
Nav1.8 and Chronic Pain: From Laboratory Animals to Clinical Patients


Abstract

1. Introduction
2. Characteristics of the Nav1.8 Channel
3. Clinical Findings of Nav1.8 in Chronic Pain
3.1. Gain-of-Function of Nav1.8 Channel
3.2. Loss-of-Function of Nav1.8 Channel
3.3. Single-Nucleotide Polymorphism of Nav1.8 Channel
3.4. Expression Changes in the Nav1.8 Channel in Patients with Chronic Pain
3.5. Clinical Trials in Treating Chronic Pain with Nav1.8 Blockers
3.6. Study of Chronic Pain with iPSCs from Patients
4. Preclinical Study of Nav1.8 in Chronic Pain
4.1. Electrophysiological Studies of Nav1.8 in Animals with Chronic Pain
4.2. Transcriptional and Translational Changes in Nav1.8 in Animals with Chronic Pain
4.3. Behavioral Studies of Nav1.8 in Animals with Chronic Pain
4.4. Studies on Mutations in Nav1.8 in Animal Models
5. Prospective Study of Nav1.8 in Chronic Pain
5.1. Differences Between Human and Animal Studies
5.2. Degeneracy
5.3. Machine Learning in Developing Nav1.8-Targeting Drugs in Chronic Pain
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Berta, T.; Qadri, Y.; Tan, P.H.; Ji, R.R. Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain. Expert Opin. Ther. Targets 2017, 21, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 199, 1079–1151. [Google Scholar] [CrossRef] [PubMed]
- Dormer, A.; Narayanan, M.; Schentag, J.; Achinko, D.; Norman, E.; Kerrigan, J.; Jay, G.; Heydorn, W. A Review of the Therapeutic Targeting of SCN9A and Nav1.7 for Pain Relief in Current Human Clinical Trials. J. Pain Res. 2023, 16, 1487–1498. [Google Scholar] [CrossRef]
- Yang, Y.; Mis, M.A.; Estacion, M.; Dib-Hajj, S.D.; Waxman, S.G. NaV1.7 as a Pharmacogenomic Target for Pain: Moving Toward Precision Medicine. Trends Pharmacol. Sci. 2018, 39, 258–275. [Google Scholar] [CrossRef]
- Yang, J.; Xie, Y.F.; Smith, R.; Ratte, S.; Prescott, S.A. Discordance between preclinical and clinical testing of NaV1.7-selective inhibitors for pain. Pain 2025, 166, 481–501. [Google Scholar] [CrossRef]
- Goodwin, G.; McMahon, S.B. The physiological function of different voltage-gated sodium channels in pain. Nat. Rev. Neurosci. 2021, 22, 263–274. [Google Scholar] [CrossRef]
- Okuse, K.; Chaplan, S.; McMahon, S.; Luo, Z.; Calcutt, N.; Scott, B.; Akopian, A.N.; Wood, J.N. Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol. Cell Neurosci. 1997, 10, 196–207. [Google Scholar] [CrossRef]
- Faber, C.G.; Lauria, G.; Merkies, I.S.; Cheng, X.; Han, C.; Ahn, H.S.; Persson, A.K.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef]
- Han, C.; Vasylyev, D.; Macala, L.J.; Gerrits, M.M.; Hoeijmakers, J.G.; Bekelaar, K.J.; Dib-Hajj, S.D.; Faber, C.G.; Merkies, I.S.; Waxman, S.G. The G1662S NaV1.8 mutation in small fibre neuropathy: Impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 2014, 85, 499–505. [Google Scholar] [CrossRef]
- Kaluza, L.; Meents, J.E.; Hampl, M.; Rosseler, C.; Hautvast, P.A.I.; Detro-Dassen, S.; Hausmann, R.; Schmalzing, G.; Lampert, A. Loss-of-function of Nav1.8/D1639N linked to human pain can be rescued by lidocaine. Pflugers Arch. 2018, 470, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- Kist, A.M.; Sagafos, D.; Rush, A.M.; Neacsu, C.; Eberhardt, E.; Schmidt, R.; Lunden, L.S.; ØRSTAVIK, k.; Kaluza, L.; Meents, J.; et al. SCN10A Mutation in a Patient with Erythromelalgia Enhances C-Fiber Activity Dependent Slowing. PLoS ONE 2016, 11, e0161789. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Narayanan, R. Ion-channel degeneracy and heterogeneities in the emergence of complex spike bursts in CA3 pyramidal neurons. J. Physiol. 2023, 601, 3297–3328. [Google Scholar] [CrossRef] [PubMed]
- Drion, G.; O’Leary, T.; Marder, E. Ion channel degeneracy enables robust and tunable neuronal firing rates. Proc. Natl. Acad. Sci. USA 2015, 112, E5361–E5370. [Google Scholar] [CrossRef]
- Yeon, J.; Takeishi a Sengupta, P. Chronic vs acute manipulations reveal degeneracy in a thermosensory neuron network. MicroPubl. Biol. 2021, 2021, 10.17912. [Google Scholar]
- Xie, Y.-F.; Yang, J.; Ratté, S.; Prescott, S.A. Similar excitability through different sodium channels and implications for the analgesic efficacy of selective drugs. eLife 2024, 12, RP90960. [Google Scholar] [CrossRef]
- Eagles, D.A.; Chow, C.Y.; King, G.F. Fifteen years of Na(V) 1.7 channels as an analgesic target: Why has excellent in vitro pharmacology not translated into in vivo analgesic efficacy? Br. J. Pharmacol. 2022, 179, 3592–3611. [Google Scholar] [CrossRef]
- Meadows, L.; Malhotra, J.D.; Stetzer, A.; Isom, L.L.; Ragsdale, D.S. The intracellular segment of the sodium channel beta 1 subunit is required for its efficient association with the channel alpha subunit. J. Neurochem. 2001, 76, 1871–1878. [Google Scholar] [CrossRef]
- Gilchrist, J.; Das, S.; Petgem, F.; Bosmans, F. Crystallographic insights into sodium-channel. Proc. Natl. Acad. Sci. USA 2013, 110, E5016–E5024. [Google Scholar] [CrossRef]
- Huang, X.; Jin, X.; Huang, G.; Huang, J.; Wu, T.; Li, Z.; Chen, J.; Kong, F.; Pan, X.; Yan, N. Structural basis for high-voltage activation and subtype-specific inhibition of human Na(v)1.8. Proc. Natl. Acad. Sci. USA 2022, 119, e2208211119. [Google Scholar] [CrossRef]
- Neumann, B.; McCarthy, S.; Gonen, S. Structural basis of inhibition of human Na(V)1.8 by the tarantula venom peptide Protoxin-I. Nat. Commun. 2025, 16, 1459. [Google Scholar] [CrossRef] [PubMed]
- Akopian, A.N.; Sivilotti, L.; Wood, J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996, 379, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sangameswaran, L.; Delgado, S.G.; Fish, L.M.; Koch, B.D.; Jakeman, L.B.; Stewart, G.R.; Sze, P.; Hunter, J.C.; Eglen, R.M.; Herman, R.C. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem. 1996, 271, 5953–5956. [Google Scholar] [CrossRef]
- Rabert, D.; Koch, B.; IInicka m Obernolte, R.; Naylor, S.; Herman, R.; Eglen, R.M.; Hunter, J.C.; Sangameswaran, L. A tetrodotoxin-resistant voltage-gated sodium channel from human dorsal root ganglia, hPN3 SCN10A. Pain 1998, 78, 107–114. [Google Scholar] [CrossRef]
- Rush, A.M.; Dib-Hajj, S.D.; Liu, S.; Cummins, T.R.; Black, J.A.; Waxman, S.G. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. USA 2006, 103, 8245–8250. [Google Scholar] [CrossRef]
- Yamada, A.; Yamada, A.I.; Ling, J.; Furue, H.; Luo, W.; Gu, J.G. Properties of Nav1.8 (ChR2)-positive and Nav1.8(ChR2)-negative afferent mechanoreceptors in the hindpaw glabrous skin of mice. Mol. Brain 2023, 16, 27. [Google Scholar] [CrossRef]
- Benn, S.; Costigan, M.; Tate, S.; Fitzgerald, M.; Woolf, C. Developmental expression of the TTX-resistant voltage-gated sodium channels Nav1.8 (SNS) and Nav1.9 (SNS2) in primary sensory neurons. J. Neurosci. 2001, 21, 6077–6085. [Google Scholar] [CrossRef]
- Amaya, F.; Decosterd, I.; Samad, T.A.; Plumpton, C.; Tate, S.; Mannion, R.J.; Costigan, M.; Woolf, C.J. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol. Cell Neurosci. 2000, 15, 331–342. [Google Scholar] [CrossRef]
- Dib-Hajj, S.; Black, B.; Cummins t kENNEYa kOCSISj, S.G.W. Rescue of alpha-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J. Neurophysiol. 1998, 79, 2668–2676. [Google Scholar] [CrossRef]
- Browne, L.E.; Clare, J.J.; Wray, D. Functional and pharmacological properties of human and rat NaV1.8 channels. Neuropharmacology 2009, 56, 905–914. [Google Scholar] [CrossRef]
- Tenza-Ferrer, H.; Collodetti, M.; Nicolau, E.S.; Birbrair, A.; Magno, L.A.V.; Romano-Silva, M.A. Transiently Nav1.8-expressing neurons are capable of sensing noxious stimuli in the brain. Front. Cell Neurosci. 2022, 16, 933874. [Google Scholar] [CrossRef] [PubMed]
- Szulczyk, B.; Pasierski, M.; Gawlak, M. Prefrontal cortex pyramidal neurons express functional Nav1.8 tetrodotoxin-resistant sodium currents. Clin. Exp. Pharmacol. Physiol. 2022, 49, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Rannals, M.D.; Hamersky, G.R.; Page, S.C.; Campbell, M.N.; Briley, A.; Gallo, R.A.; Phan, B.N.; Hyde, T.M.; Kleinman, J.E.; Shin, J.H.; et al. Psychiatric Risk Gene Transcription Factor 4 Regulates Intrinsic Excitability of Prefrontal Neurons via Repression of SCN10a and KCNQ1. Neuron 2016, 90, 43–55. [Google Scholar] [CrossRef]
- Namadurai, S.; Yereddi, N.R.; Cusdin, F.S.; Huang, C.L.; Chirgadze, D.Y.; Jackson, A.P. A new look at sodium channel beta subunits. Open Biol. 2015, 5, 140192. [Google Scholar] [CrossRef]
- Nevin, S.T.; Lawrence, N.; Nicke, A.; Lewis, R.J.; Adams, D.J. Functional modulation of the human voltage-gated sodium channel Na(V)1.8 by auxiliary beta subunits. Channels 2021, 15, 79–93. [Google Scholar] [CrossRef]
- Huang, J.; Yang, Y.; Zhao, P.; Gerrits, M.M.; Hoeijmakers, J.G.; Bekelaar, K.; Merkies, I.S.J.; Faber, C.G.; Dib-Hajj, S.D.; Waxman, S.G. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J. Neurosci. 2013, 33, 14087–14097. [Google Scholar] [CrossRef]
- Han, C.; Themistocleous, A.C.; Estacion, M.; Dib-Hajj, F.B.; Blesneac, I.; Macala, L.; Fratter, C.; Bennett, D.L.; Waxman, S.G.; Dib-Hajj, S.D. The novel activity of carbamazepine as an activation modulator extends from NaV1.7 mutations to the NaV1.8-S242T mutant channel from a patient with painful diabetic neuropathy. Mol. Pharmacol. 2018, 94, 1256–1269. [Google Scholar] [CrossRef]
- Dabby, R.; Sadeh, M.; Broitman, Y.; Yosovich, K.; Dickman, R.; Leshinsky-Silver, E. Painful small fiber neuropathy with gastroparesis: A new phenotype with a novel mutation in the SCN10A gene. J. Clin. Neurosci. 2016, 26, 84–88. [Google Scholar] [CrossRef]
- Coates, M.D.; Kim, J.S.; Carkaci-Salli, N.; Vrana, K.E.; Koltun, W.A.; Puhl, H.L.; Adhikary, S.D.; Janicki, P.K.; Ruiz-Velasco, V. Impact of the Na(V)1.8 variant, A1073V, on post-sigmoidectomy pain and electrophysiological function in rat sympathetic neurons. J. Neurophysiol. 2019, 122, 2591–2600. [Google Scholar] [CrossRef]
- Duan, G.; Sun, J.; Li, N.; Zheng, H.; Guo, S.; Zhang, Y.; Wang, Q.; Ying, Y.; Zhang, M.; Huang, P.; et al. A variant in the SCN10A enhancer may affect human mechanical pain sensitivity. Mol. Pain. 2018, 14, 1744806918763275. [Google Scholar] [CrossRef]
- Duan, G.; Han, C.; Wang, Q.; Guo, S.; Zhang, Y.; Ying, Y.; Huang, P.; Zhang, L.; Macala, L.; Shah, P.; et al. A SCN10A SNP biases human pain sensitivity. Mol. Pain. 2016, 12, 1744806916666083. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez, E.; Imamura Kawasawa, Y.; Walter, V.; Zhang, L.; Koltun, W.A.; Huang, X.; Vrana, K.E.; Coates, M.D. Homozygosity for the SCN10A Polymorphism rs6795970 Is Associated With Hypoalgesic Inflammatory Bowel Disease Phenotype. Front. Med. 2018, 5, 324. [Google Scholar] [CrossRef] [PubMed]
- Loose, S.; Lischka, A.; Kuehs, S.; Nau, C.; Heinemann, S.H.; Kurth, I.; Leipold, E. Peripheral temperature dysregulation associated with functionally altered Na(V)1.8 channels. Pflugers Archive 2023, 475, 1343–1355. [Google Scholar] [CrossRef]
- Coates, M.D.; Walter, V.; Stuart, A.; Small, J.; Dalessio, S.; Carkaci-Salli, N.; Ouyan, A.; Clarke, K.; Tinsley, A.; Williams, E.; et al. Impact of SCN10A Polymorphism on Abdominal Pain Perception and Visceral Hypoalgesia in Crohn’s Disease and Ulcerative Colitis. Clin. Transl. Gastroenterol. 2024, 15, e00778. [Google Scholar] [CrossRef]
- Christidis, N.; Kang, I.; Cairns, B.; Kumar, U.; Dong, X.; Rosen, A.; Kopp, S.; Ernberg, M. Expression of 5-HT3 receptors and TTX resistant sodium channels (Na(V)1.8) on muscle nerve fibers in pain-free humans and patients with chronic myofascial temporomandibular disorders. J. Headache Pain. 2014, 15, 63. [Google Scholar] [CrossRef]
- Bird, E.; Christmas, C.; Loescher, A.; Smith, K.; Robinson, P.; Black, J.A.; Waxman, S.G.; Boissonade, F.M. Correlation of Nav1.8 and Nav1.9 sodium channel expression with neuropathic pain in human subjects with lingual nerve neuromas. Mol. Pain. 2013, 9, 52. [Google Scholar] [CrossRef]
- Black, J.A.; Nikolajsen, L.; Kroner, K.; Jensen, T.S.; Waxman, S.G. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 2008, 64, 644–653. [Google Scholar] [CrossRef]
- Bucknill, A.; Coward, K.; Plumpton, C.; Tate, S.; Bountra, C.; Birch, R.; Sandison, A.; Hughes, S.P.F.; Anand, P. Nerve fibers in lumbar spine structures and injured spinal roots express the sensory neuron-specific sodium channels SNS PN3 and NaN SNS2. Spine 2002, 27, 135–140. [Google Scholar] [CrossRef]
- Jo, S.; Fujita, A.; Osorno, T.; Stewart, R.G.; Vaelli, P.M.; Bean, B.P. Differential state-dependent Nav1.8 inhibition by suzetrigine, LTGO-33, and A-887826. J. Gen. Physiol. 2025, 157, e202413719. [Google Scholar] [CrossRef]
- Gilchrist, J.M.; Yang, N.D.; Jiang, V.; Moyer, B.D. Pharmacologic Characterization of LTGO-33, a Selective Small Molecule Inhibitor of the Voltage-Gated Sodium Channel Na(V)1.8 with a Unique Mechanism of Action. Mol. Pharmacol. 2024, 105, 233–249. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Honore, P.; Shieh, C.C.; Chapman, M.; Joshi, S.; Zhang, X.F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Shieh, C.C.; Chapman, M.L.; Matulenko, M.A.; Hakeem, A.H.; Atkinson, R.N.; Kort, M.E.; Marron, B.E.; Joshi, S.; Honore, P.; et al. A-887826 is a structurally novel, potent and voltage-dependent Na(v)1.8 sodium channel blocker that attenuates neuropathic tactile allodynia in rats. Neuropharmacology 2010, 59, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.E.; Brown, A.R.; Theile, J.W.; Loucif, A.J.; Alexandrou, A.J.; Fuller, M.D.; Mahoney, J.H.; Antonio, B.M.; Gerlach, A.C.; Printzenhoff, D.M.; et al. A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br. J. Pharmacol. 2015, 172, 2654–2670. [Google Scholar] [CrossRef]
- Hijma, H.J.; Siebenga, P.S.; de Kam, M.L.; Groeneveld, G.J. A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Crossover Study to Evaluate the Pharmacodynamic Effects of VX-150, a Highly Selective NaV1.8 Inhibitor, in Healthy Male Adults. Pain. Med. 2021, 22, 1814–1826. [Google Scholar] [CrossRef]
- Hijma, H.J.; van Brummelen, E.M.J.; Siebenga, P.S.; Groeneveld, G.J. A phase I, randomized, double-blind, placebo-controlled, single- and multiple dose escalation study evaluating the safety, pharmacokinetics and pharmacodynamics of VX-128, a highly selective Nav 1.8 inhibitor, in healthy adults. Clin. Transl. Sci. 2021, 15, 981–993. [Google Scholar] [CrossRef]
- May, M. New approaches to opioid-free pain treatment. Nature Med. 2025. Available online: https://www.nature.com/articles/d41591-025-00024-w (accessed on 4 April 2025). [CrossRef]
- Robinson, C.L.; Schatman, M.E.; Hasoon, J.; Chung, M.; Emerick, T.; Bianco, G.L.; Ashina, S.; Yong, R.J. Suzetrigine: Is This What We Have Been Waiting for or Just the Beginning? J. Pain. Res. 2025, 18, 2047–2049. [Google Scholar] [CrossRef]
- de Greef, B.T.A.; Hoeijmakers, J.G.J.; Geerts, M.; Oakes, M.; Church, T.J.E.; Waxman, S.G.; Dib-Hajj, S.D.; Faber, C.G.; Merkies, I.S.J. Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: A randomized controlled trial. Brain 2019, 142, 263–275. [Google Scholar] [CrossRef]
- Sheets, P.L.; Heers, C.; Stoehr, T.; Cummins, T.R. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J. Pharmacol. Exp. Ther. 2008, 326, 89–99. [Google Scholar] [CrossRef]
- Maihofner, C.; Schneider, S.; Bialas, P.; Gockel, H.; Beer, K.; Bartels, M.; Kern, K.U. Successful treatment of complex regional pain syndrome with topical ambroxol: A case series. Pain. Manag. 2018, 8, 427–436. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- McDermott, L.A.; Weir, G.A.; Themistocleous, A.C.; Segerdahl, A.R.; Blesneac, I.; Baskozos, G.; Clark, A.J.; Millar, V.; Peck, L.J.; Ebner, D.; et al. Defining the Functional Role of NaV1.7 in Human Nociception. Neuron 2019, 101, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Alsaloum, M.; Waxman, S.G. iPSCs and DRGs: Stepping stones to new pain therapies. Trends Mol. Med. 2022, 28, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Alsaloum, M.; Labau, J.I.R.; Liu, S.; Estacion, M.; Zhao, P.; Dib-Hajj, F.; Waxman, S.G. Contributions of Na(V)1.8 and Na(V)1.9 to excitability in human induced pluripotent stem-cell derived somatosensory neurons. Sci. Rep. 2021, 11, 24283. [Google Scholar] [CrossRef]
- Klein, T.; Klug, K.; Henkel, L.; Kwok, C.K.; Edenhofer, F.; Klopocki, E.; Kurth, I. Üceyler, N. Generation of two induced pluripotent stem cell lines from skin fibroblasts of sisters carrying a c.1094C>A variation in the SCN10A gene potentially associated with small fiber neuropathy. Stem Cell Res. 2019, 35, 101396. [Google Scholar] [CrossRef]
- Garrison, S.R.; Weyer, A.D.; Barabas, M.E.; Beutler, B.A.; Stucky, C.L. A gain-of-function voltage-gated sodium channel 1.8 mutation drives intense hyperexcitability of A- and C-fiber neurons. Pain 2014, 155, 896–905. [Google Scholar] [CrossRef]
- Blasius, A.L.; Dubin, A.E.; Petrus, M.J.; Lim, B.K.; Narezkina, A.; Criado, J.R.; Wills, D.N.; Xia, Y.; Moresco, E.Y.; Ehlers, C.; et al. Hypermorphic mutation of the voltage-gated sodium channel encoding gene Scn10a causes a dramatic stimulus-dependent neurobehavioral phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 19413–19418. [Google Scholar] [CrossRef]
- Xiao, Y.; Barbosa, C.; Pei, Z.; Xie, W.; Strong, J.A.; Zhang, J.M.; Cummins, T.R. Increased resurgent sodium currents in Nav1.8 contribute to nociceptive sensory neuron hyperexcitability associated with peripheral neuropathies. J. Neurosci. 2019, 39, 1539–1550. [Google Scholar] [CrossRef]
- Matthews, E.A.; Wood, J.N.; Dickenson, A.H. Nav 1.8-Null Mice Show Stimulus-Dependent Deficits in Spinal Neuronal Activity. Mol. Pain 2006, 2, 1744–8069-1742-1745. [Google Scholar] [CrossRef]
- Fjell, J.; Cummins, T.; Dib-Hajj, S.; Fried, K.; Black, B. SGW Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res. Mol. Brain Res. 1999, 67, 267–282. [Google Scholar] [CrossRef]
- Li, G.; Liu, X.; Du, J.; Chen, J.; She, F.; Wu, C.; Li, C. Positive shift of Nav1.8 current inactivation curve in injured neurons causes neuropathic pain following chronic constriction injury. Mol. Med. Rep. 2015, 12, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Nascimento de Lima, A.P.; Zhang, H.; Chen, L.; Effraim, P.R.; Gomis-Perez, C.; Cheng, X.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. Nav1.8 in small dorsal root ganglion neurons contributes to vincristine-induced mechanical allodynia. Brain 2024, 147, 3157–3170. [Google Scholar] [CrossRef] [PubMed]
- Belkouch, M.; Dansereau, M.; Tetrealut, R.; Biet, M.; Beaudet, N.; Dumaine, R.; Chraibi, A.; Mélik-Parsadaniantz, S.; Sarret, P. Functional up-regulation of Nav1.8 sodium channel in Aβ afferent fibers subjected to chronic peripheral inflammation. J. Neuroinflamm. 2014, 11, 45. [Google Scholar] [CrossRef]
- Yang, F.; Zou, Y.Q.; Li, M.; Luo, W.J.; Chen, G.Z.; Wu, X.Z. Intervertebral foramen injection of plerixafor attenuates neuropathic pain after chronic compression of the dorsal root ganglion: Possible involvement of the down-regulation of Nav1.8 and Nav1.9. Eur. J. Pharmacol. 2021, 908, 174322. [Google Scholar] [CrossRef]
- Li, N.; Liu, B.; Wu, W.; Hong, Y.; Zhang, J.; Liu, Y.; Zhang, M.; Zhang, X.; Duan, G. Upregulation of transcription factor 4 downregulates NaV1.8 expression in DRG neurons and prevents the development of rat inflammatory and neuropathic hypersensitivity. Exp. Neurol. 2020, 327, 113240. [Google Scholar] [CrossRef]
- Zhang, X.L.; Cao, X.Y.; Lai, R.C.; Xie, M.X.; Zeng, W.A. Puerarin Relieves Paclitaxel-Induced Neuropathic Pain: The Role of Na(v)1.8 beta1 Subunit of Sensory Neurons. Front. Pharmacol. 2018, 9, 1510. [Google Scholar] [CrossRef]
- Liu, X.D.; Yang, J.J.; Fang, D.; Cai, J.; Wan, Y.; Xing, G.G. Functional upregulation of nav1.8 sodium channels on the membrane of dorsal root Ganglia neurons contributes to the development of cancer-induced bone pain. PLoS ONE 2014, 9, e114623. [Google Scholar] [CrossRef]
- Dib-Hajj, S.; Black, B.; Felts, P. SGW Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc. Natl. Acad. Sci. USA 1996, 93, 14950–14954. [Google Scholar] [CrossRef]
- Dib-Hajj, S.; Fjell, J.; Cummins, T.; Zheng, Z.; Fried, K.; LaMotte, R.; Black, J.A.; Waxman, S.G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [Google Scholar] [CrossRef]
- Sleeper, A.; Cummins, T.; Dib-Hajj, S.; Hormuzdiar, W.; Tyrrell, L.; Waxman, G.; Black, J.A.A. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J. Neurosci. 2000, 120, 7279–7289. [Google Scholar] [CrossRef]
- Black, J.; Langworthy, K.; Hinson, A.; Dib-Hajj, S.; Waxman, S. NGF has opposing effects on Na+ channel III and SNS gene expression in spinal sensory neurons. Neuroreport 1997, 8, 2331–2335. [Google Scholar] [CrossRef] [PubMed]
- Cédric, J.L.; Marie, P.; Marc RSr Isabelle, D. Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 2014, 10, 19. [Google Scholar]
- Chidiac, C.; Xue, Y.; Muniz Moreno, M.D.M.; Bakr Rasheed, A.A.; Lorentz, R.; Birling, M.C.; Ruff, C.G.; Herault, Y. The Human SCN10A(G1662S) Point Mutation Established in Mice Impacts on Mechanical, Heat, and Cool Sensitivity. Front. Pharmacol. 2021, 12, 780132. [Google Scholar] [CrossRef]
- Miller, R.E.; Ishihara, S.; Bhattacharyya, B.; Menichella, D.; Miller, R.J.; Malfait, A.M. Chemogenetic silencing of NAV1.8-expressing nociceptors reduces pain-related behaviors in a stage-dependent manner in experimental osteoarthritis. Osteoarthr. Cartil. 2016, 24, S9. [Google Scholar] [CrossRef]
- Lai, J.; Gold, M.S.; Kim, C.S.; Biana, D.; Ossipov, M.H.; Hunterc, J.C.; Porreca, F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain 2002, 95, 143–152. [Google Scholar] [CrossRef]
- Novakovic, S.D.T.E.; McGivern, J.G.; Haraguchi, M.; Sangameswaran, L.; Gogas, K.R.; Eglen, R.M.; Hunter, J.C. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J. Neurosci. 1998, 18, 2174–2187. [Google Scholar] [CrossRef]
- Daou, I.; Beaudry, H.; Ase, A.R.; Wieskopf, J.S.; Ribeiro-da-Silva, A.; Mogil, J.S.; Séguéla, P. Optogenetic Silencing of Nav1.8-Positive Afferents Alleviates Inflammatory and Neuropathic Pain. eNeuro 2016, 3, eneuro.0140-15.2016. [Google Scholar] [CrossRef]
- Haroun, R.; Gossage, S.J.; Luiz, A.P.; Arcangeletti, M.; Sikandar, S.; Zhao, J.; Cox, J.J.; Wood, J.N. Chemogenetic silencing of Na(V)1.8 positive sensory neurons reverses chronic neuropathic and bone cancer pain in FLEx PSAM(4)-GlyR mice. eNeuro 2023, 10, eneuro.0151-23.2023. [Google Scholar] [CrossRef]
- Yang, R.; Wang, Q.Q.; Feng, Y.; Li, X.H.; Li, G.X.; She, F.L.; Zhu, X.J.; Li, C.L. Over-expression of miR-3584-5p Represses Nav1.8 Channel Aggravating Neuropathic Pain caused by Chronic Constriction Injury. Mol. Neurobiol. 2023, 60, 5237–5255. [Google Scholar] [CrossRef]
- O’Brien, M.S.; Philpott, H.T.A.; McDougall, J.J. Targeting the Nav1.8 ion channel engenders sex-specific responses in lysophosphatidic acid-induced joint neuropathy. Pain 2019, 160, 269–278. [Google Scholar] [CrossRef]
- Messina, D.N.; Peralta, E.D.; Acosta, C.G. Complex alterations in inflammatory pain and analgesic sensitivity in young and ageing female rats: Involvement of ASIC3 and Nav1.8 in primary sensory neurons. Inflamm. Res. 2024, 73, 669–691. [Google Scholar] [CrossRef] [PubMed]
- Urru, M.; Muzzi, M.; Coppi, E.; Ranieri, G.; Buonvicino, D.; Camaioni, E.; Coppini, R.; Pugliese, A.M.; Tanaka, B.; Estacion, M. Dexpramipexole blocks Nav1.8 sodium channels and provides analgesia in multiple nociceptive and neuropathic pain models. Pain 2020, 161, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.D.; Cheng, X.; Üçeyler, N.; Sommer, C.; Dib-Hajj, S.D.; Waxman, S.G. Sodium Channel Nav1.7 Is Essential for Lowering Heat Pain Threshold after Burn Injury. J. Neurosci. 2012, 32, 10819–10832. [Google Scholar] [CrossRef] [PubMed]
- Leo, S.; D’Hooge, R.; Meert, T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain Res. 2010, 208, 149–157. [Google Scholar] [CrossRef]
- Zimmermann, K.; Leffler, A.; Babes, A.; Cendan, C.M.; Carr, R.W.; Kobayashi, J.; Nau, C.; Wood, J.N.; Reeh, P.W. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 2007, 447, 855–858. [Google Scholar] [CrossRef]
- Kerr, B.; Souslova, V.; McMahon, S.B.; Wood, J.N. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 2001, 12, 3077–3080. [Google Scholar] [CrossRef]
- Laird, J.M.A.; Souslova, V.; Wood, J.N.; Cervero, F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS PN3)-null mice. J. Neurosci. 2002, 22, 8352–8356. [Google Scholar] [CrossRef]
- Nassar, M.A.; Levato, A.; Stirling, L.C.; Wood, J.N. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol. Pain 2005, 1, 24. [Google Scholar] [CrossRef]
- Bhuiyan, S.A.; Xu, M.; Yang, L.; Semizoglou, E.; Bhatia, P.; Pantaleo, K.I.; Tochitsky, I.; Jain, A.; Erdogan, B.; Blairs, S.; et al. Harmonized cross-species cell atlases of trigeminal and dorsal root ganglia. Sci. Adv. 2024, 10, eadj9173. [Google Scholar] [CrossRef]
- Chang, W.; Berta, T.; Kim, Y.H.; Lee, S.; Lee, S.Y.; Ji, R.R. Expression and Role of Voltage-Gated Sodium Channels in Human Dorsal Root Ganglion Neurons with Special Focus on Nav1.7, Species Differences, and Regulation by Paclitaxel. Neurosci. Bull. 2018, 34, 4–12. [Google Scholar] [CrossRef]
- Rostock, C.; Schrenk-Siemens, K.; Pohle, J.; Siemens, J. Human vs. Mouse Nociceptors–Similarities and Differences. Neuroscience 2018, 387, 13–27. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, D.S.; Vardigan, J.D.; Zhou, X.; Rosahl, T.W.; Zhou, H.; Price, E.A.; Clements, M.K.; Li, Y.; Varghese, N.; Krasowska-Zoladek, A.; et al. Humanized NaV1.8 rats overcome cross-species potency shifts in developing novel NaV1.8 inhibitors. Neurobiol. Pain 2025, 18, 100182. [Google Scholar] [CrossRef] [PubMed]
- Vardigan, J.D.; Pall, P.S.; McDevitt, D.S.; Huang, C.; Clements, M.K.; Li, Y.; Kraus, R.L.; Breslin, M.J.; Bungard, C.J.; Nemenoy, K.; et al. Analgesia and peripheral c-fiber modulation by selective Na v 1.8 inhibition in rhesus. Pain 2025, 166, 631–643. [Google Scholar] [CrossRef]
- Edelman, G.M.; Gally, J.A. Degeneracy and complexity in biological systems. Proc. Natl. Acad. Sci. USA 2001, 98, 13763–13768. [Google Scholar] [CrossRef]
- Ghazisaeidi, S.; Muley, M.; Salter, M. Neuropathic Pain: Mechanisms, Sex Differences, and Potential Therapies for a Global Problem. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Ratte, S.; Prescott, S.A. Afferent hyperexcitability in neuropathic pain and the inconvenient truth about its degeneracy. Curr. Opin. Neurobiol. 2016, 36, 31–37. [Google Scholar] [CrossRef]
- Huang, J.; Estacion, M.; Zhao, P.; Dib-Hajj, F.B.; Schulman, B.; Abicht, A.; Kurth, I.; Brochmann, K.; Waxman, S.G. A Novel Gain-of-Function Nav1.9 Mutation in a Child With Episodic Pain. Front. Neurosci. 2019, 13, 918. [Google Scholar] [CrossRef]
- Huang, J.; Han, C.; Estacion, M.; Vasylyev, D.; Hoeijmakers, J.G.; Gerrits, M.M.; Tyrrel, L.; Lauria, G.; Baber, C.G.; Dib-Hajj, S.D.; et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 2014, 137, 1627–1642. [Google Scholar] [CrossRef]
- Leipold, E.; Liebmann, L.; Korenke, G.C.; Heinrich, T.; Giesselmann, S.; Baets, J.; Ebbinghaus, M.; Goral, R.O.; Stödberg, T.; Hennings, J.C.; et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 2013, 45, 1399–1404. [Google Scholar] [CrossRef]
- Phatarakijnirund, V.; Mumm, S.; McAlister, W.H.; Novack, D.V.; Wenkert, D.; Clements, K.L.; Whyte, M. Congenital insensitivity to pain: Fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9. Bone 2016, 84, 289–298. [Google Scholar] [CrossRef]
- Lolignier, S.; Amsalem, M.; Maingret, F.; Padilla, F.; Gabriac, M.; Chapuy, E.; Eschalier, A.; Delmas, P.; Busserolles, J. Nav1.9 Channel Contributes to Mechanical and Heat Pain Hypersensitivity Induced by Subacute and Chronic Inflammation. PLoS ONE 2011, 6, e23083. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Shao, J.; Cao, J.; Ren, X.; Cai, W.; Su, S.; George, S.; Tan, Z.; Zang, W.; Dong, T. Protein kinase C-alpha upregulates sodium channel Nav1.9 in nociceptive dorsal root ganglion neurons in an inflammatory arthritis pain model of rat. J. Cell Biochem. 2020, 121, 768–778. [Google Scholar] [CrossRef]
- Amsalem, M.; Poilbout, C.; Ferracci, G.; Delmas, P.; Padilla, F. Membrane cholesterol depletion as a trigger of Nav1.9 channel-mediated inflammatory pain. EMBO J. 2018, 37, e97349. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain. 2008, 12, 564–572. [Google Scholar] [CrossRef]
- Mao, J.; Gold, M.S.; Backonja, M.M. Combination drug therapy for chronic pain: A call for more clinical studies. J. Pain 2011, 12, 157–166. [Google Scholar] [CrossRef]
- Zhu, D.; Liu, K.; Wan, C.L.; Lu, J.; Zhao, H.X. Identification of novel therapeutic targets for neuropathic pain based on gene expression patterns. J. Cell Physiol. 2019, 234, 19494–19501. [Google Scholar] [CrossRef]
- Tigerholm, J.; Petersson, M.E.; Obreja, O.; Eberhardt, E.; Namer, B.; Weidner, C.; Lampert, A.; Carr, R.W.; Schmelz, M.; Fransén, E. C-Fiber Recovery Cycle Supernormality Depends on Ion Concentration and Ion Channel Permeability. Biophys. J. 2015, 108, 1057–1071. [Google Scholar] [CrossRef]
- Scheib, H.; McLay, I.; Guex, N.; Clare, J.; Blaney, F.; Dale, T.; Nate, S.; Robertson, G.M. Modeling the pore structure of voltage-gated sodium channels in closed, open, and fast-inactivated conformation reveals details of site 1 toxin and local anesthetic binding. J. Mol. Model. 2006, 12, 813–822. [Google Scholar] [CrossRef]
- Kan, P.; Zhu, Y.F.; Ma, J.; Singh, G. Computational modeling to study the impact of changes in Nav1.8 sodium channel on neuropathic pain. Front. Comput. Neurosci. 2024, 18, 1327986. [Google Scholar] [CrossRef]
- Petersson, M.E.; Obreja, O.; Lampert, A.; Carr, R.W.; Schmelz, M.; Fransen, E. Differential axonal conduction patterns of mechano-sensitive and mechano-insensitive nociceptors--a combined experimental and modelling study. PLoS ONE 2014, 9, e103556. [Google Scholar] [CrossRef]
- Chen, L.; Jiang, J.; Dou, B.; Feng, H.; Liu, J.; Zhu, Y.; Zhang, B.; Zhou, T.; Wei, G.W. Machine Learning Study of the Extended Drug-target Interaction Network informed by Pain Related Voltage-Gated Sodium Channels. Pain 2024, 165, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Waxman, S.G. Physiological interactions between Nav1.7 and Nav1.8 sodium channels: A computer simulation study. J. Neurophysiol. 2011, 106, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Correll, D.J.; Lechner, S.M.; Jazic, I.; Miao, X.; Shaw, D.; Simard, C.; Osteen, J.D.; Hare, B.; Beaton, A.; et al. Selective Inhibition of NaV1.8 with VX-548 for Acute Pain. N. Engl. J. Med. 2023, 389, 393–405. [Google Scholar] [CrossRef]
- Medlock, L.; Sekiguchi, K.; Hong, S.; Dura-Bernal, S.; Lytton, W.W.; Prescott, S.A. Multiscale Computer Model of the Spinal Dorsal Horn Reveals Changes in Network Processing Associated with Chronic Pain. J. Neurosci. 2022, 42, 3133–3149. [Google Scholar] [CrossRef]
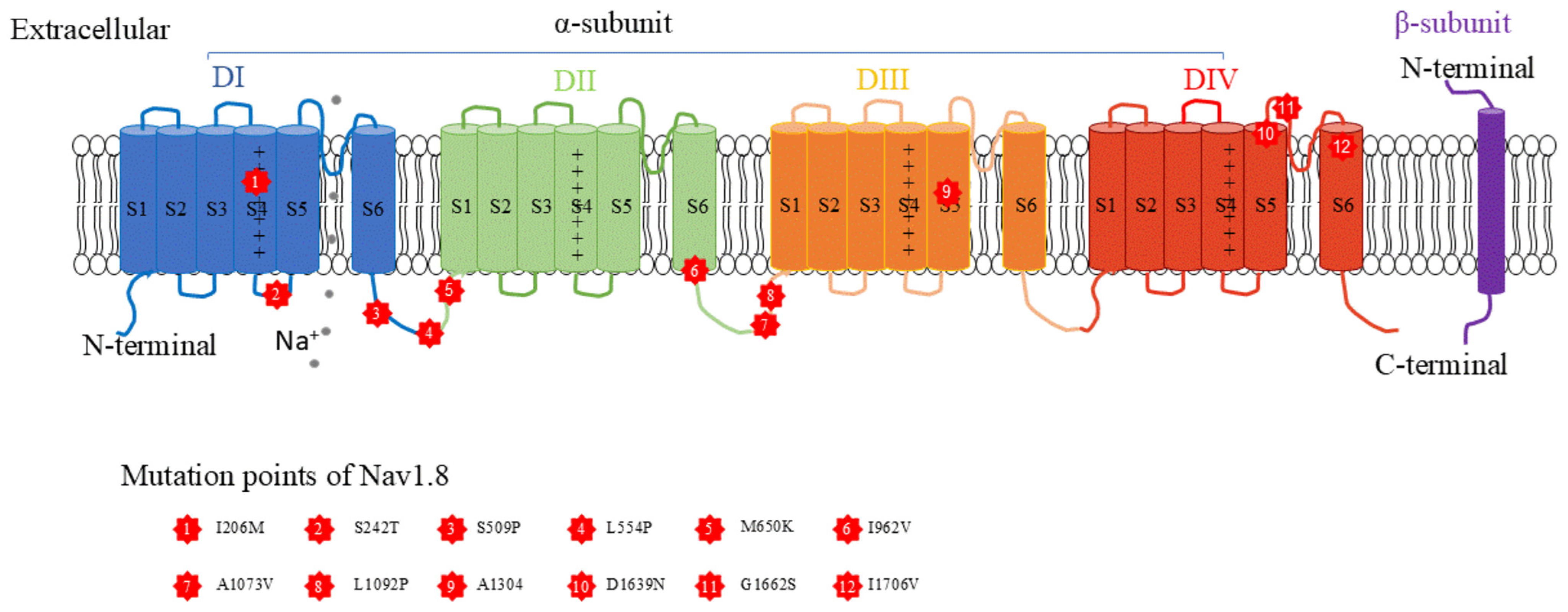

{kind=link}
{kind=link}
| References | PMID | Age (yrs) | Sex | Mutation Focus | Function | Clinical | E-Phys |
|---|---|---|---|---|---|---|---|
| [11] | 30099632 | 32/37 | F/F | D1639N | Loss | SFN | |
| [10] | 24006052 | 24/62 | F | G1662S | Gain | SFN | |
| [36] | 23986244 | 61 | M | I1706V | Loss | SFN | Transfected DRG neuron, 40–55 h, CsCL-based, small |
| [9] | 23115331 | 67, 39, 69 | 2 M/1 F | L554P, A1304T | Gain | iSFN | |
| [37] | 30135145 | 67 | M | S242T | Gain | SFN | Transfected DRG NGF/GDNF, 40–48 h |
| [38] | 26711856 | 37 | F | D1639N | na | SFN: severe progressive gastroparesis and diffuse painful small fiber sensory neuropathy | |
| [39] | 31642403 | 166 | M/F | A1073V | Lower abdominal pain scores | Male SD rat, SCG no trophic factors, 16–24 h, NMG-based, small | |
| [40] | 29448912 | rs6801957-G/A | decreased experimental mechanical pain sensitivity | ||||
| [41] | 27590072 | 22.6 | 187 M/309 F | A1073V | Loss | Higher thresholds for mechanical pain | Shifts channel activation by −4.3 mV and accelerates inactivation, reduces repetitive firing of DRG neurons, and lowers mechanical pain sensitivity |
| [42] | 30538988 | 41.7 | 58 M/63 F | A1073V | Loss | Hypoalgesic IBD patients | |
| [12] | 27598514 | 53 | M | p.M650K | Loss | Erythromelalgia: increased activity-dependent slowing in CMi and less spontaneous firing in peripheral nerve fibers than non-mutant erythromelalgia | P3-6d Wistar rat, f/m, small DRG neurons, culture 1 day. Shifted steady-state fast inactivation of Nav1.8 to hyperpolarization, increased AP duration, and reduced AP rate |
| NCT | Drug | Clinical Trial | Pain Model | Gender | Age | Sample Size | Dose | Routine | Status |
|---|---|---|---|---|---|---|---|---|---|
| 015121608 | PF-04531083 | Phase II | Post-surgical dental pain | M | 18–55 | 90 | 1–2 g, single | Oral | Terminated for futility based on results of internal analysis |
| 03304522 | VX-150 | Phase II | SFN | M/F | 18–80 | 89 | 1.25 g, daily, 6 wks | Oral | |
| 06176196 | VX-548 | Phase II | Painful lumbosacral radiculopathy | M/F | 18–70 | Estimate 200 | 12 wks | Oral | Recruiting |
| 05660538 | VX-548 | Phase II | Diabetic peripheral neuropathy | M/F | 18–80 | 192 | 23, 46, 69 mg, qd, 12 wks | Oral | Significantly reduced pain |
| 06628908, 06696443 | Suzetrigine | Phase III | Diabetic peripheral neuropathy | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.-F. Nav1.8 and Chronic Pain: From Laboratory Animals to Clinical Patients. Biomolecules 2025, 15, 694. https://doi.org/10.3390/biom15050694
Xie Y-F. Nav1.8 and Chronic Pain: From Laboratory Animals to Clinical Patients. Biomolecules. 2025; 15(5):694. https://doi.org/10.3390/biom15050694
Chicago/Turabian StyleXie, Yu-Feng. 2025. "Nav1.8 and Chronic Pain: From Laboratory Animals to Clinical Patients" Biomolecules 15, no. 5: 694. https://doi.org/10.3390/biom15050694
APA StyleXie, Y.-F. (2025). Nav1.8 and Chronic Pain: From Laboratory Animals to Clinical Patients. Biomolecules, 15(5), 694. https://doi.org/10.3390/biom15050694