
Examining the Effects of the RUNX1 p.Leu43Ser Variant on FPD/AML Phenotypes Using a CRISPR/Cas9-Generated Knock-In Murine Model

 , , , , , , , , ,
, , , , , , , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Murine Model and Experimental Design
2.3. Characterization of Hematopoietic Cells by Flow CytometryPanel by Thermo Fisher
2.4. Histopathology
2.5. RNA-Seq from Bone Marrow Samples
2.6. Statistical Analysis
3. Results
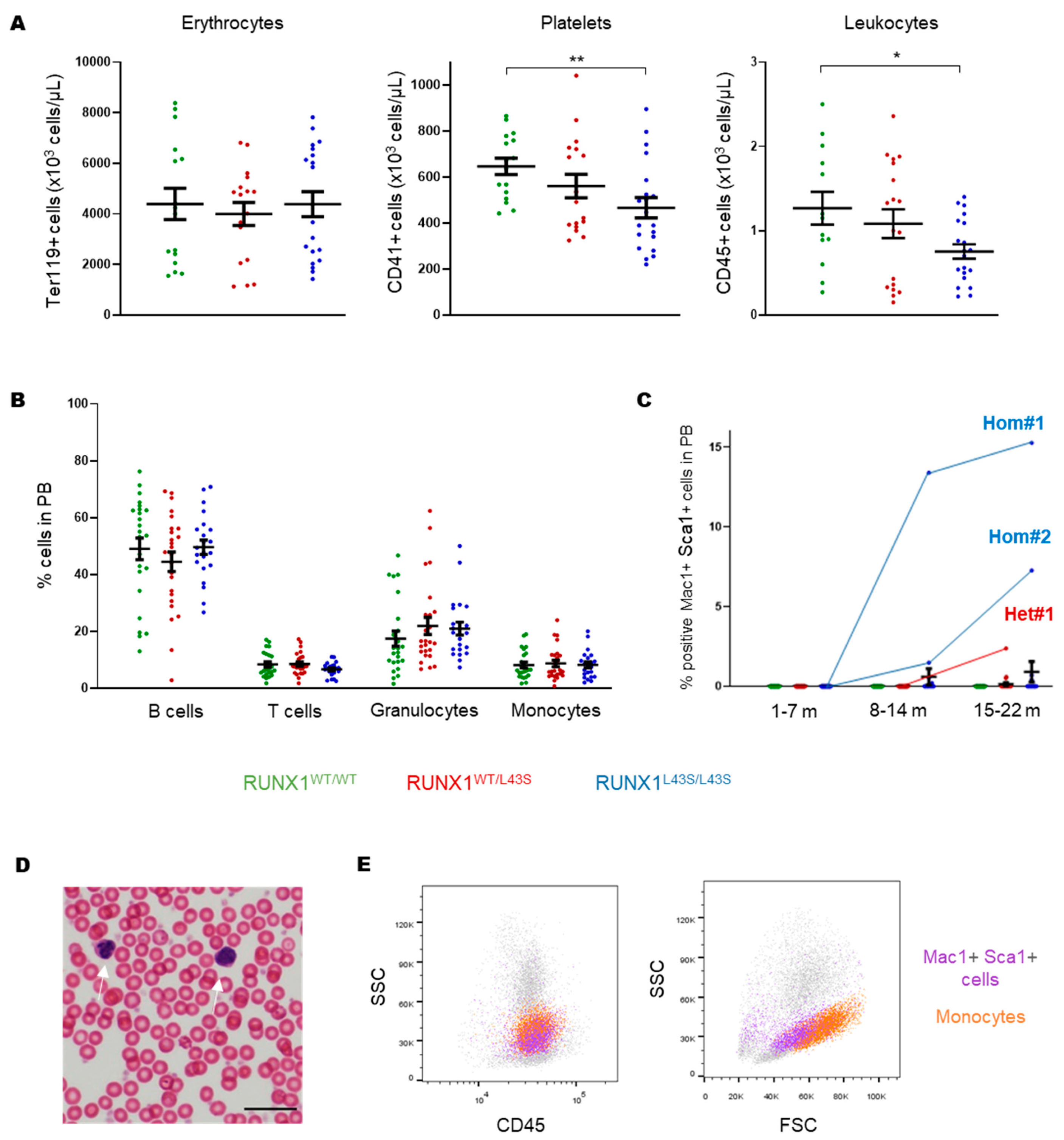
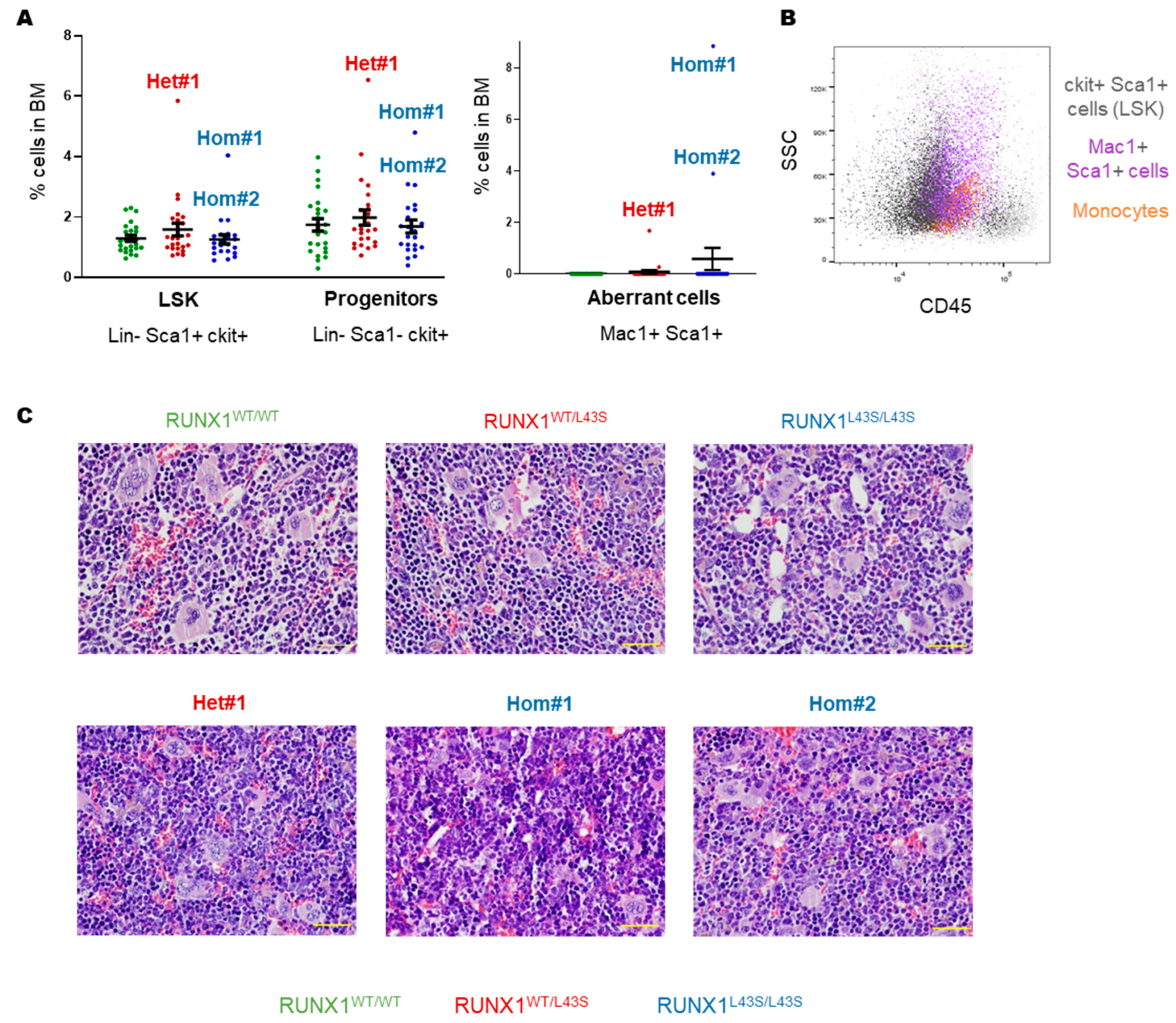
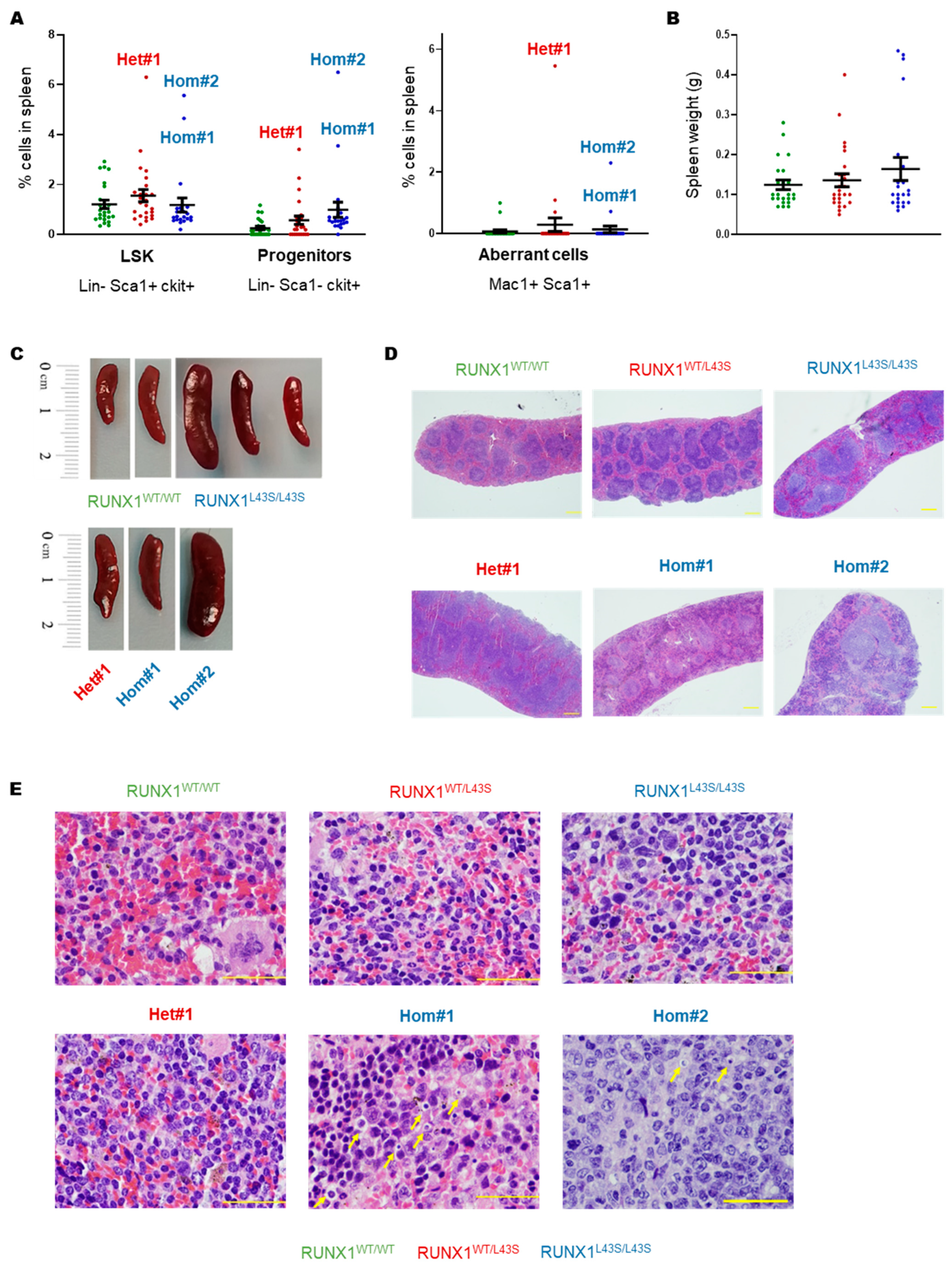
3.1. Phenotyping of Aberrant Cells in Peripheral Blood, Bone Marrow, and Spleen
3.2. Irradiation Increases the Risk of Aberrant Cell Proliferation in Mice Carrying the RUNX1 p.Leu43Ser Variant
3.3. RNA-Seq Revealed Aberrant Gene Expression in Hom#1 and Hom#2 Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| BM | Bone marrow |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| FC | Flow cytometry |
| FPD/AML | Familial Platelet Disorder with a predisposition to Acute Myeloid Leukemia |
| HTS | High-throughput sequencing |
| LSK | Lin− ckit+ sca1+ |
| MAF | Minor allele frequency |
| MM-VCEP | Myeloid Malignancy Variant Curation Expert Panel |
| PB | Peripheral blood |
| RUNX1 | Runt-related transcription factor 1 |
| WHO | World Health Organization |
References
- Simon, L.; Spinella, J.F.; Yao, C.Y.; Lavallée, V.P.; Boivin, I.; Boucher, G.; Audemard, E.; Bordeleau, M.E.; Lemieux, S.; Hébert, J.; et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood 2020, 135, 1882–1886. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, D.C.; Speck, N.A. RUNX1 Mutations in Inherited and Sporadic Leukemia. Front. Cell Dev. Biol. 2017, 5, 111. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.P.T.; Kavelaars, F.G.; Löwenberg, B.; Valk, P.J.M.; Raaijmakers, M.H.G.P. RUNX1 germline variants in RUNX1-mutant AML: How frequent? Blood 2021, 137, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Arts, P.; Carmichael, C.L.; Babic, M.; Dobbins, J.; Chong, C.E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Cho, B.S.; Kim, H.J.; Han, E.; Jang, W.; Han, K.; Lee, J.W.; Chung, N.G.; Cho, B.; Kim, M.; et al. Reclassification of acute myeloid leukemia according to the 2016 who classification. Ann. Lab. Med. 2019, 39, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Koeffler, H.P.; Leong, G. Preleukemia: One name, many meanings. Leukemia 2017, 31, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.P.T.; Versluis, J.; Valk, P.J.M.; Bierings, M.; Tamminga, R.Y.J.; Hooimeijer, L.H.; Döhner, K.; Gresele, P.; Tawana, K.; Langemeijer, S.M.C.; et al. Disease characteristics and outcome of acute myeloid leukemia in familial platelet disorder with associated myeloid malignancy. Hemasphere 2025, 9, e70057. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Renneville, A.; Bourdon, V.; Philippe, N.; Roche-Lestienne, C.; Boissel, N.; Dhedin, N.; André, J.M.; Cornillet-Lefebvre, P.; Baruchel, A.; et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009, 113, 5583–5587. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debré, I.; Duployez, N.; Bucci, M.; Geffroy, S.; Micol, J.B.; Renneville, A.; Boissel, N.; Dhédin, N.; Réa, D.; Nelken, B.; et al. Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia 2016, 30, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Feurstein, S.; Mohan, S.; Porter, C.C.; Jackson, S.A.; Keel, S.; Chicka, M.; Brown, A.L.; Kesserwan, C.; Agarwal, A.; et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019, 3, 2962–2979. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Luo, X.; Feurstein, S.; Kesserwan, C.; Mohan, S.; Pineda-Alvarez, D.E.; Godley, L.A. How I curate: Applying American Society of Hematology-Clinical Genome Resource Myeloid Malignancy Variant Curation Expert Panel rules for RUNX1 variant curation for germline predisposition to myeloid malignancies. Haematologica 2020, 105, 870–887. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Fenwarth, L. Controversies about germline RUNX1 missense variants. Leuk. Lymphoma 2020, 61, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.; Lammens, T.; Ferster, A.; Erlacher, M.; Yoshimi, A.; Niemeyer, C.M.; Ernst, M.P.T.; Raaijmakers, M.H.G.P.; Duployez, N.; Flaum, A.; et al. Functional classification of RUNX1 variants in familial platelet disorder with associated myeloid malignancies. Leukemia 2021, 35, 3304–3308. [Google Scholar] [CrossRef] [PubMed]
- Borst, S.; Nations, C.C.; Klein, J.G.; Pavani, G.; Maguire, J.A.; Camire, R.M.; Drazer, M.W.; Godley, L.A.; French, D.L.; Poncz, M.; et al. Study of inherited thrombocytopenia resulting from mutations in ETV6 or RUNX1 using a human pluripotent stem cell model. Stem Cell Rep. 2021, 16, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Lamolda, M.; Montes, R.; Simón, I.; Perales, S.; Martínez-Navajas, G.; Lopez-Onieva, L.; Ríos-Pelegrina, R.; Del Moral, R.G.; Griñan-Lison, C.; Marchal, J.A.; et al. GENYOi005-A: An induced pluripotent stem cells (iPSCs) line generated from a patient with Familial Platelet Disorder with associated Myeloid Malignancy (FPDMM) carrying a p.Thr196Ala variant. Stem Cell Res. 2019, 41, 101603. [Google Scholar] [CrossRef] [PubMed]
- Palma-Barqueros, V.; Bastida, J.M.; López-Andreo, M.J.; Zámora-Cánovas, A.; Zaninetti, C.; Ruiz-Pividal, J.F.; Bohdan, N.; Padilla, J.; Teruel-Montoya, R.; Marín-Quilez, A.; et al. Platelet transcriptome analysis in patients with germline RUNX1 mutations. J. Thromb. Haemost. 2023, 21, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Marín-Quílez, A.; García-Tuñón, I.; Fernández-Infante, C.; Hernández-Cano, L.; Palma-Barqueros, V.; Vuelta, E.; Sánchez-Martín, M.; González-Porras, J.R.; Guerrero, C.; Benito, R.; et al. Characterization of the Platelet Phenotype Caused by a Germline RUNX1 Variant in a CRISPR/Cas9-Generated Murine Model. Thromb. Haemost. 2021, 121, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Cobaleda, C.; Gutiérrez-Cianca, N.; Pérez-Losada, J.; Flores, T.; García-Sanz, R.; González, M.; Sánchez-García, I. A primitive hematopoietic cell is the target for the leukemic transformation in human Philadelphia-positive acute lymphoblastic leukemia. Blood 2000, 95, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Herrero, S.; Maia, V.; Gutiérrez-Berzal, J.; Calzada, N.; Sanz, M.; González-Manchón, C.; Pericacho, M.; Ortiz-Rivero, S.; González-Porras, J.R.; Arechederra, M.; et al. C3G transgenic mouse models with specific expression in platelets reveal a new role for C3G in platelet clotting through its GEF activity. Biochim. Biophys. Acta. 2012, 1823, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Herrero, S.; Fernández-Infante, C.; Hernández-Cano, L.; Ortiz-Rivero, S.; Guijas, C.; Martín-Granado, V.; González-Porras, J.R.; Balsinde, J.; Porras, A.; Guerrero, C. C3G contributes to platelet activation and aggregation by regulating major signaling pathways. Signal Transduct. Target. Ther. 2020, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Martín-Granado, V.; Ortiz-Rivero, S.; Carmona, R.; Gutiérrez-Herrero, S.; Barrera, M.; San-Segundo, L.; Sequera, C.; Perdiguero, P.; Lozano, F.; Martín-Herrero, F.; et al. C3G promotes a selective release of angiogenic factors from activated mouse platelets to regulate angiogenesis and tumor metastasis. Oncotarget 2017, 8, 110994–111011. [Google Scholar] [CrossRef] [PubMed]
- Carabias, A.; Guerrero, C.; de Pereda, J.M. C3G self-regulatory mechanism revealed: Implications for hematopoietic malignancies. Mol. Cell Oncol. 2021, 8, 1837581. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Berzal, J.; Castellano, E.; Martín-Encabo, S.; Gutiérrez-Cianca, N.; Hernández, J.M.; Santos, E.; Guerrero, C. Characterization of p87C3G, a novel, truncated C3G isoform that is overexpressed in chronic myeloid leukemia and interacts with Bcr-Abl. Exp. Cell Res. 2006, 312, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Harada, Y.; Huang, G.; Harada, H. Myeloid neoplasms with germ line RUNX1 mutation. Int. J. Hematol. 2017, 106, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.K.; Lee, J.; Ng, C.; Kosmo, B.; Chiu, L.; Seah, E.; Mok, M.M.H.; Tan, K.; Osato, M.; Chng, W.J.; et al. Preleukemic and second-hit mutational events in an acute myeloid leukemia patient with a novel germline RUNX1 mutation. Biomark. Res. 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Toya, T.; Kawazu, M.; Ueno, T.; Tsukamoto, A.; Iizuka, H.; Nakagawa, M.; Nannya, Y.; Arai, S.; Harada, H.; et al. Recurrent CDC25C mutations drive malignant transformation in FPD/AML. Nat. Commun. 2014, 5, 4770. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Kasahara, H.; Yoshida, K.; Yoshimi, A.; Kunimoto, H.; Watanabe, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Harada, Y.; et al. Genetic basis of myeloid transformation in familial platelet disorder/acute myeloid leukemia patients with haploinsufficient RUNX1 allele. Blood Cancer J. 2016, 6, e392. [Google Scholar] [CrossRef] [PubMed]
- Growney, J.D.; Shigematsu, H.; Li, Z.; Lee, B.H.; Adelsperger, J.; Rowan, R.; Curley, D.P.; Kutok, J.L.; Akashi, K.; Williams, I.R.; et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005, 106, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.; Muselman, A.; Du, R.; Harada, Y.; Scholl, A.G.; Yan, M.; Matsuura, S.; Weng, S.; Harada, H.; Zhang, D.E. Hmga2 is a direct target gene of RUNX1 and regulates expansion of myeloid progenitors in mice. Blood 2014, 124, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Kunimoto, H.; Watanabe, N.; Fukuchi, Y.; Yuasa, S.; Yamazaki, S.; Nishimura, T.; Sadahira, K.; Fukuda, K.; Okano, H.; et al. Impaired hematopoietic differentiation of RUNX1-mutated induced pluripotent stem cells derived from FPD/AML patients. Leukemia 2014, 28, 2344–2354. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.P.; Wang, C.Q.; Ng, C.E.; Ito, Y.; Araki, M.; Tergaonkar, V.; Huang, G.; Osato, M. RUNX1 meets MLL: Epigenetic regulation of hematopoiesis by two leukemia genes. Leukemia 2013, 27, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Bath, M.L.; Turner, M.L.; Vandenberg, C.J.; Bouillet, P.; Metcalf, D.; Scott, C.L.; Cory, S. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood 2010, 116, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debré, I.; Manchev, V.T.; Balayn, N.; Bluteau, D.; Tomowiak, C.; Legrand, C.; Langlois, T.; Bawa, O.; Tosca, L.; Tachdjian, G.; et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood 2015, 125, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef] [PubMed]
- Olofsen, P.A.; Fatrai, S.; van Strien, P.M.H.; Obenauer, J.C.; de Looper, H.W.J.; Hoogenboezem, R.M.; Erpelinck-Verschueren, C.A.J.; Vermeulen, M.P.W.M.; Roovers, O.; Haferlach, T.; et al. Malignant Transformation Involving CXXC4 Mutations Identified in a Leukemic Progression Model of Severe Congenital Neutropenia. Cell Rep. Med. 2020, 1, 100074. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.T.; Poole, A.W. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J. Thromb. Haemost. 2010, 8, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Kometani, K.; Ishida, D.; Hattori, M.; Minato, N. Rap1 and SPA-1 in hematologic malignancy. Trends Mol. Med. 2004, 10, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Minato, N.; Kometani, K.; Hattori, M. Regulation of Immune Responses and Hematopoiesis by the Rap1 Signal. Adv. Immunol. 2007, 93, 229–264. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin-Quilez, A.; García-Tuñón, I.; Benito, R.; Ordoñez, J.L.; Díaz-Ajenjo, L.; Lama-Villanueva, A.; Guerrero, C.; Pérez-Losada, J.; González-Porras, J.R.; Hernández-Rivas, J.M.; et al. Examining the Effects of the RUNX1 p.Leu43Ser Variant on FPD/AML Phenotypes Using a CRISPR/Cas9-Generated Knock-In Murine Model. Biomolecules 2025, 15, 708. https://doi.org/10.3390/biom15050708
Marin-Quilez A, García-Tuñón I, Benito R, Ordoñez JL, Díaz-Ajenjo L, Lama-Villanueva A, Guerrero C, Pérez-Losada J, González-Porras JR, Hernández-Rivas JM, et al. Examining the Effects of the RUNX1 p.Leu43Ser Variant on FPD/AML Phenotypes Using a CRISPR/Cas9-Generated Knock-In Murine Model. Biomolecules. 2025; 15(5):708. https://doi.org/10.3390/biom15050708
Chicago/Turabian StyleMarin-Quilez, Ana, Ignacio García-Tuñón, Rocío Benito, José Luis Ordoñez, Lorena Díaz-Ajenjo, Ana Lama-Villanueva, Carmen Guerrero, Jesús Pérez-Losada, José Ramón González-Porras, Jesús María Hernández-Rivas, and et al. 2025. "Examining the Effects of the RUNX1 p.Leu43Ser Variant on FPD/AML Phenotypes Using a CRISPR/Cas9-Generated Knock-In Murine Model" Biomolecules 15, no. 5: 708. https://doi.org/10.3390/biom15050708
APA StyleMarin-Quilez, A., García-Tuñón, I., Benito, R., Ordoñez, J. L., Díaz-Ajenjo, L., Lama-Villanueva, A., Guerrero, C., Pérez-Losada, J., González-Porras, J. R., Hernández-Rivas, J. M., del Rey, M., & Bastida, J. M. (2025). Examining the Effects of the RUNX1 p.Leu43Ser Variant on FPD/AML Phenotypes Using a CRISPR/Cas9-Generated Knock-In Murine Model. Biomolecules, 15(5), 708. https://doi.org/10.3390/biom15050708