
Diagnosis of Inherited Platelet Disorders: Clinical Evaluation and Functional and Molecular Assays



Abstract
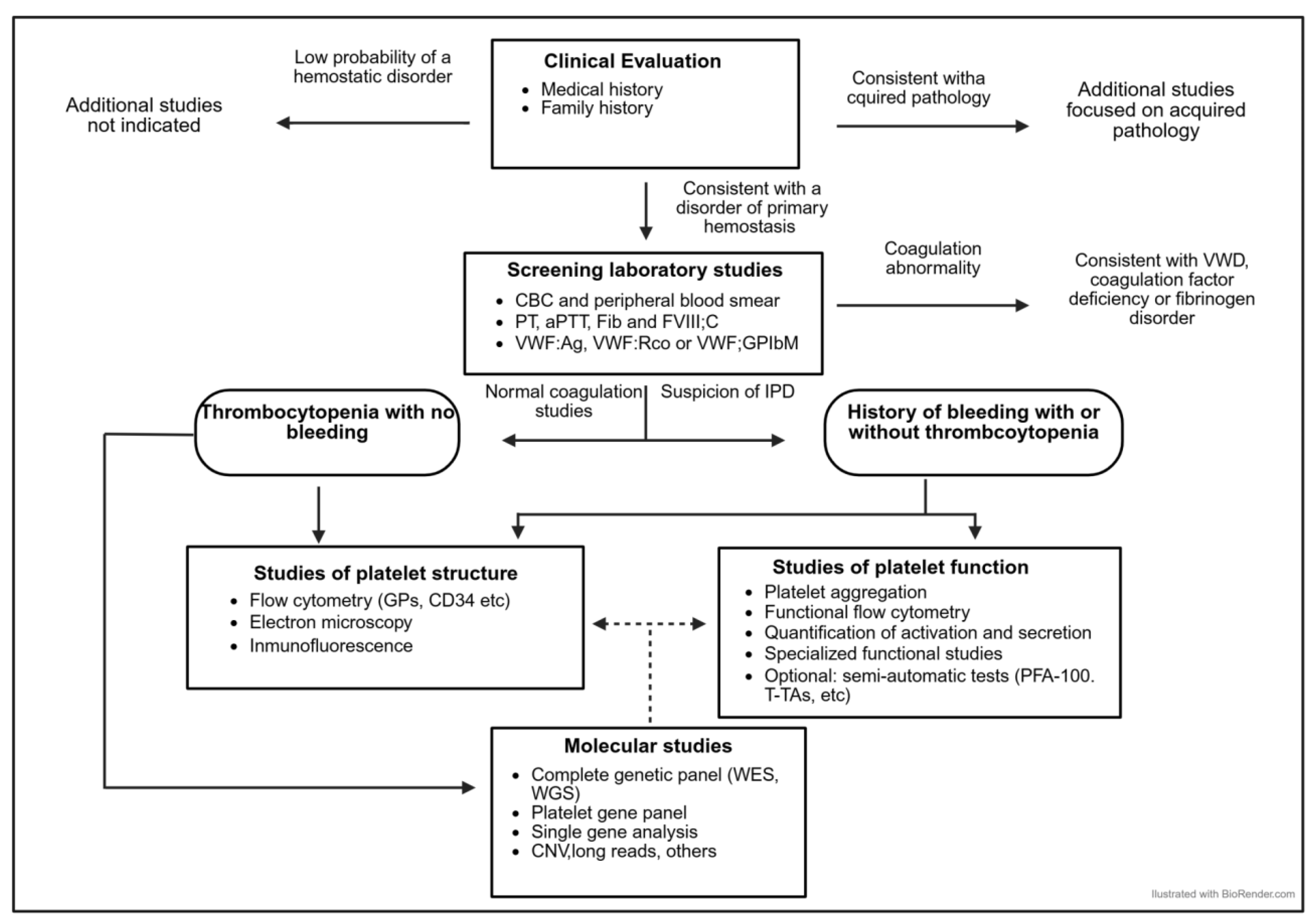
1. Background and Objectives
2. Personal and Family History of the Patients with Suspected IPD
2.1. Personal History of Patients with Suspected IPD
2.1.1. The Patients with Bleeding With or Without Thrombocytopenia
- ▪
- A physical examination of the patient, looking for external signs of bleeding.
- ▪
- Evaluate whether the patient’s current and/or previous bleeding symptoms meet the typical characteristics of a primary hemostatic disorder: mucocutaneous bleeding, immediate bleeding after trauma or in the immediate postoperative period. It should be noted that these signs are also typical of other disorders such as von Willebrand disease (VWD) or endothelium-related pathology. In some severe IPFDs, such as Glanzmann thrombasthenia (GT), there may also be urinary, muscular bleeding and hemarthrosis, which are generally more typical of secondary hemostatic disorders such as hemophilia.
- ▪
- Objectively assess bleeding using validated tools such as the bleeding assessment tool developed by the International Society on Thrombosis and Hemostasis (ISTH-BAT) [13,14,15] (Table 1). The absolute value of the ISTH-BAT (bleeding score) that is considered pathological, and therefore, suggestive of platelet dysfunction is ≥3 points in children, ≥4 in men, and ≥6 in women. An ISTH-BAT ≥ 6 has been described to have a positive predictive value of 99% for an IPFD [15], but a lower score does not exclude the existence of a possible mild or moderate IPFD. Its main limitations are the pediatric population and/or young adults without hemostatic challenges, recall bias, the saturation of the score with recurrent symptoms, and its inability to distinguish different types of bleeding disorders.
- ▪
- In addition to the severity of bleeding in terms of the absolute ISTH-BAT value, it is also important to assess the age of onset, persistence, and location of bleeding.
- ▪
- In cases of patients with thrombocytopenia, concomitant platelet dysfunction is suspected if bleeding is disproportionate to the patient’s platelet count. In other systemic diseases or in ITP, spontaneous or even severe bleeding is usually not observed if the platelet count exceeds 10 × 109/L [16,17].
- ▪
- In some IPDs with mild impairment of platelet function, bleeding is not an essential feature, and a lack of bleeding manifestations does not rule out the diagnosis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symptom | Score | ||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |
| Epistaxis | No/Trivial | >5/Year or >10 min | Consultation only a | Packing or cauterization or antifibrinolytic | BT/RT/DDAVP |
| Cutaneous | No/Trivial | For bruises 5 or more [>1 cm] in exposed areas | Consultation only a | Extensive | Spontaneous hematoma requiring BT |
| Bleeding from Minor Wounds | No/Trivial | >5/y or more than 10 min | Consultation only a | Surgical hemostasis | BT/RT/DDAVP |
| Oral Cavity | No/Trivial | Present | Consultation only a | Surgical hemostasis or antifibrinolytic | BT/RT/DDAVP |
| GI Bleeding | No/Trivial | Present (not associated with GI lesions) | Consultation only a | Surgical hemostasis or antifibrinolytic | BT/RT/DDAVP |
| Hematuria | No/Trivial | Present (macroscopic) | Consultation only a | Surgical hemostasis iron therapy | BT/RT/DDAVP |
| Tooth Extractions | No/Trivial or not carried out | Reported in ≤25% of all procedures, no interventions | Reported in >25% of procedures, no interventions | Resuturing or packing | BT/RT/DDAVP |
| Surgery | No/Trivial or not carried out | Reported in ≤25% of all procedures, no interventions | Reported in >25% of procedures, no interventions | Surgical hemostasis or antifibrinolytic | BT/RT/DDAVP |
| Menorrhagia | No/Trivial | Consultation only a or hanging pads more frequently than every 2 h; or clot sand flooding; or PBAC score > 100 | Time of work/school > 2/y or requiring antifibrinolytic or hormonal therapy or iron therapy | Requiring combined treatment with antifibrinolytic agents and hormonal therapy or present since menarche and >12 months | Acute menorrhagia requiring hospital admission and emergency treatment or BT/RT/DDAVP or D&C or endometrial ablation or hysterectomy |
| Post-partum hemorrhage | No/Trivial or no deliveries | Consultation only a or use of syntocin or lochia > 6 weeks | Iron therapy or antifibrinolytic | BT/RT/DDAVP or requiring examination under anesthesia and/or the use of uterine balloon package to tamponade the uterus | Any surgical procedure requiring critical care or surgical intervention (e.g., hysterectomy, iliac artery ligation, uterine artery embolization, uterine brace sutures) |
| Muscle hematomas | No/Trivial | Post trauma, no therapy | Spontaneous, no therapy | Spontaneous or traumatic requiring DDAVP or RT | Spontaneous or traumatic requiring surgical intervention or BT |
| Hemarthrosis | No/Trivial | Post trauma, no therapy | Spontaneous, no therapy | Spontaneous or traumatic requiring DDAVP or RT | Spontaneous or traumatic requiring surgical intervention or BT |
| CNS bleeding | Never | - | - | Subdural, any intervention | Intracerebral, any intervention |
| Other bleeding | No/Trivial | Present | Consultation only a | Surgical hemostasis or antifibrinolytic | BT/RT/DDAVP |
2.1.2. The Patients with Thrombocytopenia
- ▪
- Spectrum of clinical manifestations:
- -
- Thrombocytopenia of variable severity and clinically significant bleeding (see above).
- -
- Thrombocytopenia, usually mild–moderate, asymptomatic, or associated with minimal/absent bleeding
- -
- Syndromic thrombocytopenia
- -
- Thrombocytopenia with predisposition or risk to other hematologic or extra-hematologic complications
- ▪
- Platelet count and platelet morphology:
- ▪
- Associated systemic diseases (syndromes or predisposition):
2.1.3. Patients with Other Atypical Presentations
- ▪
- Patients with a previous diagnosis of ITP who do not respond to standard treatments, especially pediatric or young adult patients, and/or cases defined as familial ITP. It should be noted that, historically, a significant proportion of patients with ITs were/are erroneously diagnosed and treated with ITP, due to the higher incidence of this disease and the fact that general practitioners and hematologists are more familiar with this acquired pathology [5,20].
- ▪
- Patients presenting with hematologic neoplasm without other predisposing factors may previously have had long-standing, unrecognized thrombocytopenia. Up to 30% of RUNX1 disease-causing genetic variants in adult patients with acute myeloblastic leukemia have been reported to be germline [21]. Hence, patients could have previously been diagnosed with RUNX1-related thrombocytopenia (RUNX1-RT or RUNX1-FPD, or FPD/AML, or FPDMM).
- ▪
- People with disproportionate thrombocytopenia in the context of pregnancy or “familial” heavy menstrual bleeding.
2.2. Family History of Patients with Suspected IPD
- ▪
- The presence of relatives with clinical characteristics like those of the index patient, which increases the likelihood of them having an IPD.
- ▪
- If affected relatives belong to several generations, an autosomal dominant (AD) IPD may be suspected.
- ▪
- If there are no affected relatives, or the few that are belong to different generations, an autosomal recessive (AR) IPD is more likely. This also applies to families with known consanguinity.
- ▪
- ▪
- The absence of other affected family members should not rule out the existence of an IPD considering that:
- ▪
- In cases of AR inherited IPDs in which only homozygous or compound heterozygous carriers manifest the disease, heterozygous subjects are usually asymptomatic.
- -
- -
- Patients with IPD show high variability in their clinical presentation, due to variable penetrance and the presence of other elements (congenital or acquired, known or unknown) that modulate clinical and laboratory phenotypes.
2.3. Other General Recommendations
3. Laboratory Studies
3.1. Phase 1: Initial Laboratory Studies
- (a)
- Coagulation studies:
- -
- Prothrombin time (PT);
- -
- Activated partial thromboplastin time (APTT);
- -
- FVIII activity;
- -
- Fibrinogen activity.
- ○
- Prolonged screening of clotting times should prompt additional evaluation to identify specific coagulation factor deficiencies.
- (b)
- Study of von Willebrand Factor (VWF):
- -
- VWF antigen level;
- -
- Functional activity of VWF.
- ○
- ○
- When interpreting the results, it is essential to take into account the ABO blood type, since individuals with type O usually have lower levels of VWF compared to other blood types [29].
- ○
- APTT may be prolonged if plasma FVIII levels are sufficiently low, especially in VWD types 2N and 3.
- ○
- In some cases, a coagulation disorder may coexist with platelet dysfunction. Therefore, platelet function studies may be recommended in patients with mild VWF deficiency or in those with known coagulation factor disorders who present with bleeding manifestations that are disproportionate or unusual to those expected in these conditions [30,31].
- (c)
- Complete blood count (CBC):
- ○
- Platelet count and platelet size:
- ○
- Red blood cell and leukocyte count:
- ○
- Other parameters of interest:
- (d)
- Conventional peripheral blood (PB) smear:
- ○
- Platelet clumping and satellitism should be evaluated as a cause of pseudothrombocytopenia, an artifactual thrombocytopenia, usually in blood anticoagulated with EDTA, which occurs in up to 0.3% of cases [37].
- ○
- Platelet aggregates can also occur in platelet-type VWD (PT-VWD), and its phenocopy VWD-type 2B.
- ○
- Identification possible morphological and granular abnormalities in platelets:
- ▪
- Platelet size determined with the mean platelet diameter (MPD) can guide the diagnostic process of a IT, as it allows for the identification of platelets that are considered large (MPD > 3.2 μm), normal (MPD 2.6–3.2 μm) or small (MPD < 2.6 μm) [38]. Microthrombocytopenia can be related to molecular alterations in the FYB, ARCP1B, PTPRJ (CD148), or WAS genes. In contrast, macrothrombocytopenia, even with giant platelets, is typical of alterations in the GP1BA, GP1B, GP9, MYH9, SRC, and SLFN14 genes, and in the majority of ITs from variants in genes that encode cytoskeletal proteins or participate in glycosylation [1,2,4].
- ▪
- The presence of poorly stained platelets in the smear is generally suggestive of a defect in the number or content of alpha granules. Gray or pale or highly vacuolated platelets associated with an α-granule deficiency caused by molecular abnormalities in genes such as NBEAL2, GATA-1, or GFI1B, among others, can be identified [1,2,4]. δ granules are not visible in MGG staining and must be evaluated by other methods such as “whole mount” electron microscopy (see below).
- ○
- Morphologic abnormalities in other cell lines:
- ▪
- Erythrocytes: Morphological abnormalities of red blood cells occur in cases of GATA1-RT (anisocytosis and poikilocytes), Stormorken syndrome (Howell-Jolly bodies caused by asplenia), or sitosterolemia due to variants in the ABCG5/ABCG8 genes of sterol metabolism (stomatocytes and possible signs of hemolysis) [1,2,4]. The presence of schistocytes together with thrombocytopenia, hemolysis, and renal failure suggests the diagnosis of thrombotic microangiopathy, an entity with which the differential diagnosis should be made.
- ▪
- Leukocytes: It is essential to rule out the presence of dysplastic features or the presence of immature elements that suggest a possible underlying myeloid neoplasm. In some, but not all, patients with MYH9-RD, blue-stained cytoplasmic inclusion bodies, commonly called Döhle-like bodies, are observed in neutrophils. Immunofluorescent staining with antibodies against MYH9 is a much more sensitive alternative for detecting these MYH9 aggregates in neutrophils that are present in virtually all cases of MYH9-RD [8,39]. The presence of giant granules in leukocytes suggests the diagnosis of Chediak–Higashi syndrome (CHS).
- (e)
- Bone marrow study:
- ○
- In patients with IPD and high risk of bone marrow failure (CAMT, thrombocytopenia with radioulnar synostosis, or thrombocytopenia with absence of radius), bone marrow biopsy is recommended.
- ○
- In patients with an IPD with a risk of developing hematological malignancies, a baseline bone marrow biopsy can be considered. If there are significant clinical changes such as splenomegaly, or relevant alterations in the complete blood count/peripheral blood smear (cytopenias or in cases of bone marrow fibrosis), a bone marrow biopsy is recommended.
3.2. Phase 2: Global Hemostasis and/or Platelet Function Assays
- ○
- Semi-automated global hemostasis tests, such as PFA-100 tests, are only optional in the diagnosis of IPD.
- ○
- They can be useful to quickly exclude severe platelet dysfunction. For example, in the PFA-100 test, the closure times with both cartridges (Col-ADP and Col-EPI) are always abnormal (≥300 s) in cases of BSS or GT.
- ○
- A normal or minimally abnormal result does not exclude mild to moderate platelet dysfunction. If the suspicion is high enough by the patient’s clinical symptoms, further testing is recommended.
3.3. Phase 3: Basic Studies of the Platelet Phenotype
- (a)
- Platelet aggregation
- ○
- The general recommendations for LTA established by ISTH [48] (currently under review) should be followed.
- ○
- A healthy control should always be analyzed in parallel with the patient’s sample (using identical sample extraction and handling).
- ○
- It is not necessary for the patients to fast before testing, but they must not have consumed meals with very high fat content (highly lipemic plasma) within 12 h of sample collection.
- ○
- Blood should be drawn from patients and controls, preferably in the morning, after a short rest (15–30 min), without having smoked (30 min), and without having taken supplements or medications that potentially inhibit platelets or NSAIDs (3 days), or irreversible antiplatelet agents such as acetylsalicylic acid (10 days).
- ○
- Blood should be drawn with minimal compression and a 21 g needle; first, a sample in EDTA (2–4 mL) for complete blood count or other purposes, and then blood for LTA and other functional studies (usually 20–30 mL) in citrate tubes (9:1), preferably 3.2% (109 mM) and buffered, although 3.7% citrate, 129 mM is acceptable.
- ○
- If commercial vacuum tubes with a fixed blood volume are used (the ones commonly found in hospitals), it is ideal to use those with a larger volume (10 or 5 mL, preferably over tubes of <3 mL), ensure that they are filled correctly, and that the blood mixes well with the anticoagulant.
- ○
- Samples should be stored at room temperature (do not store in refrigerators or in coolers).
- ○
- Avoid vigorously shaking the tubes. If mixing is necessary, do so by gentle inversion or using a tube rotator at low speed.
- ○
- If samples must be transported to another laboratory or center, they must be transported in suitable containers with the tubes properly protected. This is especially important if the transport is long-distance and involves courier companies (instruct them not to refrigerate and handle carefully).
- ○
- ○
- The LTA study on 12 to 24 h blood samples sent by courier may be considered if there is no other option; for example, if the hospital does not have the methodology and/or the patient cannot travel to another center. In this case, it is essential to analyze samples from a healthy control collected and shipped in parallel, as well as samples from another healthy control collected just before the LTA test is performed (as a normal response control).
- ○
- In our experience, in blood aged ≈ 24 h, there is an almost complete loss of aggregation with weak agonists such as epinephrine (any dose) or with low doses of ADP, TRAP, or others; with medium/high doses of these agonists, a moderate reduction in aggregation is observed (≈25–30%, sometimes the loss of the second wave); and with potent agonists such as CRP, arachidonic acid, or ristocetin, LTA is usually only slightly altered.
- ○
- PRP is generally obtained by centrifuging the original tubes (usually 4–5 mL) at room temperature for 10 min at 150–200× g, with minimal or no brake. Transferring to other tubes, checking for lipemia, icterus, or hemolysis, and taking a sample for a complete blood count. The platelet count should be higher than that of the original blood.
- ○
- In patients with large or giant platelets (such as those with various macrothrombocytopenias or BSS), standard centrifugation may result in the loss of a significant number of platelets, particularly large platelets. In these cases, it is possible to obtain PRP at a slower speed (below 100× g) or by gravity sedimentation after allowing the tubes to rest (≈1 h) at an inclination angle of 45 degrees. It should be noted that these PRPs may be more contaminated with leukocytes and red blood cells; this contamination can be reduced by allowing the PRP to rest again or by centrifuging at low speed (50× g, 5 min).
- ○
- After separating the PRP, the platelet-poor plasma (PPP) obtained will be used as a blank by centrifuging the tubes at room temperature for 15 min at 1200–1500× g.
- ○
- The recommended platelet count in PRP is 150–400 × 109/L. The ISTH guidelines recommend not adjusting the platelet count of the PRP obtained, but if adjusted, using a suitable buffer [48] such as Tyrode’s buffer. However, many experts prefer to adjust the PRP count with PPP to 200–250 × 109/L. 10 Our practice is to not adjust the PRP if it is in the range of 150–400 × 109/L, but to adjust it with PPP to 250–300 × 109/L if the count is >400 × 109/L.
- ○
- In IT patients, PRP may be lower than 150 × 109/L; hence, adjusting the control PRP to the same level is recommended. Current aggregometers, such as the Stago TA 4-V3, can reproduce platelet aggregation with PRP levels of approximately 50 × 109/L.
- ○
- The LTA assay is performed at 37 °C. The equipment is thermostatically controlled. Recording is typically carried out for 5 min; for some uncommonly used agonists like PMA, it is ideal to extend the time to 10 min, as the aggregation response is slow. It may also take 10–15 min to observe disaggregation with agonists like epinephrine.
- ○
- Ideally, each laboratory should have its own normal LTA range for both normal-count PRP (150–400 × 109/L) and thrombocytopenic PRP (≈50–100 × 109/L). Where appropriate, a normal range should also be available for PRP from blood samples from ≈24 h apart. The recommended number of controls for establishing these normal ranges is >40.
- ○
- Table 3 shows the panel of agonists that we recommended in the first LTA study.
- ○
- If the volume of PRP obtained is insufficient for the proposed panel, a more basic study is recommended with 1.5 mM arachidonic acid, 5 μM ADP, 25 μM TRAP, 1.25 mg/mL ristocetin, and 2 μg/mL collagen. This study may be sufficient to guide an IPFD in a first LTA study.
- ○
- In each LTA study, it is advisable to repeat the test with any agonist if it gives an abnormal result, both in the patient and in the control.
- ○
- Depending on the results of LTA obtained, consideration should be given to testing other doses and/or other agonists (PAR-4, A23187, PMA, convulxin, PAF, or others) in the same study, or in a subsequent study according to the diagnostic suspicion.
- ○
- If a patient’s LTA study is abnormal, it should be repeated after at least one month to confirm the persistence of the abnormality.
- ○
- In the global interpretation of the aggregation curves, the following must be assessed:
- ▪
- Change in platelet shape: This is reflected by a reduction in light transmission at the beginning (10–30 s) of the aggregation curve. This is not evident with all agonists.
- ▪
- Delay in the onset of aggregation (lag): with some agonists such as collagen, a lag of ≈30–50 s is typical.
- ▪
- Aggregation slope: usually directly proportional to maximum aggregation. It is usually greater with strong agonists.
- ▪
- Second wave of aggregation: Typical of epinephrine and weak agonists/low doses. Not evident with strong agonists/high doses.
- ▪
- Maximum aggregation (%).
- ▪
- Disaggregation: Reflects the formation of loose aggregates. If it occurs, final aggregation is less than maximum. It usually occurs with weak/low-dose agonists (ADP, TRAP, U46619) in moderate IPFD signaling/secretion, or due to platelet desensitization, for example, in PRP from non-fresh blood.
- ○
- Some aggregation patterns in response to the recommended agonists are characteristic of specific types of IPFD and allow for their diagnosis. Others are indicative of a category of defects but are not specific and do not allow for the diagnosis of a specific type. There are multiple expert guides and publications with recommendations for interpreting aggregation patterns (Table 4) [10,51].
| Agonist | Concentration | |
|---|---|---|
| PRP Fresh Blood | PRP Blood 12–24 h * | |
| Arachidonic Ac. | 1.5 mM | 1.5 mM |
|
ADP
If altered at low dose | 2.5 μM 10 μM | 5 μM 10 μM |
|
Epinephrine
If altered at low dose | 5 μM 10 μM | - |
|
TRAP (PAR-1)
If altered at low dose | 12.5 μM 25 μM | 25 μM |
|
Ristocetin
If platelet type VWD is suspected | 1.25 mg/mL 0.5–0.8 mg/mL | 1.25 mg/mL 0.8 mg/mL |
|
Collagen
If altered at low dose | 2 μg/mL 5 μg/mL | 5 μg/mL 10 μg/mL |
|
CRP (If altered with collagen)
If altered at low dose | 2 μg/mL 5 μg/mL | 5 μg/mL 10 μg/mL |
|
U46616 (If altered with arachidonic acid)
If altered at low dose | 2 μM 5 μM | 5 μM 10 μM |
| TPC | LTA Pattern |
|---|---|
|
BSS
(GPIb/IX defect) | Aggregation absent/severely decreased with ristocetin; normal or reduced, but not absent, with other agonists (arachidonic acid, ADP, collagen, TRAP). |
|
GT
(defect of αIIbβ3) | Aggregation absent/severely decreased with all agonists (arachidonic acid, ADP, collagen, TRAP), except ristocetin (reduced but not absent). This pattern, perhaps less severe, can be seen in cases of dominant thrombocytopenia due to mutations in the Glanzmann genes (ITGA2B and ITGB3). |
|
RASGRP2 defect
(CalDAG-GEF) | Aggregation absent/severely decreased with low-moderate doses of most agonists (ADP, epinephrine, collagen, TRAP, etc.), but normal or minimally affected with high doses of these agonists, arachidonic acid, ristocetin or PMA (PKC activator). |
| GPVI deficiency | Aggregation absent/severely decreased with CRP (even high doses) and low doses of collagen; normal/less affected with other agonists. |
| P2Y12 receptor deficiency | Severely decreased and reversible aggregation with ADP even at high doses (>10 μM). Aggregation is also reduced with low–moderate doses of other agonists such as collagen, but normal or minimally affected with high doses of epinephrine (5–10 μM). The P2Y12 defect can be confirmed by other assays such as PGE1-induced cAMP formation in the presence and absence of ADP or epinephrine. |
| TxA2 receptor deficiency | Aggregation absent/severely decreased with arachidonic acid and U46619. Variably reduced with moderate–low doses of other agonists such as collagen or TRAP. |
|
TxA2 generation deficiency
(COX1 or Tx-synthetase defects) | Aggregation absent/severely decreased with arachidonic acid, but normal or minimally affected with U46619. Variably reduced/reversible with weak/low dose agonists (epinephrine, ADP, TRAP, collagen), reflecting the lack of secondary potentiation by TxA2. |
| Defect of dense granules (δ) or their secretion. | Aggregation absent/severely decreased with low–moderate doses of multiple agonists, in particular collagen at low doses, due to a lack of feedback by secreted ADP. Conversely, normal or minimally affected aggregation with high doses of ADP (5–10 μM). If lumiaggregometry is performed, ATP secretion is absent/severely decreased even with strong agonists/high doses. The granule defect may be confirmed by secretion assays (serotonin, CD63, etc.) or electron microscopy (whole mount; see below). |
| Signaling defect via Gi (ADP [P2Y12] or epinephrine receptors) | Aggregation absent or severely decreased and/or reversible with high doses of epinephrine and ADP (10 μM). Variably reduced with moderate–low doses of other agonists such as collagen or TRAP; normal or minimally affected with arachidonic acid. |
| Signaling defect via Gq/G12/13 pathway (thrombin receptors, TxA2) | Aggregation absent or decreased and/or reversible with moderate–low doses of TRAP or U46616, normal or minimally affected with ADP, epinephrine, CRP, or PMA. |
| Defects of transcription factors such as RUNX1 or ETV6 | Variably reduced aggregation with moderate–low doses of multiple agonists. Less severe pattern than in GT or RASGRP2 defect. |
- (b)
- Platelet structural and functional studies by flow cytometry
- ○
- Follow the ISTH guidelines to the application of FC in the study and research of IPDs, which describes the consensus among experts regarding clinical settings in which it is useful, pre-analytical variables, the standardization of instruments and reagents, methods of performance, and the reporting of results and quality control [53].
- ○
- The FC study of any IPD should include the evaluation of the main GPs. Based on scientific evidence [10,11,53,54,55] and our experience, we recommend:
- ▪
- Performing the test in whole blood (EDTA or citrate) diluted 1/10 with a buffer (1/5 if thrombocytopenic with platelets < 25 × 109/L).
- ▪
- Identify the platelet population with a tracer antibody against a major GP (CD41*PE; anti-IIb). For example, in cases of GT (lacking GPIIb), an anti-CD42B*PE (anti-GPIb) could be used. Alternatively, the platelet population can be identified by its size and scatter pattern.
- ▪
- Fixating the sample with 2% PFA upon completion of labeling, and dilution of the sample before analysis.
- ▪
- If the analysis of the samples is not carried out in within 2 h, samples should be stored at 4 °C for up to 18–24 h, and re-mixed well (gentle vortex before analyzing them in the cytometer.
- ▪
- Table 5 shows a recommended GP analysis panel and reference intervals for each in healthy controls.
- ○
- The FC study of a patient with suspected IPFD, with or without associated thrombocytopenia, should also include activation testing and agonist-induced granule secretion if functional FC is available. Based on our experience and scientific evidence [10,11,53,54,55], we recommend:
- ▪
- Performing the assay on diluted PRP (≈10–20 × 109/L) with Tyrode buffer. The assay is feasible on diluted citrated whole blood (1/10, or 1/5 if thrombocytopenic with platelets < 25 × 109/L). In a basic test, the following tubes are prepared:
- ○
- Diluted PRP.
- ○
- Tracer antibody to identify platelets (CD41*APC; or a CD42B*APC or if the patient has GT]; alternatively, platelets could be identified by the size and scatter pattern.
- ○
- Active integrin αIIbβ3: fibrinogen (Fg)*FITC, PAC-1*FITC antibody, or an anti-Fg Ab *FITC (would attach to the fibrinogen that binds the integrin).
- ○
- P-selectin release ((α granules), or anti-CD63 *PE antibody (δ granules).
- ○
- Platelet agonist at the appropriate final concentration.
- ▪
- Incubate (30 min at room temperature), fix (PFA 2%), dilute (1/10) the sample with buffer, and run it through the cytometer. Capturing at least 5000–10,000 platelets (CD41*APC+ events) is recommended, assessing the % platelets+ and the median fluorescence in FL1 (Fg*488, PAC-1*FITC or anti-Fg*FITC) and FL2 (CD62*PE or CD63*PE). Establish the corresponding compensations or adjustments in the different fluorescence channels, depending on the technical characteristics of the cytometer used.
- ▪
- Similarly to the analysis of GP, if testing is not performed within 2 h, storing the samples at 4 °C for up to 18–24 h is recommended.
- ▪
- Table 6 shows a recommended panel of agonists for performing FC activation and secretion assays, and the reference intervals of each in healthy controls.
- ○
- (c)
- Electron microscopy—whole mount.
- ○
- Whole-mount grids should be prepared as soon as possible (ideally <24 h)
- ○
- The number of δ granules should be counted to be about 25–50 platelets, using a magnification scale of 4000 to 10,000X. Normal platelets have about 2–5 δ granules (although the reference varies between performing laboratories) compared to 0–2 in subjects with severe defects (HPS, SCH), or even >100 in giant platelets.
- ○
- Repeating the test at least twice is recommended in patients with moderate granule deficiency to verify the consistency of findings.
3.4. Phase 4: Expanded Platelet Phenotype Studies
- (a)
- Analysis of Peripheral Blood Smear by Immunofluorescence
- ○
- Given the complexity of performing and interpreting immunofluorescence results, which requires significant expertise and the need for a broad array of antibodies and sophisticated equipment, we recommend this test in the last phase of the IPD diagnostic process. However, specialized centers with the necessary equipment and experience in this methodology may consider performing it in earlier phases.
- ○
- Non-specialized centers can consider this test in phases 1–3 if their work is limited to preparing smears and sending them by conventional mail to a specialized center with experience in this methodology.
- ○
- Using antibodies already validated for this procedure, as well as a well-standardized methodology for performing the smears, is recommended [60].
- (b)
- Platelet ultrastructure by transmission electron microscopy (TEM)
- ○
- Anticoagulated whole blood can be used for TEM; we recommend using PRP where the platelet concentration is higher and there is less contamination with other cells.
- ○
- Expertise is required to assess platelet ultrastructure and document potential abnormalities. Central imaging services supporting research often have software tools useful for these tasks.
- (c)
- Platelet Aggregation Studies and/or Flow Cytometry
- •
- Extended platelet aggregation studies with less conventional agonists (PAR-4, PAF, A23187, DTT, rhodocetin, CD9, combinations of agonists with inhibitors, or others).
- •
- Extended FC studies to detect other membrane receptors (P2Y1/P2Y12, Clec-2, GPIV, CD36, or others); calcium mobilization; intraplatelet proteins (VASP, others); procoagulant activity (Annexin-V binding); ristocetin-induced VWF binding; etc.
- •
- The methodology to be applied is like that described above, but it is advisable to have experience in the tests to be performed, hence why they are preferably performed in specialized centers.
- (d)
- Clot retraction studies
- (e)
- Quantification of second messengers, cytokines or other bioactive substances, granule proteins, membrane receptors or intraplatelet proteins
- •
- Obtaining and storing serum to quantify TxB2 (stable metabolite of TxA2) synthesis by ELISA or other alternative method. This compound is primarily synthesized by platelets and is essential for feedback on platelet reactivity to multiple agonists. Abnormal TxA2 synthesis may suggest functional/genetic alterations in enzymes such as cyclooxygenase and/or thromboxane synthetase.
- •
- Obtaining and preserving plasma for potential quantification (ELISA, HPLC, others) of bioactive substances or antiplatelet antibodies.
- •
- Obtaining and preserving supernatants from platelets stimulated with agonists, with or without simultaneous recording of aggregation, for the quantification (ELISA, HPLC, others) of second messengers (cAMP, cGMP, or others, in samples from lysed platelets by freeze–thaw or platelets, or using a platelet lysis agent), or proteins secreted from α granules (VWF, b-GT, PAF, Factor V, P-selectin, etc.) or δ granules (serotonin, ADP, ATP, etc.).
- •
- Preparing washed platelets and from them platelet lysates for the quantification of specific platelet proteins (membrane, cytosolic, granular), by electrophoresis and Western blot (WB) with specific antibodies
- •
- (f)
- Other research studies:
3.5. Phase 5: Molecular Studies: Genetic Diagnosis
- ▪
- Pre-selected gene panels (variable between 10 and 300 genes).
- ▪
- Whole exome (WES, coding portion [exons] and flanking regions of all genes; 1–2% of the genome).
- ▪
- Whole genome (WGS, complete sequence of all genes).
- ○
- If the suspected diagnosis of IT is well supported by the clinical phenotype, available laboratory data, and/or family history, molecular testing should always be considered from the beginning of the diagnostic process. This applies to all types of IT, even those that can be unequivocally diagnosed with other methods without the need for molecular testing (such as BSS and GT, among others).
- ○
- It is essential to inform patients and their families of the objectives of molecular testing and its possible results and consequences, including the possibility of not reaching a definitive diagnosis due to technical limitations and the possibility of unexpected or difficult-to-interpret findings (uncertain significance). They should also be informed that in many cases, genetic diagnosis can aid in clinical management and allow for family screening and genetic counseling, but that it will not always provide a direct benefit in the treatment or progression of their disease.
- ○
- It is essential to obtain informed consent (IC) from the patient before performing the molecular studies, including specific options for patients to decide whether they wish to receive information about incidental findings, variants associated with risk of malignancy or other serious diseases, including carrier status for recessive disorders.
- ○
- When informing patients and obtaining the IC, applicable legislation (data protection, etc.) and the recommendations of scientific societies such as ISTH should be observed [77].
- ○
- The type of methodology to be used and the most appropriate time to perform the molecular study depends on the type of suspected IPD, the importance of the molecular study both in the diagnosis of the disease (whether or not it can be diagnosed with other laboratory studies), as well as in its prognosis and clinical management, the accessibility of the methodology, the ability to cover its cost, and the experience of the group or laboratory genetic diagnosis in general.
- ○
- Possible methodological approaches include:
- ▪
- Familial screening for specific genetic variants: If the pathogenic variant is known in a patient, their relatives can be screened using Sanger sequencing, targeted Nanopore sequencing, or potentially other targeted methods.
- ▪
- Single-gene Sanger sequencing: This may be a valid option for monogenic IPD with very specific clinical and phenotypic features. Examples include: BSS (GP1BA, GP1BB, GP9 genes); platelet-type von Willebrand disease (GP1B1); GT (ITGA2B and ITGB3); MYH9-RD if there is macrothrombocytopenia, Döhle bodies, and/or typical extra hematologic manifestations (MYH9). However, considering the current costs of HTS, and that the genes to be sequenced can be large and complex, our recommendation is to use HTS directly, allowing for the relatively rapid sequencing of multiple genes.
- ▪
- HTS sequencing with gene panels: This is the most common, practical, and least expensive approach currently available. When selecting genes for these panels, we recommend following the recommendations of expert groups such as the ISTH [79] (https://www.isth.org/page/GinTh_GeneLists, accessed on 29 May 2025). A negative result, assuming the disorder is inherited, may be due to methodological limitations or to the fact that the causal gene is not included in the panel. In practice, some laboratories sequence the whole exome, but the analysis is restricted, as a first option, to panels of these selected genes.
- ▪
- WES and WGS sequencing: This is appropriate for cases with a clear suspicion of IPD, where there is no candidate gene, and HTS gene panel testing is negative. WGS offers the additional advantage of exploring the non-coding DNA region, allowing for the detection of variants in distal and proximal regulatory elements. The chances of successful diagnosis with WES or WGS increase considerably if DNA is analyzed not only from the patient but also from other family members (affected and non-affected at minimum using a trio approach).
- ▪
- Single-gene sequencing, WES or WGS by long-sequence sequencing (4th generation sequencing): currently reserved for cases with unequivocal suspicion of a specific IPD, in which conventional HTS sequencing (of short sequences) has been negative, perhaps due to its inability to detect complex structural variants [71,78].
- ○
- The validation and interpretation of the molecular data obtained is a critical aspect for the success of genetic diagnosis and should always include the patient’s clinical and laboratory context. Therefore, either directly or through appropriate bioinformatics and/or genetics services, the following should be considered:
- ▪
- Applying well-established sequencing quality criteria (depth, read quality, sequence alignment with the reference genome, etc.)
- ▪
- Applying efficient variant filtering (eliminating common or low-frequency variants based on population databases such as gnomAD; adjusting frequency thresholds according to the dominant or recessive inheritance pattern; prioritizing according to the probable effect on proteins, etc.).
- ▪
- Consulting databases of specific disorders and molecular variants such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 29 May 2025), ClinGen (https://clinicalgenome.org/, accessed on 29 May 2025) or ISTH resources such as Gold Variant database [80], to establish associations of previously known variants with IPD phenotypes.
- ▪
- Applying internationally recognized criteria, such as those in the ACMG/AMP guidelines, to classify variants as pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign [81]. ClinGen includes expert panels that have adapted the ACMG/AMP rules for certain types of IT, such as GT [82] RUNX1-RT [83].
- ▪
- Using web platforms such as Varsome (https://varsome.com/, accessed on 29 May 2025) that provide extensive information on genetic variants and propose a preliminary classification of their pathogenicity according to ACMG/AMP criteria.
- ○
- Working as an interdisciplinary team in the final evaluation of the candidate variants to be reported, considering the experimental data, the possible co -segregation, and the strength of the genotype–phenotype relationship.
- ○
- Avoiding reporting variants in genes without sufficient information to support a gene–disease relationship.
- ○
- Reviewing previously identified VUS variants if new relevant evidence emerges.
- ○
- Incorporating advances in technology and genetic discoveries to improve diagnosis.
- ○
- Include patients with a confirmed genetic diagnosis of a IPD in a patient registry such as the Spanish Registry of Inherited Platelet Disorders (RETPLAC) (https://retplac.imib.es/, accessed on 29 May 2025).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palma-Barqueros, V.; Revilla, N.; Sánchez, A.; Cánovas, A.Z.; Rodriguez-Alén, A.; Marín-Quílez, A.; González-Porras, J.R.; Vicente, V.; Lozano, M.L.; Bastida, J.M.; et al. Inherited Platelet Disorders: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 4521. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T.; Nurden, P. Inherited thrombocytopenias: History, advances and perspectives. Haematologica 2020, 105, 2004–2019. [Google Scholar] [CrossRef] [PubMed]
- Nurden, P.; Stritt, S.; Favier, R.; Nurden, A.T. Inherited platelet diseases with normal platelet count: Phenotypes, genotypes and diagnostic strategy. Haematologica 2021, 106, 337–350. [Google Scholar] [CrossRef]
- Pecci, A.; Balduini, C.L. Inherited thrombocytopenias: An updated guide for clinicians. Blood Rev. 2021, 48, 100784. [Google Scholar] [CrossRef]
- Sánchez-Guiu, I.; I Antón, A.; Padilla, J.; Velasco, F.; Lucia, J.F.; Lozano, M.; Cid, A.R.; Sevivas, T.; Lopez-Fernandez, M.F.; Vicente, V.; et al. Functional and molecular characterization of inherited platelet disorders in the Iberian Peninsula: Results from a collaborative study. Orphanet J. Rare Dis. 2014, 9, 213. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef]
- Gresele, P.; Harrison, P.; Gachet, C.; Hayward, C.; Kenny, D.; Mezzano, D.; Mumford, A.; Nugent, D.; Nurden, A.; Cattaneo, M. Diagnosis of inherited platelet function disorders: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 314–322. [Google Scholar] [CrossRef]
- Zaninetti, C.; Wolff, M.; Greinacher, A. Diagnosing Inherited Platelet Disorders: Modalities and Consequences. Hamostaseologie 2021, 41, 475–488. [Google Scholar] [CrossRef]
- Bastida Bermejo, J.M.B.; Hernández-Rivas, J.M.; González-Porras, J.R. Novel approaches for diagnosing inherited platelet disorders. Med. Clin. 2017, 148, 71–77. [Google Scholar] [CrossRef]
- Bourguignon, A.; Tasneem, S.; Hayward, C.P. Screening and diagnosis of inherited platelet disorders. Crit. Rev. Clin. Lab. Sci. 2022, 59, 405–444. [Google Scholar] [CrossRef]
- Rivera, J.P.-B.V.; Vicente, V.; Lozano, M.L. Pruebas de laboratorio para el estudio de la función plaquetaria y diagnóstico de los trastornos plaquetarios congénitos. HEMATOLOGÍA Vol 22 • Nº Extra; XIII Congreso del Grupo CAHT. 2018. Available online: https://www.sah.org.ar/revistasah/numeros/vol22/sup/39_Pruebas_laboratorio_estudio_funcion_plaquetaria_diagnostico_trastornos_plaquetarios_cong.pdf (accessed on 29 May 2025).
- Casari, C.; Bergmeier, W. Acquired platelet disorders. Thromb. Res. 2016, 141 (Suppl. 2), S73–S75. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Falcinelli, E.; Bury, L.; Alessi, M.-C.; Guglielmini, G.; Falaise, C.; Podda, G.; Fiore, M.; Mazziotta, F.; Sevivas, T.; et al. Association of laboratory test results with the bleeding history in patients with inherited platelet function disorders (the Bleeding Assesment Tool-LABoratory tests substudy): Communication from the Platelet Physiology ISTH-SSC. Res. Pract. Thromb. Haemost. 2024, 8, 102305. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Falcinelli, E.; Bury, L.; Pecci, A.; Alessi, M.; Borhany, M.; Heller, P.G.; Santoro, C.; Cid, A.R.; Orsini, S.; et al. The ISTH bleeding assessment tool as predictor of bleeding events in inherited platelet disorders: Communication from the ISTH SSC Subcommittee on Platelet Physiology. J. Thromb. Haemost. 2021, 19, 1364–1371. [Google Scholar] [CrossRef]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the Platelet Physiology SSC. J. Thromb. Haemost. 2020, 18, 732–739. [Google Scholar] [CrossRef]
- Piel-Julian, M.-L.; Mahévas, M.; Germain, J.; Languille, L.; Comont, T.; Lapeyre-Mestre, M.; Payrastre, B.; Beyne-Rauzy, O.; Michel, M.; Godeau, B.; et al. Risk factors for bleeding, including platelet count threshold, in newly diagnosed immune thrombocytopenia adults. J. Thromb. Haemost. 2018, 16, 1830–1842. [Google Scholar] [CrossRef]
- Triulzi, D.J. How well do platelets prevent bleeding? Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 518–522. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lillicrap, D.; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef] [PubMed]
- Balduini, C.L. Treatment of inherited thrombocytopenias. Haematologica 2022, 107, 1278–1292. [Google Scholar] [CrossRef]
- Balduini, C.L.; Savoia, A.; Seri, M. Inherited thrombocytopenias frequently diagnosed in adults. J. Thromb. Haemost. 2013, 11, 1006–1019. [Google Scholar] [CrossRef]
- Simon, L.; Spinella, J.-F.; Yao, C.-Y.; Lavallée, V.-P.; Boivin, I.; Boucher, G.; Audemard, E.; Bordeleau, M.-E.; Lemieux, S.; Hébert, J.; et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood 2020, 135, 1882–1886. [Google Scholar] [CrossRef]
- Šrámek, A.; Eikenboom, J.C.J.; Briët, E.; Vandenbroucke, J.P.; Rosendaal, F.R. Usefulness of Patient Interview in Bleeding Disorders. Arch. Intern. Med. 1995, 155, 1409–1415. [Google Scholar] [CrossRef]
- Bury, L.; Falcinelli, E.; Gresele, P. Inherited Platelet Function Disorders: Algorithms for Phenotypic and Genetic Investigation. Semin. Thromb. Hemost. 2016, 42, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Klersy, C.; Gresele, P.; Lee, K.J.; De Rocco, D.; Bozzi, V.; Russo, G.; Heller, P.G.; Loffredo, G.; Ballmaier, M.; et al. MYH9-Related Disease: A Novel Prognostic Model to Predict the Clinical Evolution of the Disease Based on Genotype-Phenotype Correlations. Hum. Mutat. 2014, 35, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Savoia, A.; De Rocco, D.; Pecci, A. MYH9 gene mutations associated with bleeding. Platelets 2017, 28, 312–315. [Google Scholar] [CrossRef]
- Monard, A.L.; Mussert, C.M.; van Duijl, T.T.; Kruip, M.J.; Henskens, Y.M.; Biggelaar, M.v.D.; Schutgens, R.E.; Schols, S.E.; Fijnvandraat, K.J.; Meijer, K.; et al. Bleeding disorder of unknown cause: An illustrated review on current practice, knowledge gaps, and future perspectives. Res. Pract. Thromb. Haemost. 2024, 8, 102625. [Google Scholar] [CrossRef]
- Perez Botero, J.; Di Paola, J. Diagnostic approach to the patient with a suspected inherited platelet disorder: Who and how to test. J. Thromb. Haemost. 2021, 19, 2127–2136. [Google Scholar] [CrossRef]
- James, P.D.; Connell, N.T.; Ameer, B.; Di Paola, J.; Eikenboom, J.; Giraud, N.; Haberichter, S.; Jacobs-Pratt, V.; Konkle, B.; McLintock, C.; et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021, 5, 280–300. [Google Scholar] [CrossRef]
- James, P.; Leebeek, F.; Casari, C.; Lillicrap, D. Diagnosis and treatment of von Willebrand disease in 2024 and beyond. Haemophilia 2024, 30 (Suppl. 3), 103–111. [Google Scholar] [CrossRef]
- Quiroga, T.; Goycoolea, M.; Panes, O.; Aranda, E.; Martínez, C.; Belmont, S.; Muñoz, B.; Zúñiga, P.; Pereira, J.; Mezzano, D. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica 2007, 92, 357–365. [Google Scholar] [CrossRef]
- Pontara, E.; Gresele, P.; Cattini, M.G.; Daidone, V.; Barbon, G.; Girolami, A.; Zanon, E.; Casonato, A. Spontaneous hemarthrosis in combined Glanzmann thrombasthenia and type 2N von Willebrand disease. Blood Coagul. Fibrinolysis 2014, 25, 401–404. [Google Scholar] [CrossRef]
- Noris, P.; Klersy, C.; Gresele, P.; Giona, F.; Giordano, P.; Minuz, P.; Loffredo, G.; Pecci, A.; Melazzini, F.; Civaschi, E.; et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: A multicentric, real life study. Br. J. Haematol. 2013, 162, 112–119. [Google Scholar] [CrossRef]
- Cheves, T.A.; DeMarinis, S.; Sweeney, J.D. Laboratory Methods in the Assessment of Hereditary Hemostatic Disorders. Hematol. Oncol. Clin. N. Am. 2021, 35, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Benlachgar, N.; Doghmi, K.; Masrar, A.; Mahtat, E.M.; Harmouche, H.; Mezalek, Z.T. Immature platelets: A review of the available evidence. Thromb. Res. 2020, 195, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Koike, Y.; Kunishima, S.; Ishii, R.; Danbara, M.; Horie, R.; Yatomi, Y.; Higashihara, M. Immature platelet fraction measurement is influenced by platelet size and is a useful parameter for discrimination of macrothrombocytopenia. Hematology 2015, 20, 587–592. [Google Scholar] [CrossRef]
- Ferreira, F.L.B.; Colella, M.P.; Medina, S.S.; Costa-Lima, C.; Fiusa, M.M.L.; Costa, L.N.G.; Orsi, F.A.; Annichino-Bizzacchi, J.M.; Fertrin, K.Y.; Gilberti, M.F.P.; et al. Evaluation of the immature platelet fraction contribute to the differential diagnosis of hereditary, immune and other acquired thrombocytopenias. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Lardinois, B.; Favresse, J.; Chatelain, B.; Lippi, G.; Mullier, F. Pseudothrombocytopenia-A Review on Causes, Occurrence and Clinical Implications. J. Clin. Med. 2021, 10, 594. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Noris, P.; Biino, G.; Pecci, A.; Civaschi, E.; Savoia, A.; Seri, M.; Melazzini, F.; Loffredo, G.; Russo, G.; Bozzi, V.; et al. Platelet diameters in inherited thrombocytopenias: Analysis of 376 patients with all known disorders. Blood 2014, 124, e4–e10. [Google Scholar] [CrossRef]
- Zaninetti, C.; Greinacher, A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. J. Clin. Med. 2020, 9, 539. [Google Scholar] [CrossRef]
- Mingot-Castellano, M.E.; Hirnyk, M.C.; Sánchez-González, B.; Álvarez-Román, M.T.; Bárez-García, A.; Bernardo-Gutiérrez, Á.; Bernat-Pablo, S.; Bolaños-Calderón, E.; Butta-Coll, N.; Caballero-Navarro, G.; et al. Recommendations for the Clinical Approach to Immune Thrombocytopenia: Spanish ITP Working Group (GEPTI). J. Clin. Med. 2023, 12, 6422. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Pasalic, L.; Lippi, G. Towards 50 years of platelet function analyser (PFA) testing. Clin. Chem. Lab. Med. 2023, 61, 851–860. [Google Scholar] [CrossRef]
- Mehic, D.; Assinger, A.; Gebhart, J. Utility of Global Hemostatic Assays in Patients with Bleeding Disorders of Unknown Cause. Hamostaseologie 2024, 44, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Karczmarska-Wódzka, A.; Bugieda, J.; Sobczak, P. The Use of Total Thrombus Formation Analysis System as a Tool to Assess Platelet Function in Bleeding and Thrombosis Risk—A Systematic Review. Int. J. Mol. Sci. 2021, 22, 8605. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Hvas, A.-M.; Hojbjerg, J.A. Platelet Function Testing: Update and Future Directions. Semin. Thromb. Hemost. 2023, 49, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Hayward, C.P.M.; Harrison, P.; Cattaneo, M.; Ortel, T.L.; Rao, A.K. Platelet function analyzer (PFA)-100R closure time in the evaluation of platelet disorders and platelet function. J. Thromb. Haemost. 2006, 4, 312–319. [Google Scholar] [CrossRef]
- Lecchi, A.; La Marca, S.; Padovan, L.; Boscarino, M.; Peyvandi, F.; Tripodi, A. Flow-chamber device (T-TAS) to diagnose patients suspected of platelet function defects. Blood Transfus. 2024, 22, 55–64. [Google Scholar] [CrossRef]
- Stépanian, A.; Fischer, F.; Flaujac, C.; Eschwège, V.; Delassasseigne, C.; Leflem, L.; Loridon, F.; Voisin, S.; Lasne, D. Light transmission aggregometry for platelet function testing: Position paper on current recommendations and French proposals for accreditation. Platelets 2024, 35, 2427745. [Google Scholar] [CrossRef]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.M.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the standardization of light transmission aggregometry: A consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J. Thromb. Haemost. 2013, 11, 1183–1189. [Google Scholar] [CrossRef]
- Alessi, M.-C.; Coxon, C.; Ibrahim-Kosta, M.; Bacci, M.; Voisin, S.; Rivera, J.; Greinacher, A.; Raster, J.; Pulcinelli, F.; Devreese, K.M.; et al. Multicenter evaluation of light transmission platelet aggregation reagents: Communication from the ISTH SSC Subcommittee on Platelet Physiology. J. Thromb. Haemost. 2023, 21, 2596–2610. [Google Scholar] [CrossRef]
- Alessi, M.-C.; Sié, P.; Payrastre, B. Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders. J. Clin. Med. 2020, 9, 763. [Google Scholar] [CrossRef]
- Dawood, B.B.; Lowe, G.C.; Lordkipanidzé, M.; Bem, D.; Daly, M.E.; Makris, M.; Mumford, A.; Wilde, J.T.; Watson, S.P. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonist panel. Blood 2012, 120, 5041–5049. [Google Scholar] [CrossRef]
- Palma-Barqueros, V.; Revilla, N.; Zaninetti, C.; Galera, A.M.; Sánchez-Fuentes, A.; Zámora-Cánovas, A.; Bohdan, N.; Padilla, J.; Marín-Quilez, A.; Rodriguez-Alen, A.; et al. Src-related thrombocytopenia: A fine line between a megakaryocyte dysfunction and an immune-mediated disease. Blood Adv. 2022, 6, 5244–5255. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frelinger, A.L.; Rivera, J.; Connor, D.E.; Freson, K.; Greinacher, A.; Harrison, P.; Kunishima, S.; Lordkipanidzé, M.; Michelson, A.D.; Ramström, S.; et al. Consensus recommendations on flow cytometry for the assessment of inherited and acquired disorders of platelet number and function: Communication from the ISTH SSC Subcommittee on Platelet Physiology. J. Thromb. Haemost. 2021, 19, 3193–3202. [Google Scholar] [CrossRef]
- Rand, M.L.; Reddy, E.C.; Israels, S.J. Laboratory diagnosis of inherited platelet function disorders. Transfus. Apher. Sci. 2018, 57, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, T.; Jain, K.; Gu, S.X.; Qiu, M.; Gu, V.W.; Melchinger, H.; Rinder, H.; Martin, K.A.; Gardiner, E.E.; Lee, A.I.; et al. A guide to molecular and functional investigations of platelets to bridge basic and clinical sciences. Nat. Cardiovasc. Res. 2022, 1, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Frelinger, A.L. Flow Cytometry and Platelets. Clin. Lab. Med. 2024, 44, 511–526. [Google Scholar] [CrossRef]
- Ramstrom, A.S.; Fagerberg, I.H.; Lindahl, T.L. A flow cytometric assay for the study of dense granule storage and release in human platelets. Platelets 1999, 10, 153–158. [Google Scholar] [CrossRef]
- Dupuis, A.; Gachet, C. Inherited platelet disorders: Management of the bleeding risk. Transfus. Clin. Biol. 2018, 25, 228–235. [Google Scholar] [CrossRef]
- Chen, D.; Uhl, C.B.; Bryant, S.C.; Krumwiede, M.; Barness, R.L.; Olson, M.C.; Gossman, S.C.; Damgard, S.E.; Gamb, S.I.; Cummins, L.A.; et al. Diagnostic laboratory standardization and validation of platelet transmission electron microscopy. Platelets 2018, 29, 574–582. [Google Scholar] [CrossRef]
- Zaninetti, C.; Leinøe, E.; Lozano, M.L.; Rossing, M.; Bastida, J.M.; Zetterberg, E.; Rivera, J.; Greinacher, A. Validation of immunofluorescence analysis of blood smears in patients with inherited platelet disorders. J. Thromb. Haemost. 2023, 21, 1010–1019. [Google Scholar] [CrossRef]
- Scandola, C.; Erhardt, M.; Rinckel, J.-Y.; Proamer, F.; Gachet, C.; Eckly, A. Use of electron microscopy to study megakaryocytes. Platelets 2020, 31, 589–598. [Google Scholar] [CrossRef]
- Jansen, E.E.; Hartmann, M. Clot Retraction: Cellular Mechanisms and Inhibitors, Measuring Methods, and Clinical Implications. Biomedicines 2021, 9, 1064. [Google Scholar] [CrossRef]
- Lozano, M.L.; Cook, A.; Bastida, J.M.; Paul, D.S.; Iruin, G.; Cid, A.R.; Adan-Pedroso, R.; González-Porras, J.R.; Hernández-Rivas, J.M.; Fletcher, S.J.; et al. Novel mutations in RASGRP2, which encodes CalDAG-GEFI, abrogate Rap1 activation, causing platelet dysfunction. Blood 2016, 128, 1282–1289. [Google Scholar] [CrossRef]
- Feghhi, S.; Sniadecki, N.J. Mechanobiology of Platelets: Techniques to Study the Role of Fluid Flow and Platelet Retraction Forces at the Micro- and Nano-Scale. Int. J. Mol. Sci. 2011, 12, 9009–9030. [Google Scholar] [CrossRef] [PubMed]
- Gibbins, J.M.; Mahaut-Smith, M.P. (Eds.) Platelets and Megakaryocytes; Humana Press: Totowa, NJ, USA, 2012. [Google Scholar]
- Gibbins, J.M.; Mahaut-Smith, M.P. (Eds.) Platelets and Megakaryocytes: Volume 2: Perspectives and Techniques; Humana Press: Totowa, NJ, USA, 2004; ISBN 978-1-58829-011-3. [Google Scholar]
- Bastida, J.M.; Benito, R.; Lozano, M.L.; Marín-Quilez, A.; Janusz, K.; Martín-Izquierdo, M.; Hernández-Sánchez, J.; Palma-Barqueros, V.; Hernández-Rivas, J.M.; Rivera, J.; et al. Molecular Diagnosis of Inherited Coagulation and Bleeding Disorders. Semin. Thromb. Hemost. 2019, 45, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Mumford, A.D.; Westbury, S.K. Genetic Techniques Used in the Diagnosis of Inherited Platelet Disorders. Semin. Thromb. Hemost. 2019, 45, 685–694. [Google Scholar] [CrossRef]
- Donck, F.V.; Downes, K.; Freson, K. Strengths and limitations of high-throughput sequencing for the diagnosis of inherited bleeding and platelet disorders. J. Thromb. Haemost. 2020, 18, 1839–1845. [Google Scholar] [CrossRef]
- Donck, F.V.; Labarque, V.; Freson, K. Hemostatic phenotypes and genetic disorders. Res. Pract. Thromb. Haemost. 2021, 5, e12637. [Google Scholar] [CrossRef]
- Cuenca-Guardiola, J.; de la Morena-Barrio, B.; García, J.L.; Sanchis-Juan, A.; Corral, J.; Fernández-Breis, J.T. Improvement of large copy number variant detection by whole genome nanopore sequencing. J. Adv. Res. 2022, 50, 145–158. [Google Scholar] [CrossRef]
- Downes, K.; Megy, K.; Duarte, D.; Vries, M.; Gebhart, J.; Hofer, S.; Shamardina, O.; Deevi, S.V.V.; Stephens, J.; Mapeta, R.; et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood 2019, 134, 2082–2091. [Google Scholar] [CrossRef]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2017, 103, 148–162. [Google Scholar] [CrossRef]
- Leinøe, E.; Zetterberg, E.; Kinalis, S.; Østrup, O.; Kampmann, P.; Norström, E.; Andersson, N.; Klintman, J.; Qvortrup, K.; Nielsen, F.C.; et al. Application of whole-exome sequencing to direct the specific functional testing and diagnosis of rare inherited bleeding disorders in patients from the Öresund Region, Scandinavia. Br. J. Haematol. 2017, 179, 308–322. [Google Scholar] [CrossRef]
- Heremans, J.; Freson, K. High-throughput sequencing for diagnosing platelet disorders: Lessons learned from exploring the causes of bleeding disorders. Int. J. Lab. Hematol. 2018, 40 (Suppl. 1), 89–96. [Google Scholar] [CrossRef]
- Johnson, B.; Lowe, G.C.; Futterer, J.; Lordkipanidze, M.; MacDonald, D.; Simpson, M.A.; Sanchez-Guiu, I.; Drake, S.; Bem, D.; Leo, V.; et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica 2016, 101, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Downes, K.; Borry, P.; Ericson, K.; Gomez, K.; Greinacher, A.; Lambert, M.; Leinoe, E.; Noris, P.; Van Geet, C.; Freson, K. Subcommittee on Genomics in Thrombosis, Hemostasis. Clinical management, ethics and informed consent related to multi-gene panel-based high throughput sequencing testing for platelet disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2020, 18, 2751–2758. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zamora-Cánovas, A.; de la Morena-Barrio, B.; Marín-Quilez, A.; Sierra-Aisa, C.; Male, C.; Fernández-Mosteirin, N.; Trapero-Marugán, M.; Padilla, J.; Garrido-Rodriguez, P.; Sánchez-Fuentes, A.; et al. Targeted long-read sequencing identifies and characterizes structural variants in cases of inherited platelet disorders. J. Thromb. Haemost. 2023, 22, 851–859. [Google Scholar] [CrossRef]
- Megy, K.; Downes, K.; Simeoni, I.; Bury, L.; Morales, J.; Mapeta, R.; Bellissimo, D.B.; Bray, P.F.; Goodeve, A.C.; Gresele, P.; et al. Curated disease-causing genes for bleeding, thrombotic, and platelet disorders: Communication from the SSC of the ISTH. J Thromb. Haemost. 2019, 17, 1253–1260. [Google Scholar] [CrossRef]
- Megy, K.; Downes, K.; Morel-Kopp, M.; Bastida, J.M.; Brooks, S.; Bury, L.; Leinoe, E.; Gomez, K.; Morgan, N.V.; Othman, M.; et al. GoldVariants, a resource for sharing rare genetic variants detected in bleeding, thrombotic, and platelet disorders: Communication from the ISTH SSC Subcommittee on Genomics in Thrombosis and Hemostasis. J. Thromb. Haemost. 2021, 19, 2612–2617. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ross, J.E.; Zhang, B.M.; Lee, K.; Mohan, S.; Branchford, B.R.; Bray, P.; Dugan, S.N.; Freson, K.; Heller, P.G.; Kahr, W.H.A.; et al. Specifications of the variant curation guidelines for ITGA2B/ITGB3: ClinGen Platelet Disorder Variant Curation Panel. Blood Adv. 2021, 5, 414–431. [Google Scholar] [CrossRef]
- Luo, X.; Feurstein, S.; Mohan, S.; Porter, C.C.; Jackson, S.A.; Keel, S.; Chicka, M.; Brown, A.L.; Kesserwan, C.; Agarwal, A.; et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019, 3, 2962–2979. [Google Scholar] [CrossRef]




| System | Clinical Manifestation | IPD (Genes) | Type of IPD | Platelet Size |
|---|---|---|---|---|
| Cutaneous | Eczema | Wiskott–Aldrich Syndrome (WAS) | IT | Small |
| Xanthomas in tendons and xanthelasmas | Sitosterolemia (ABCG5, ABCG8) | IT | Large | |
| Oculo-cutaneous albinism | Hermansky–Pudlak Syndrome (HPS-1 to 11; 11 subtypes) Chediak–Higashi Syndrome (LYST) | IPFD | Normal | |
| Ichthyosis | Progressive variable erythroderma (KDSR) Stormorken Syndrome (STIM1) | IT IPFD | Large Normal | |
| Sensory Organs | Hearing loss | DIAPH1-RT (DIAPH1) MYH9-RD (MYH9) MECOM-RD (MECOM) | IT | Large Large Normal |
| Waterfalls | MYH9-RD (MYH9) | IT | Large | |
| Immunological | Immunodeficiency | Leukocyte adhesion deficiency (LAD) type III (FERMT3) Chediak–Higashi Syndrome (LYST) Wiskott–Aldrich Syndrome (WAS) | IPFD IPFD IT | Normal Normal Small |
| Hypogammaglobulinemia | Roifman Syndrome (RNU4ATAC) | IT | Normal | |
| Hematological | Hemolytic anemia | Sitosterolemia (ABCG5, ABCG8) | IT | Large |
| Erythrocytosis | ANKRD26-RT (ANKRD26) | IT | Normal | |
| Hematopoietic neoplasia | ANKRD26-RT (ANKRD26) ETV6-RT (ETV6) RUNX1-RT (RUNX1) ERG-RT (ERG) | IT | Normal | |
| Hemoglobinopathy | GATA1-RT (GATA1) | IT | Normal | |
| Bone marrow failure | MECOM-RD (MECOM) Congenital amegakaryocytic thrombocytopenia (CAMT) (MPL) THPO-RT (THPO) | IT | Normal | |
| Neutropenia | Hermansky–Pudlak Syndrome types 2 and 10 (HPS2, HPS10) Wiskott–Aldrich Syndrome (WAS) DIAPH1-RT (DIAPH1) | IPFD IT IT | Normal Small Large | |
| Myelofibrosis | G6b-B-RT (MPIG6B) Gray platelet syndrome (GPS) (NBEAL2) SRC-RT (SRC) | IT | Large | |
| Splenomegaly | Gray platelet syndrome (GPS) (NBEAL2) | IT | Large | |
| Musculoskeletal | Radius and/or ulna malformation | Radioulnar–ulnar synostosis with thrombocytopenia (RUSAT1) (HOXA11) MECOM-RD (MECOM) Thrombocytopenia with absent radii (TAR) (RBM8A) | IT | Normal |
| Myopathy | GNE-RT (GNE) Stormorken Syndrome (STIM1) | IT IPFD | Large Normal | |
| Facial dysmorphia | Baraitser–Winter Syndrome (BWCA) Takenouchi–Kosaki Syndrome (CDC42) Roifman Syndrome (RNU4ATAC) | IT IT IT | Large Large Normal | |
| Delayed growth | Roifman Syndrome (RNU4ATAC) Stormorken Syndrome (STIM1) | IT IPFD | Normal Normal | |
| Arthrogryposis | Arthrogryposis syndrome, renal dysfunction and cholestasis (ARC) 1 and 2 (VIPAS39, VPS33B) | IPFD | Normal | |
| Cardiovascular | Atherosclerosis | Sitosterolemia (ABCG5, ABCG8) | IT | Large |
| Structural congenital heart disease | MECOM-RT (MECOM) DiGeorge syndrome (22q11.2 deletion) | IT IT | Normal Large | |
| Digestive | Cholestasis | Arthrogryposis syndrome, renal dysfunction and cholestasis (ARC) 1 and 2 (VIPAS39, VPS33B) | IPFD | Normal |
| Granulomatous colitis | Hermansky–Pudlak syndrome types 1 and 4 (HPS1, HPS4) | IPFD | Normal | |
| Duodenal ulcers | Phospholipase A2c-RD (PLA2G4A) | IPFD | Normal | |
| Respiratory | Pulmonary fibrosis | Hermansky–Pudlak Syndrome type 1, 2, 4 (HPS1, HPS2, HPS4) | IPFD | Normal |
| Neurological | Intellectual disability | Baraitser–Winter syndrome (BWCA), Takenouchi–Kosaki syndrome (CDC42) Roifman Syndrome (RNU4ATAC) | IT IT IT | Large Large Normal |
| Periventricular heterotopia | FLNA-RT (FLNA) | IT | Large | |
| Lymphatic | Lymphedema | Takenouchi–Kosaki Syndrome (CDC42) | IT | Large |
| Genitourinary | Congenital renal-urinary malformation | MECOM-RD Arthrogryposis syndrome, renal dysfunction and cholestasis (ARC) 1 and 2 (VIPAS39, VPS33B) | IT IPFD | Normal Normal |
| Nephropathy | MYH9-RD | IT | Large |
| GPs | Antibody | Median Fluorescence of Healthy Controls in the Accuri C6 Cytometer (Mean ± SD) | n |
|---|---|---|---|
| Ib | CD42b*FITC | 15,647 ± 3935 | 109 |
| IX | CD42a*FITC | 17,578 ± 6764 | 113 |
| IIb | CD41a*PE | 58,245 ± 15,374 | 113 |
| IIIa | CD61*FITC | 39,339 ± 7859 | 101 |
| Ia | CD49b*FITC | 1545 ± 845 | 113 |
| VI | GPVI-APC | 3877 ± 919 | 111 |
| - | Nonspecific Ig-FITC | 290 ± 172 | 88 |
| CD34 | CD34*APC | 231 ± 58 | 40 |
| - | FSC (size) | 127,773 ± 29,148 | 121 |
| - | SSC (granularity) | 11,906 ± 2694 | 120 |
| Median Fluorescence in Healthy Controls (n = 74–116). Accuri C6 Cytometer | |||
|---|---|---|---|
| Agonist | Fg*FITC αIIbβ3 active | CD62*PE (α granules) | CD63*PE (δ granules) |
| Tyrode Buffer | 467 ± 147 | 273 ± 71 | 510 ± 123 |
| ADP—10 μM | 1494 ± 728 | 1395 ± 697 | - |
| TRAP—25 μM | 2891 ± 1779 | 4719 ± 2033 | 3268 ± 1433 |
| PAR-4—1250 μM | 5208 ± 3975 | 6321 ± 3237 | - |
| CRP—10 μg/mL | 9154 ± 6984 | 6726 ± 106 | 4059 ± 1341 |
| U46619—5 μM + ADP—2,5 μM | 8819 ± 6154 | 7263 ± 2775 | - |
| PMA-100 nM | 4823 ± 4179 | 4222 ± 1996 | - |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Fuentes, A.; Pérez-Botero, J.; Bastida, J.M.; Rivera, J. Diagnosis of Inherited Platelet Disorders: Clinical Evaluation and Functional and Molecular Assays. Biomolecules 2025, 15, 846. https://doi.org/10.3390/biom15060846
Sánchez-Fuentes A, Pérez-Botero J, Bastida JM, Rivera J. Diagnosis of Inherited Platelet Disorders: Clinical Evaluation and Functional and Molecular Assays. Biomolecules. 2025; 15(6):846. https://doi.org/10.3390/biom15060846
Chicago/Turabian StyleSánchez-Fuentes, Ana, Juliana Pérez-Botero, José M. Bastida, and José Rivera. 2025. "Diagnosis of Inherited Platelet Disorders: Clinical Evaluation and Functional and Molecular Assays" Biomolecules 15, no. 6: 846. https://doi.org/10.3390/biom15060846
APA StyleSánchez-Fuentes, A., Pérez-Botero, J., Bastida, J. M., & Rivera, J. (2025). Diagnosis of Inherited Platelet Disorders: Clinical Evaluation and Functional and Molecular Assays. Biomolecules, 15(6), 846. https://doi.org/10.3390/biom15060846