
Multiomics-Based Profiling of the Fecal Microbiome Reveals Potential Disease-Specific Signatures in Pediatric IBD (PIBD) †

 , ,
, , 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Nucleic Acid Extraction
2.3. 16S Microbiome Library Preparation
2.4. Sequencing Quality Control Analysis
2.5. Metagenomics Library Preparation
2.6. Transcriptomics Library Preparation
2.7. Fecal Sample Preparation for LC-MS
2.8. Untargeted LC-MS Metabolomics
2.9. 16S Amplicon Analysis
2.10. Metagenomic and Metatranscriptomic Sequence QC
2.11. Assembly-Free Taxonomic Classification
2.12. Assembly-Free Functional Pathway Analysis
2.13. Assembly-Based Analysis
2.14. MetaboAnalyst Analysis
3. Results
3.1. Microbiome Population Analysis with 16S rRNA Sequencing
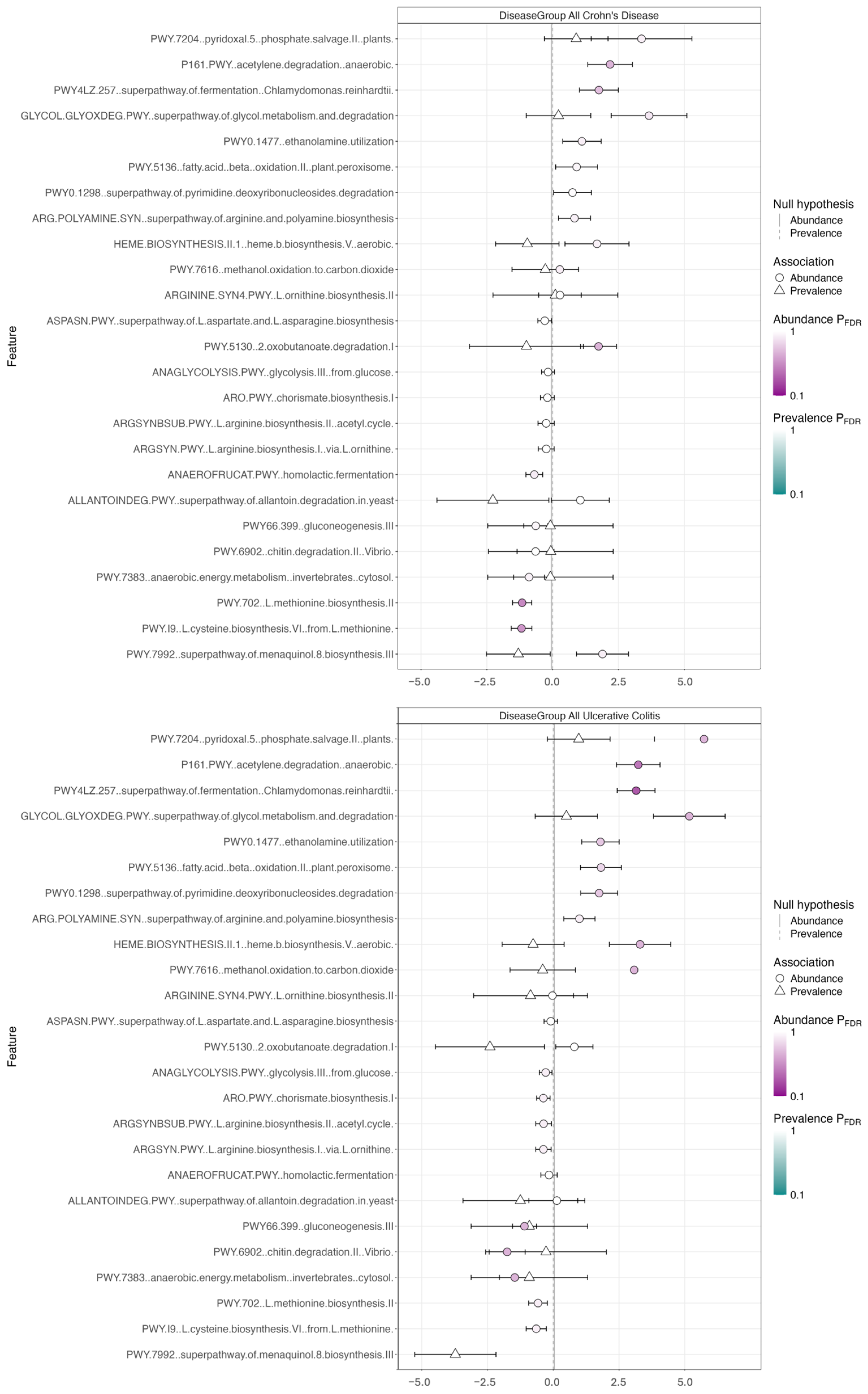
3.2. Metagenomics and Functional Pathway Analysis
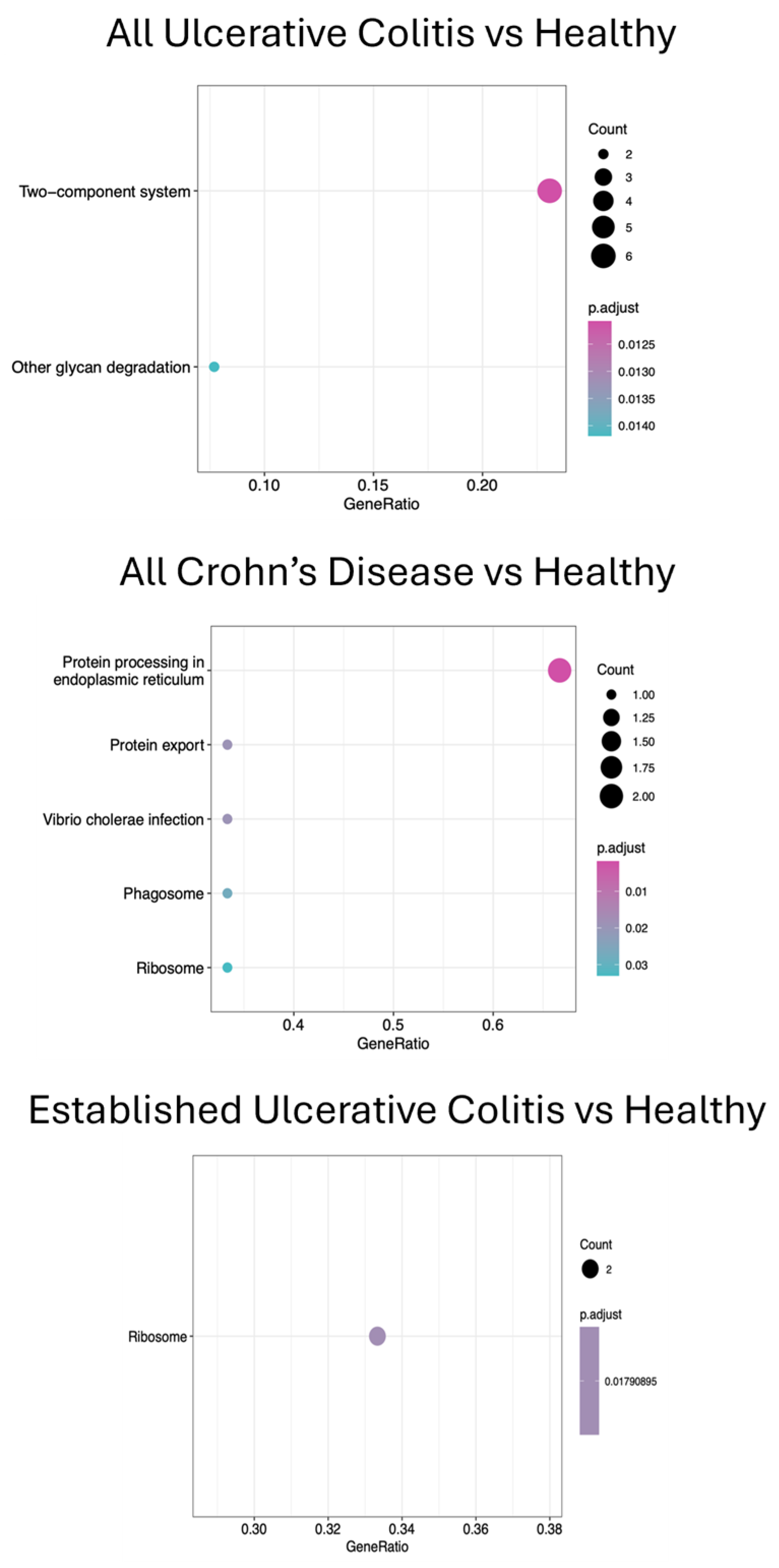
3.3. Metatranscriptomics and KEGG Pathway Analyses
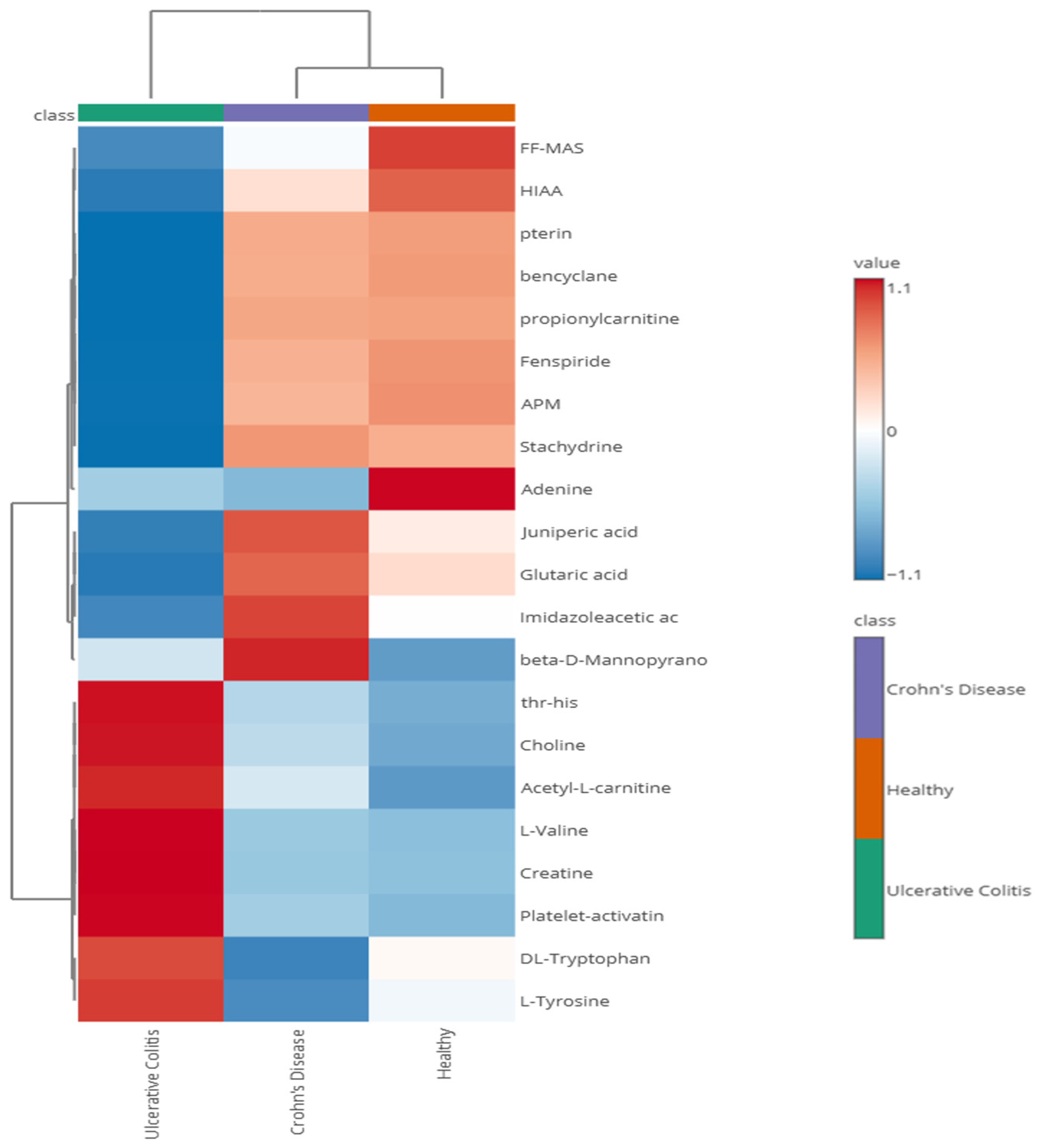
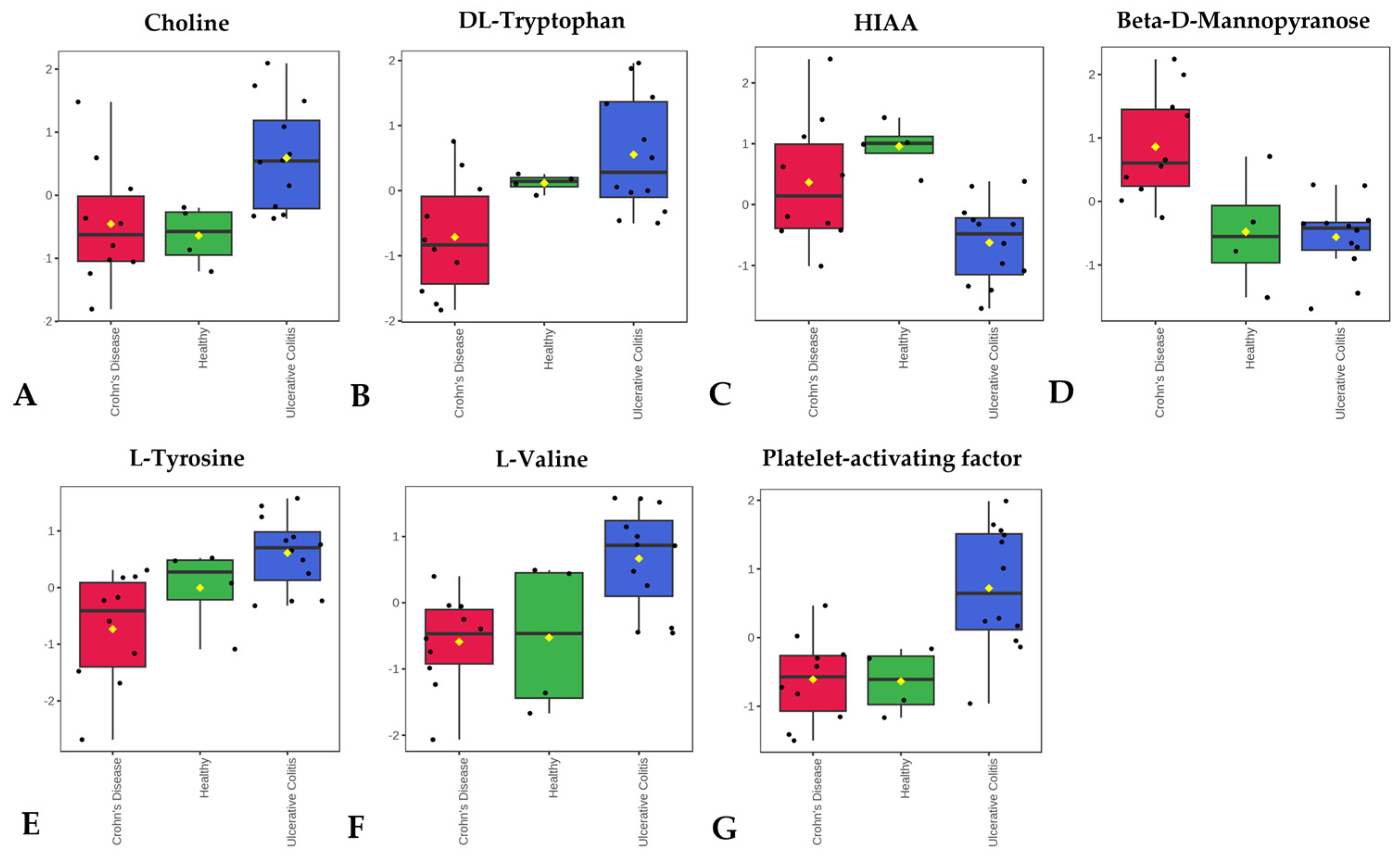
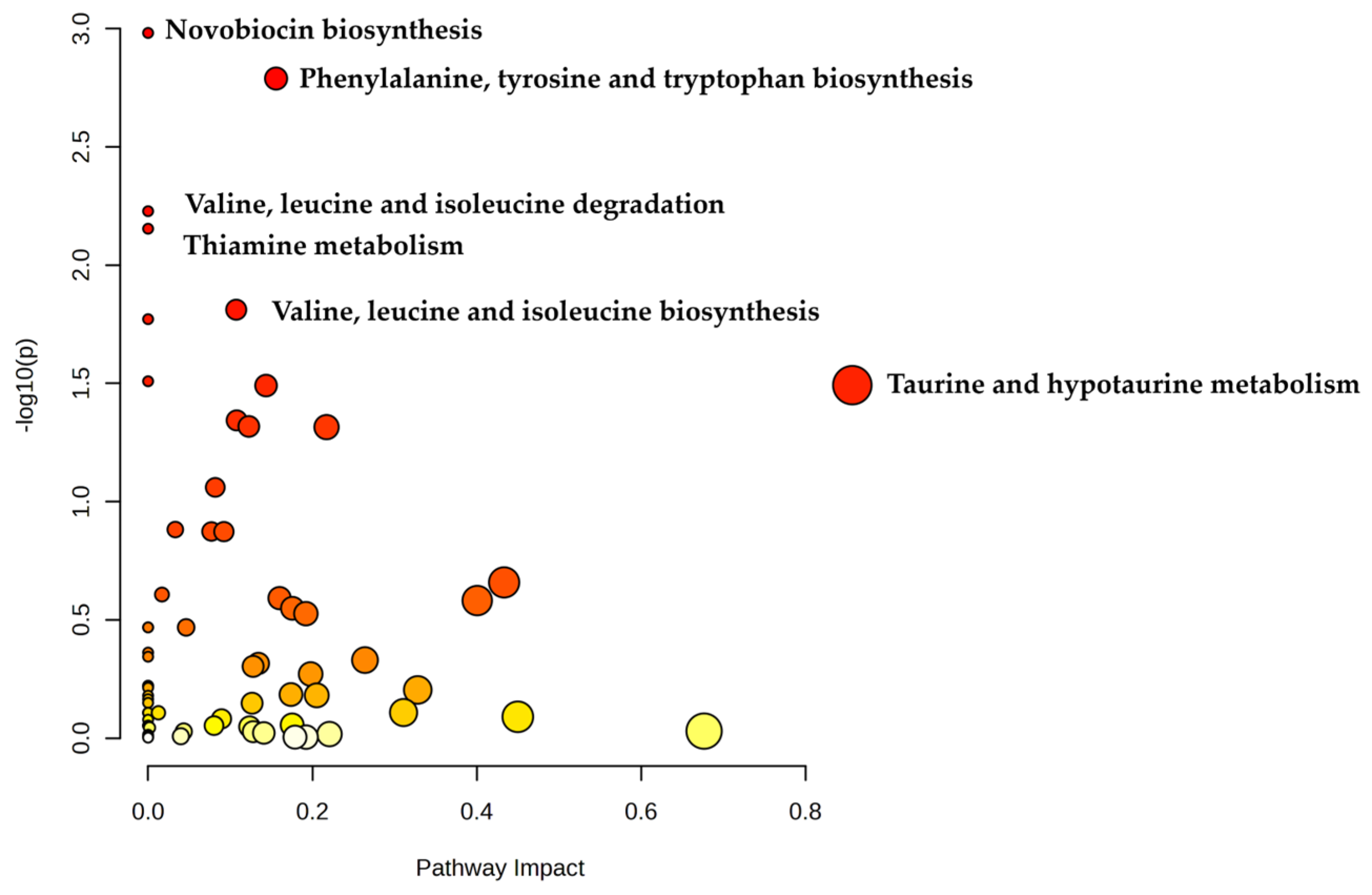
3.4. Metabolomics Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| PIBD | Pediatric Inflammatory Bowel Disease |
| IBD | Inflammatory Bowel Disease |
| GI | Gastrointestinal |
| CD | Crohn’s Disease |
| UC | Ulcerative Colitis |
References
- Sýkora, J.; Pomahačová, R.; Kreslová, M.; Cvalínová, D.; Štych, P.; Schwarz, J. Current global trends in the incidence of pediatric-onset inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 2741–2763. [Google Scholar] [CrossRef]
- Bouhuys, M.; Lexmond, W.S.; Van Rheenen, P.F. Pediatric Inflammatory Bowel Disease. Pediatrics 2023, 151, e2022058037. [Google Scholar] [CrossRef] [PubMed]
- Wiczynska-Ryl, J.; Krogulska, A. Incidence of Inflammatory Bowel Disease in Children. Gastroenterol. Res. 2025, 18, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Tartamus, G.V.; Serban, D.E.; Fogas, C.R.; Tantau, M.V. Pediatric Inflammatory Bowel Disease in Romania: The First Epidemiological Study of the North-West Region (2000–2020). Children 2025, 12, 403. [Google Scholar] [CrossRef]
- Conrad, M.A.; Rosh, J.R. Pediatric Inflammatory Bowel Disease. Pediatr. Clin. 2017, 64, 577–591. [Google Scholar] [CrossRef]
- Cococcioni, L.; Panelli, S.; Varotto-Boccazzi, I.; Di Carlo, D.; Pistone, D.; Leccese, G.; Zuccotti, G.V.; Comandatore, F. IBDs and the pediatric age: Their peculiarities and the involvement of the microbiota. Dig. Liver Dis. 2021, 53, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2016, 67, 108–119. [Google Scholar] [CrossRef]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Massaro, C.A.; Meade, S.; Lemarié, F.L.; Kaur, G.; Bressler, B.; Rosenfeld, G.; Leung, Y.; Williams, A.J.; Lunken, G. Gut microbiome predictors of advanced therapy response in Crohn’s disease: Protocol for the OPTIMIST prospective, longitudinal, observational pilot study in Canada. BMJ Open 2025, 15, e094280. [Google Scholar] [CrossRef]
- Moon, J.S. Clinical Aspects and Treatments for Pediatric Inflammatory Bowel Diseases. Pediatr. Gastroenterol. Hepatol. Nutr. 2019, 22, 50–56. [Google Scholar] [CrossRef]
- Kugathasan, S.; Cohen, S. Searching for New Clues in Inflammatory Bowel Disease: Tell Tales From Pediatric IBD Natural History Studies. Gastroenterology 2008, 135, 1038–1041. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Hracs, L.; Windsor, J.W.; Gorospe, J.; Cummings, M.; Coward, S.; Buie, M.J.; Quan, J.; Goddard, Q.; Caplan, L.; Markovinović, A.; et al. Global evolution of inflammatory bowel disease across epidemiologic stages. Nature 2025, 1–9. [Google Scholar] [CrossRef]
- Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms 2021, 9, 977. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Peloquin, J.M.; Goel, G.; Villablanca, E.J.; Xavier, R.J. Mechanisms of Pediatric Inflammatory Bowel Disease. Annu. Rev. Immunol. 2016, 34, 31–64. [Google Scholar] [CrossRef]
- Fiocchi, C. Omics and Multi-Omics in IBD: No Integration, No Breakthroughs. Int. J. Mol. Sci. 2023, 24, 14912. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Booth, A. It’s the song, not the singer: An exploration of holobiosis and evolutionary theory. Biol. Philos. 2016, 32, 5–24. [Google Scholar] [CrossRef]
- Baskaran, K.; Moshkovich, M.; Hart, L.; Shah, N.; Chowdhury, F.; Shanmuganathan, M.; Britz-McKibbin, P.; Pai, N. The role of urine metabolomics in the diagnosis and management of adult and pediatric Crohn’s disease and ulcerative colitis. Biomarkers 2024, 30, 104–113. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.-O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef]
- Imhann, F.; Van der Velde, K.J.; Barbieri, R.; Alberts, R.; Voskuil, M.D.; Vila, A.V.; Collij, V.; Spekhorst, L.M.; Van der Sloot, K.W.J.; Peters, V.; et al. The 1000IBD project: Multi-omics data of 1000 inflammatory bowel disease patients; data release 1. BMC Gastroenterol. 2019, 19, 5. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Spencer, E.A.; Dubinsky, M.C. Precision Medicine in Pediatric Inflammatory Bowel Disease. Pediatr. Clin. North Am. 2021, 68, 1171–1190. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Ackermann, G.; Apprill, A.; Bauer, M.; Berg-Lyons, D.; Betley, J.; Fierer, N.; Fraser, L.; Fuhrman, J.A.; Gilbert, J.A.; et al. EMP 16S Illumina Amplicon Protocol V.1. 2018. Available online: https://www.protocols.io/view/emp-16s-illumina-amplicon-protocol-nuudeww (accessed on 31 May 2023).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 August 2019).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zhang, P.; Zhu, J.; Raftery, D. Globally Optimized Targeted Mass Spectrometry: Reliable Metabolomics Analysis with Broad Coverage. Anal. Chem. 2015, 87, 12355–12362. [Google Scholar] [CrossRef]
- Wei, Y.; Jasbi, P.; Shi, X.; Turner, C.; Hrovat, J.; Liu, L.; Rabena, Y.; Porter, P.; Gu, H. Early Breast Cancer Detection Using Untargeted and Targeted Metabolomics. J. Proteome Res. 2021, 20, 3124–3133. [Google Scholar] [CrossRef]
- Jin, Y.; Chi, J.; LoMonaco, K.; Boon, A.; Gu, H. Recent review on selected xenobiotics and their impacts on gut microbiome and metabolome. TrAC Trends Anal. Chem. 2023, 166, 117155. [Google Scholar] [CrossRef]
- Garcia, M.M.; Romero, A.S.; Merkley, S.D.; Meyer-Hagen, J.L.; Forbes, C.; El Hayek, E.; Sciezka, D.P.; Templeton, R.; Gonzalez-Estrella, J.; Jin, Y.; et al. In Vivo Tissue Distribution of Polystyrene or Mixed Polymer Microspheres and Metabolomic Analysis after Oral Exposure in Mice. Environ. Health Perspect. 2024, 132, 47005. [Google Scholar] [CrossRef]
- Scieszka, D.P.; Garland, D.; Hunter, R.; Herbert, G.; Lucas, S.; Jin, Y.; Gu, H.; Campen, M.J.; Cannon, J.L. Multi-omic assessment shows dysregulation of pulmonary and systemic immunity to e-cigarette exposure. Respir. Res. 2023, 24, 138. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Li, H.; Jin, Y.; Armad, J.; Gu, H.; Mani, S.; Cui, J.Y. Maternal PBDE exposure disrupts gut microbiome and promotes hepatic proinflammatory signaling in humanized PXR-transgenic mouse offspring over time. Toxicol. Sci. 2023, 194, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.E.; Sweazea, K.L.; Bowes, D.A.; Jasbi, P.; Whisner, C.M.; Sears, D.D.; Krajmalnik-Brown, R.; Jin, Y.; Gu, H.; Klein-Seetharaman, J.; et al. Gut microbiome remodeling and metabolomic profile improves in response to protein pacing with intermittent fasting versus continuous caloric restriction. Nat. Commun. 2024, 15, 4155. [Google Scholar] [CrossRef]
- Hemmer, S.; Manier, S.K.; Wagmann, L.; Meyer, M.R. Comparison of reversed-phase, hydrophilic interaction, and porous graphitic carbon chromatography columns for an untargeted toxicometabolomics study in pooled human liver microsomes, rat urine, and rat plasma. Metabolomics 2024, 20, 49. [Google Scholar] [CrossRef]
- Dedon, L.R.; Yuan, H.; Chi, J.; Gu, H.; Arias, A.J.; Covault, J.M.; Zhou, Y. Baseline gut microbiome and metabolites are correlated with changes in alcohol consumption in participants in a randomized Zonisamide clinical trial. Sci. Rep. 2025, 15, 10486. [Google Scholar] [CrossRef]
- Chi, J.; Patterson, J.S.; Jin, Y.; Kim, K.J.; Lalime, N.; Hawley, D.; Lewis, F.; Li, L.; Wang, X.; Campen, M.J.; et al. Metabolic Reprogramming in Gut Microbiota Exposed to Polystyrene Microplastics. Biomedicines 2025, 13, 446. [Google Scholar] [CrossRef]
- Tang, D.-Q.; Zou, L.; Yin, X.-X.; Ong, C.N. HILIC-MS for metabolomics: An attractive and complementary approach to RPLC-MS. Mass Spectrom. Rev. 2016, 35, 574–600. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Liu, C.; Zhan, S.; Tian, Z.; Li, N.; Mao, R.; Zeng, Z.; Chen, M. Gut Microbiota Profile in Pediatric Patients With Inflammatory Bowel Disease: A Systematic Review. Front. Pediatr. 2021, 9, 626232. [Google Scholar] [CrossRef]
- Maukonen, J.; Kolho, K.-L.; Paasela, M.; Honkanen, J.; Klemetti, P.; Vaarala, O.; Saarela, M. Altered Fecal Microbiota in Paediatric Inflammatory Bowel Disease. J. Crohn’s Colitis 2015, 9, 1088–1095. [Google Scholar] [CrossRef]
- Schwiertz, A.; Jacobi, M.; Frick, J.S.; Richter, M.; Rusch, K.; Köhler, H. Microbiota in Pediatric Inflammatory Bowel Disease. J. Pediatr. 2010, 157, 240–244.e1. [Google Scholar] [CrossRef]
- Michail, S.; Durbin, M.; Turner, D.; Griffiths, A.M.; Mack, D.R.; Hyams, J.; Leleiko, N.; Kenche, H.; Stolfi, A.; Wine, E. Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 1799–1808. [Google Scholar] [CrossRef]
- Papa, E.; Docktor, M.; Smillie, C.; Weber, S.; Preheim, S.P.; Gevers, D.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Ingram, J.; et al. Non-Invasive Mapping of the Gastrointestinal Microbiota Identifies Children with Inflammatory Bowel Disease. PLoS ONE 2012, 7, e39242. [Google Scholar] [CrossRef]
- Jacobs, J.P.; Goudarzi, M.; Singh, N.; Tong, M.; McHardy, I.H.; Ruegger, P.; Asadourian, M.; Moon, B.-H.; Ayson, A.; Borneman, J.; et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 750–766. [Google Scholar] [CrossRef]
- Malham, M.; Lilje, B.; Houen, G.; Winther, K.; Andersen, P.S.; Jakobsen, C. The microbiome reflects diagnosis and predicts disease severity in paediatric onset inflammatory bowel disease. Scand. J. Gastroenterol. 2019, 54, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Fite, A.; Macfarlane, S.; Furrie, E.; Bahrami, B.; Cummings, J.H.; Steinke, D.T.; Macfarlane, G.T. Longitudinal Analyses of Gut Mucosal Microbiotas in Ulcerative Colitis in Relation to Patient Age and Disease Severity and Duration. J. Clin. Microbiol. 2013, 51, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Aplakidou, E.; Vergoulidis, N.; Chasapi, M.; Venetsianou, N.K.; Kokoli, M.; Panagiotopoulou, E.; Iliopoulos, I.; Karatzas, E.; Pafilis, E.; Georgakopoulos-Soares, I.; et al. Visualizing metagenomic and metatranscriptomic data: A comprehensive review. Comput. Struct. Biotechnol. J. 2024, 23, 2011–2033. [Google Scholar] [CrossRef]
- Knoll, R.L.; Forslund, K.; Kultima, J.R.; Meyer, C.U.; Kullmer, U.; Sunagawa, S.; Bork, P.; Gehring, S. Gut microbiota differs between children with Inflammatory Bowel Disease and healthy siblings in taxonomic and functional composition: A metagenomic analysis. Am. J. Physiol. Liver Physiol. 2017, 312, G327–G339. [Google Scholar] [CrossRef]
- Thomann, A.K.; Wüstenberg, T.; Wirbel, J.; Knoedler, L.-L.; Thomann, P.A.; Zeller, G.; Ebert, M.P.; Lis, S.; Reindl, W. Depression and fatigue in active IBD from a microbiome perspective—A Bayesian approach to faecal metagenomics. BMC Med. 2022, 20, 366. [Google Scholar] [CrossRef]
- Zuo, W.; Wang, B.; Bai, X.; Luan, Y.; Fan, Y.; Michail, S.; Sun, F. 16S rRNA and metagenomic shotgun sequencing data revealed consistent patterns of gut microbiome signature in pediatric ulcerative colitis. Sci. Rep. 2022, 12, 6421. [Google Scholar] [CrossRef]
- Zhang, Y.; Thompson, K.N.; Branck, T.; Yan, Y.; Nguyen, L.H.; Franzosa, E.A.; Huttenhower, C. Metatranscriptomics for the Human Microbiome and Microbial Community Functional Profiling. Annu. Rev. Biomed. Data Sci. 2021, 4, 279–311. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Filimoniuk, A.; Daniluk, U.; Samczuk, P.; Wasilewska, N.; Jakimiec, P.; Kucharska, M.; Lebensztejn, D.M.; Ciborowski, M. Metabolomic profiling in children with inflammatory bowel disease. Adv. Med. Sci. 2020, 65, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Jagt, J.Z.; Verburgt, C.M.; de Vries, R.; de Boer, N.K.H.; A Benninga, M.; de Jonge, W.J.; E van Limbergen, J.; de Meij, T.G.J. Faecal Metabolomics in Paediatric Inflammatory Bowel Disease: A Systematic Review. J. Crohn’s Colitis 2022, 16, 1777–1790. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.-P.; Ezri, J.; Cominetti, O.; Da Silva, L.; Kussmann, M.; Godin, J.-P.; Nydegger, A. Urinary Metabolic Phenotyping Reveals Differences in the Metabolic Status of Healthy and Inflammatory Bowel Disease (IBD) Children in Relation to Growth and Disease Activity. Int. J. Mol. Sci. 2016, 17, 1310. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.-P.; Su, M.-M.; Xie, G.-X.; Guiraud, S.P.; Kussmann, M.; Godin, J.-P.; Jia, W.; Nydegger, A. Urinary metabolic insights into host-gut microbial interactions in healthy and IBD children. World J. Gastroenterol. 2017, 23, 3643–3654. [Google Scholar] [CrossRef]
- Patel, N.; Alkhouri, N.; Eng, K.; Cikach, F.; Mahajan, L.; Yan, C.; Grove, D.; Rome, E.S.; Lopez, R.; Dweik, R.A. Metabolomic analysis of breath volatile organic compounds reveals unique breathprints in children with inflammatory bowel disease: A pilot study. Aliment. Pharmacol. Ther. 2014, 40, 498–507. [Google Scholar] [CrossRef]
- Monasta, L.; Pierobon, C.; Princivalle, A.; Martelossi, S.; Marcuzzi, A.; Pasini, F.; Perbellini, L. Inflammatory bowel disease and patterns of volatile organic compounds in the exhaled breath of children: A case-control study using Ion Molecule Reaction-Mass Spectrometry. PLoS ONE 2017, 12, e0184118. [Google Scholar] [CrossRef]
- Daniluk, U.; Daniluk, J.; Kucharski, R.; Kowalczyk, T.; Pietrowska, K.; Samczuk, P.; Filimoniuk, A.; Kretowski, A.; Lebensztejn, D.; Ciborowski, M. Untargeted Metabolomics and Inflammatory Markers Profiling in Children With Crohn’s Disease and Ulcerative Colitis—A Preliminary Study. Inflamm. Bowel Dis. 2019, 25, 1120–1128. [Google Scholar] [CrossRef]
- Kolho, K.-L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and serum metabolomics in paediatric inflammatory bowel disease. J. Crohn’s Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef]
- Zhang, F.; Guo, L.; Shi, J.; Jiang, H.; Zhou, F.; Zhou, Y.; Lv, B.; Xu, M. Choline metabolism in regulating inflammatory bowel disease-linked anxiety disorders: A multi-omics exploration of the gut-brain axis. Neurobiol. Dis. 2023, 191, 106390. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Kennelly, J.P.; Jacobs, R.L.; Willing, B.P. Insufficient dietary choline aggravates disease severity in a mouse model of Citrobacter rodentium-induced colitis. Br. J. Nutr. 2020, 125, 50–61. [Google Scholar] [CrossRef]
- Guo, L.; Chen, Q.; Gao, Y.; Jiang, H.; Zhou, F.; Zhang, F.; Xu, M. CDP-choline modulates cholinergic signaling and gut microbiota to alleviate DSS-induced inflammatory bowel disease. Biochem. Pharmacol. 2023, 217, 115845. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, J.T.; Wang, Y.; Hao, F.; Coskun, M.; Ludwig, C.; Günther, U.; Nielsen, O.H. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabolomics 2014, 11, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Tsuji, A.; Ikeda, Y.; Yoshikawa, S.; Taniguchi, K.; Sawamura, H.; Morikawa, S.; Nakashima, M.; Asai, T.; Matsuda, S. The Tryptophan and Kynurenine Pathway Involved in the Development of Immune-Related Diseases. Int. J. Mol. Sci. 2023, 24, 5742. [Google Scholar] [CrossRef]
- Ala, M. Tryptophan metabolites modulate inflammatory bowel disease and colorectal cancer by affecting immune system. Int. Rev. Immunol. 2021, 41, 326–345. [Google Scholar] [CrossRef]
- Dussik, C.M.; Hockley, M.; Grozić, A.; Kaneko, I.; Zhang, L.; Sabir, M.S.; Park, J.; Wang, J.; A Nickerson, C.; Yale, S.H.; et al. Gene Expression Profiling and Assessment of Vitamin D and Serotonin Pathway Variations in Patients With Irritable Bowel Syndrome. J. Neurogastroenterol. Motil. 2018, 24, 96–106. [Google Scholar] [CrossRef]
- Grozić, A.; Coker, K.; Dussik, C.M.; Sabir, M.S.; Sabir, Z.; Bradley, A.; Zhang, L.; Park, J.; Yale, S.; Kaneko, I.; et al. Identification of putative transcriptomic biomarkers in irritable bowel syndrome (IBS): Differential gene expression and regulation of TPH1 and SERT by vitamin D. PLoS ONE 2022, 17, e0275683. [Google Scholar] [CrossRef]
- Nielsen, O.H.; Irgens Hansen, T.; Gubatan, J.M.; Jensen, K.B.; Rejnmark, L. Managing vitamin D deficiency in inflammatory bowel disease. Front. Gastroenterol. 2019, 10, 394–400. [Google Scholar] [CrossRef]
- Nassif, A.; Longo, W.E.; Mazuski, J.E.; Vernava, A.M.; Kaminski, D.L. Role of cytokines and platelet-activating factor in inflammatory bowel disease: Implications for therapy. Dis. Colon Rectum 1996, 39, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Eliakim, R.; Karmeli, F.; Razin, E.; Rachmilewitz, D. Role of platelet-activating factor in ulcerative colitis: Enhanced production during active disease and inhibition by sulfasalazine and prednisolone. Gastroenterology 1988, 95, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Oshimoto, H.; Okamura, S.; Iida, T.; Ishikawa, T.; Hosaka, K.; Mori, M. Diagnostic Value of the Serum Platelet-Activating Factor Acetylhydrolase Activity in Inflammatory Bowel Disease. Tohoku J. Exp. Med. 2005, 207, 65–71. [Google Scholar] [CrossRef]
- Lu, Y.; Xiong, Y.; Zhang, S.; Wang, B.; Feng, Y.; Pu, Z.; Wei, K.; Chen, J.; Chen, D.; Zhang, P. D-mannose reduces oxidative stress, inhibits inflammation, and increases treg cell proportions in mice with ulcerative colitis. Front. Pharmacol. 2024, 15, 1454713. [Google Scholar] [CrossRef]
- Dong, L.; Xie, J.; Wang, Y.; Jiang, H.; Chen, K.; Li, D.; Wang, J.; Liu, Y.; He, J.; Zhou, J.; et al. Mannose ameliorates experimental colitis by protecting intestinal barrier integrity. Nat. Commun. 2022, 13, 4804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bhosle, A.; Bae, S.; McIver, L.J.; Pishchany, G.; Accorsi, E.K.; Thompson, K.N.; Arze, C.; Wang, Y.; Subramanian, A.; et al. Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature 2022, 606, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Butcher, J.; Schramm, L.; Mack, D.R.; Stintzi, A. Multiomic spatial analysis reveals a distinct mucosa-associated virome. Gut Microbes 2023, 15, 2177488. [Google Scholar] [CrossRef]
- Taylor, H.; Serrano-Contreras, J.I.; McDonald, J.A.K.; Epstein, J.; Fell, J.; Seoane, R.C.; Li, J.V.; Marchesi, J.R.; Hart, A.L. Multiomic features associated with mucosal healing and inflammation in paediatric Crohn’s disease. Aliment. Pharmacol. Ther. 2020, 52, 1491–1502. [Google Scholar] [CrossRef]
- Sauceda, C.; Bayne, C.; Sudqi, K.; Gonzalez, A.; Dulai, P.S.; Knight, R.; Gonzalez, D.J.; Gonzalez, C.G. Stool multi-omics for the study of host–microbe interactions in inflammatory bowel disease. Gut Microbes 2022, 14, 2154092. [Google Scholar] [CrossRef]
- Wang, S.; Kaur, S.; Kunath, B.J.; May, P.; Richardson, L.; Rogers, A.B.; Wilmes, P.; Finn, R.D.; Vizcaíno, J.A. An Approach to Integrate Metagenomics, Metatranscriptomics and Metaproteomics Data in Public Data Resources. Proteomics 2025, e202500002. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Multi ‘omic data integration: A review of concepts, considerations, and approaches. Semin. Perinatol. 2021, 45, 151456. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Levitte, S.; Patel, A.; Balabanis, T.; Wei, M.T.; Sinha, S.R. Artificial intelligence applications in inflammatory bowel disease: Emerging technologies and future directions. World J. Gastroenterol. 2021, 27, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Dovrolis, N.; Moschoviti, A.; Fessatou, S.; Karamanolis, G.; Kolios, G.; Gazouli, M. Identifying Microbiome Dynamics in Pediatric IBD: More than a Family Matter. Biomedicines 2023, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.A.; Bittinger, K.; Ren, Y.; Kachelries, K.; Vales, J.; Li, H.; Wu, G.D.; Bushman, F.D.; Devoto, M.; Baldassano, R.N.; et al. The intestinal microbiome of inflammatory bowel disease across the pediatric age range. Gut Microbes 2024, 16, 2317932. [Google Scholar] [CrossRef]
- Zhang, X.; Deeke, S.A.; Ning, Z.; Starr, A.E.; Butcher, J.; Li, J.; Mayne, J.; Cheng, K.; Liao, B.; Li, L.; et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 2018, 9, 2873. [Google Scholar] [CrossRef]
- Sandrin, T.; Pasternak, B.; Jurutka, P.; Buss, K.; Gu, H.; DeSantis, A.; Coker, K. A Multiomics-based Analysis of the Fecal Microbiome of Pediatric Inflammatory Bowel Disease (IBD) Patients Reveals Potential Disease-specific Features More Effectively Than Microbiome Community Composition Sequence Data. Gastroenterology 2025, 168, S84. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individuals | Sex: F, M (% F) | Average Age (Range) | Race: White, Hispanic, Asian (% White) | |
|---|---|---|---|---|
| Diagnosis | ||||
| Crohn’s Disease—Naive | 2 | 1, 1 (50%) | 17 | 2, 0, 0 (100%) |
| Crohn’s Disease—Established | 8 | 5, 3 (62.5%) | 13.25 (7–17) | 8, 0, 0 (100%) |
| Ulcerative Colitis—Naive | 7 | 4, 3 (57.1%) | 13.43 (8–16) | 6, 1, 0 (85.7%) |
| Ulcerative Colitis—Established | 5 | 3, 2 (60%) | 12.4 (6–18) | 3, 1, 1 (60%) |
| Healthy | 4 | 3, 1 (75%) | 11.25 (6–14) | 3, 1, 0 (75%) |
| Disease Group | ||||
| All Crohn’s Disease | 10 | 6, 4 (60%) | 14 (7–17) | 10, 0, 0 (100%) |
| All Ulcerative Colitis | 12 | 7, 5 (58.3%) | 13 (6–18) | 9, 2, 1 (75%) |
| All Healthy | 4 | 3, 1 (75%) | 9.75 (5–17) | 3, 1, 0 (75%) |
| All Samples | 26 | 16, 10 (61.5%) | 13 (5–18) | 22, 3, 1 (84.6%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeSantis, A.H.; Buss, K.; Coker, K.M.; Pasternak, B.A.; Chi, J.; Patterson, J.S.; Gu, H.; Jurutka, P.W.; Sandrin, T.R. Multiomics-Based Profiling of the Fecal Microbiome Reveals Potential Disease-Specific Signatures in Pediatric IBD (PIBD). Biomolecules 2025, 15, 746. https://doi.org/10.3390/biom15050746
DeSantis AH, Buss K, Coker KM, Pasternak BA, Chi J, Patterson JS, Gu H, Jurutka PW, Sandrin TR. Multiomics-Based Profiling of the Fecal Microbiome Reveals Potential Disease-Specific Signatures in Pediatric IBD (PIBD). Biomolecules. 2025; 15(5):746. https://doi.org/10.3390/biom15050746
Chicago/Turabian StyleDeSantis, Anita H., Kristina Buss, Keaton M. Coker, Brad A. Pasternak, Jinhua Chi, Jeffrey S. Patterson, Haiwei Gu, Peter W. Jurutka, and Todd R. Sandrin. 2025. "Multiomics-Based Profiling of the Fecal Microbiome Reveals Potential Disease-Specific Signatures in Pediatric IBD (PIBD)" Biomolecules 15, no. 5: 746. https://doi.org/10.3390/biom15050746
APA StyleDeSantis, A. H., Buss, K., Coker, K. M., Pasternak, B. A., Chi, J., Patterson, J. S., Gu, H., Jurutka, P. W., & Sandrin, T. R. (2025). Multiomics-Based Profiling of the Fecal Microbiome Reveals Potential Disease-Specific Signatures in Pediatric IBD (PIBD). Biomolecules, 15(5), 746. https://doi.org/10.3390/biom15050746