Griffin, C.T.; Cai, J.; Ma, J.X.; Takahashi, Y. Deficient RPE mitochondrial energetics leads to subretinal fibrosis in age-related neovascular macular degeneration. Commun. Biol. 2024, 7, 1075. [Google Scholar] [CrossRef] [PubMed]
- Sripathi, S.R.; Hu, M.W.; Liu, M.M.; Wan, J.; Cheng, J.; Duan, Y.; Mertz, J.L.; Wahlin, K.J.; Maruotti, J.; Berlinicke, C.A.; et al. Transcriptome landscape of epithelial to mesenchymal transition of human stem cell-derived RPE. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1. [Google Scholar] [CrossRef]
- Sripathi, S.R.; Hu, M.W.; Turaga, R.C.; Mertz, J.; Liu, M.M.; Wan, J.; Maruotti, J.; Wahlin, K.J.; Berlinicke, C.A.; Qian, J.; et al. Proteome landscape of epithelial-to-mesenchymal transition (EMT) of retinal pigment epithelium shares commonalities with malignancy-associated EMT. Mol. Cell. Proteom. MCP 2021, 20, 100131. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-mesenchymal transition: Epigenetic reprogramming driving cellular plasticity. Trends Genet. TIG 2017, 33, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Liang, J.; Jiang, M.; Zhang, T.; Wan, X.; Wu, J.; Li, X.; Chen, J.; Sun, J.; et al. METTL3-mediated m6A modification of HMGA2 mRNA promotes subretinal fibrosis and epithelial-mesenchymal transition. J. Mol. Cell Biol. 2023, 15, mjad005. [Google Scholar] [CrossRef]
- Rhee, K.D.; Yu, J.; Zhao, C.Y.; Fan, G.; Yang, X.J. Dnmt1-dependent DNA methylation is essential for photoreceptor terminal differentiation and retinal neuron survival. Cell Death Dis. 2012, 3, e427. [Google Scholar] [CrossRef]
- Gugnoni, M.; Ciarrocchi, A. Long noncoding RNA and epithelial mesenchymal transition in cancer. Int. J. Mol. Sci. 2019, 20, 1924. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Kannan, R.; Felszeghy, S.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. The regulation of NFE2L2 (NRF2) signalling and epithelial-to-mesenchymal transition in age-related macular degeneration pathology. Int. J. Mol. Sci. 2019, 20, 5800. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 2019, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sheng, X.; Ding, Q.; Wang, Y.; Zhao, J.; Zhang, J. Subretinal fibrosis secondary to neovascular age-related macular degeneration: Mechanisms and potential therapeutic targets. Neural Regen. Res. 2025, 20, 378–393. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Mechanism | Research Object | Study Model | Target Subtype | Possible Pathways | Major Roles | References |
|---|---|---|---|---|---|---|
| Oxidative Stress | KRT8 | ARPE-19 cells; human primary RPE cells | N/A | MAPK1/ERK2, MAPK3/ERK1 | Induce EMT | [31] |
| PIWIL4 | ARPE-19 cells | N/A | AKT—Phosphorylation, PIWIL4 chelates into cytoplasmic stress granules | Induce EMT | [32] | |
| MnSOD | ARPE-19 cells | N/A | Snail-MnSOD axis | Inhibit EMT | [34] | |
| Inhibit Smurf1 | ARPE-19 cells; NaIO3-induced C57BL/6J mice | Dry AMD | TGF-β pathway and NF-κβ pathway. | Inhibit EMT | [36] | |
| PTEN deficiency | PTENflox mice | N/A | PI3K-Akt | Induce EMT | [37] | |
| Kallistatin | ARPE-19 cells; NaIO3-induced C57BL/6J mice | Dry AMD | Downregulate the transcription factor Snail | Inhibit EMT | [38] | |
| Nrf2/PGC-1α deficiency | Double knock-out C57BL/6J mice | Dry AMD | Weakened antioxidant defenses caused by lack of two genes, Mitochondria/autophagy deficiency | Induce EMT | [41] | |
| Hypoxia | HIF-1α | ARPE-19 cells; C57BL/6J mice CNV model | N/A | HIF-1α/p53/miRNA-34a/Klotho axis | Induce EMT | [44] |
| PGF | ARPE-19 cells | Wet AMD | NF-κB | Induce EMT | [46] | |
| Combined silencing of TGF-β2 and Snail genes | ARPE-19 cells | Wet AMD | knockdown of both inhibited EMT to a greater extent than knockdown of either gene individually | Inhibit EMT | [47] | |
| Autophagy | Loss of Prom1 | Isolated RPE cells from C57/BL6J mice | Dry AMD | Impaired autophagy; Prom1-mTORC1-TFEB axis | Promote EMT | [49] |
| PINK1 deficiency | Human autopsy eyes; C57BL/6J mice | Early AMD | RMNS, Nrf2, TXNRD1, PI3K/AKT | Induce EMT | [51] | |
| PGC-1α deficiency | ARPE-19 cells; C57BL/6J mice | N/A | Impaired autophagy | Induce EMT | [52] | |
| CXCR5 deficiency | C57BL/6J mice | N/A | CXCL13/CXCR5, PI3K/AKT/FOXO1 signal axis, Impaired autophagy | Induce EMT | [53,54,55] | |
| CeO2-NP | ARPE-19 cells; Light-damaged SD albino rats | Dry AMD | Interference Autophagy Pathway | Inhibit EMT | [56] | |
| Inflammation | Proinflammatory cytokines | ARPE-19 cells | N/A | Gene Expression Regulation | Induce EMT | [58] |
| Nurr1 | Primary human RPE cells; ARPE-19 cells; C57BL/6J mice | N/A | Regulate the expression of EMT-related genes and proteins | Inhibit EMT | [60] | |
| Wnt5a | hTERT-PRE-1cells; ARPE-19 cells | N/A | Antagonistic Wnt/β-catenin Pathway, TNF-α/NF-κB | Inhibit EMT | [62] | |
| AM2 | ARPE-19 cells; Laser-induced C57BL/6J mice | Wet AMD | Upregulate Meox2, Suppress Inflammation | Inhibit EMT | [64] | |
| Complement Activation | The genetic ablation of C3 | iPSC-RPE | N/A | Inhibit EMT | [66] | |
| C5a | Human eye samples; Laser-induced C57BL/6J mice | Wet AMD | Smad2/3, ERK1/2, C5aR | Induce EMT | [67] | |
| MicroRNA | miRNA-204 | hfRPE | N/A | Inhibition Of TGF-β Pathway | Inhibit EMT | [26,69] |
| miRNA-211 | hfRPE | N/A | Inhibition Of TGF-β Pathway | Inhibit EMT | [26,69] | |
| miRNA-34a | ARPE-19 cells; C57BL/6J mice CNV model | N/A | Inhibition Of TGF-β Pathway | Inhibit EMT | [44] | |
| miR-302d | ARPE-19 cells | Wet AMD | Inhibition Of TGF-β Pathway | Inhibit EMT | [70] | |
| miR-93 | ARPE-19 cells | Wet AMD | Inhibition Of TGF-β Pathway | Inhibit EMT | [70] | |
| miR-27b | Human skin fibroblasts; ARPE-19 cells | Wet AMD | miR-27b/HOXC6 axis | Inhibit EMT | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Wang, Y.; Zhang, L.; Yang, W.; Fu, S. Molecular Mechanisms of Epithelial–Mesenchymal Transition in Retinal Pigment Epithelial Cells: Implications for Age-Related Macular Degeneration (AMD) Progression. Biomolecules 2025, 15, 771. https://doi.org/10.3390/biom15060771
Wang N, Wang Y, Zhang L, Yang W, Fu S. Molecular Mechanisms of Epithelial–Mesenchymal Transition in Retinal Pigment Epithelial Cells: Implications for Age-Related Macular Degeneration (AMD) Progression. Biomolecules. 2025; 15(6):771. https://doi.org/10.3390/biom15060771
Chicago/Turabian StyleWang, Na, Yaqi Wang, Lei Zhang, Wenjing Yang, and Songbo Fu. 2025. "Molecular Mechanisms of Epithelial–Mesenchymal Transition in Retinal Pigment Epithelial Cells: Implications for Age-Related Macular Degeneration (AMD) Progression" Biomolecules 15, no. 6: 771. https://doi.org/10.3390/biom15060771
APA StyleWang, N., Wang, Y., Zhang, L., Yang, W., & Fu, S. (2025). Molecular Mechanisms of Epithelial–Mesenchymal Transition in Retinal Pigment Epithelial Cells: Implications for Age-Related Macular Degeneration (AMD) Progression. Biomolecules, 15(6), 771. https://doi.org/10.3390/biom15060771