1. Introduction
Newborn calf diarrhea (NCD) is a significant cause of morbidity and mortality in calves under 7 days of age, resulting in a reduced growth rate, increased risk of infection with other pathogens, and serious mortality and economic losses [
1]. In 1969, the “reo”-like virus was first identified in the feces of calves with NCD [
2]. In 1974, after observing the virus with an electron microscope, Flewett suggested naming the virus “rotavirus” as the virus particles look like wheels (given that “rota” is Latin for “wheel”). Four years later, the name was officially recognized by the International Committee on the Classification of Viruses [
3]. In 1976, the virus was found in many other animals and was found to lead to acute gastroenteritis and cause serious effects in humans and animals [
4].
Bovine rotavirus (BRV) is one of the main pathogens that causes acute diarrhea in calves under the age of one month [
5,
6]. BRV belongs to the genus rotavirus in the family
Reoviridae and is a double-stranded ribonucleic acid virus [
7]. The majority of BRVs exhibit species specificity and genetic variability arising from genetic transfer, gene rearrangements, and fragment exchanges. Its intact and infectious virions have a three-layer capsid structure [
8]. The virus structural proteins include core proteins (VP1, VP2, and VP3), inner capsid proteins (VP6), and outer capsid proteins [
9]. Among them, outer capsid proteins, including VP7 and VP4, can induce the production of neutralizing antibodies in animals and play an important role in the body’s immunity [
10].
Passive immunization is an effective way to prevent diarrhea in newborn calves. By vaccinating pregnant cows with a BRV vaccine to induce the production of antibodies against BRV, the antibodies can be transferred to calves through lactation. To date, several BRV vaccines against the main pathogens causing calf diarrhea have been developed and used on cattle farms. The vaccines mainly contain a mixture of inactivated BRV, bovine coronavirus (BCV), and
Escherichia coli (
E. coli) [
11]. However, the quality of bovine colostrum, especially the BRV-specific antibody titer in the colostrum, has a significant impact on the passive immunity obtained by calves. When cows are immunocompromised, the levels of key bioactive components in their colostrum may fall significantly below the established industry benchmarks or anticipated thresholds [
12].
Meanwhile, an egg yolk IgY antibody against the VP8 capsid protein of bovine group A rotavirus is one of the strategies used to eliminate rotavirus infection in the animal environment and protect livestock herds. Formula milk containing an egg yolk antibody against bovine rotavirus can increase the response of mucosal antibody-secreting cells, reduce virus shedding, and reduce the severity and duration of diarrhea [
13]. Vega C and others found that feeding uninfected newborn calves colostrum containing anti-BRV-specific IgY for 14 days could reduce the severity of diarrhea when the calves were challenged with BRV [
14]. In 2020, a new avian IgY antibody product named IgY-DNT that can effectively prevent diarrhea in newborn calves was introduced to supplement passive immunity. IgY-DNT can regulate the intestinal mucosal immune response, resulting in an increase in antibody-secreting cells in the duodenum and ileum [
15]. Nevertheless, the susceptibility of IgY to proteolysis is one of the limitations to the oral use of IgY for passive immunotherapy. The lack of standardization of the production, extraction, and purification processes for IgY antibodies from laboratory animals has been one of the major challenges in the product licensing, regulation, and approval of IgY-based bioproducts. More safety studies are needed to verify their safety as human and veterinary treatments [
16].
Recent studies have revealed that approximately 10% of bovine antibodies possess a special ultra-long complementarity-determining region (CDR) H3 structure in the antibody heavy chains. This structure was first found in bovine IgM antibodies and was later also found in other bovine antibodies, such as IgG. The bovine ultra-long CDR H3 structure consists of a “stalk” structure on the surface of the antibody (a double chain with an ascending chain and descending chain) facing away from the beta ribbon, as well as a disulfide-rich “knob” structure located on the β-banded “stalk” [
15]. This structure is similar to the “handle” and “cap” of mushrooms and is different from the typical antibody CDR H3 structure [
17].
To understand the structure of the ultra-long CDR H3 in antibodies, Haakenson K et al. analyzed the two Fab fragments from the bovine antibodies BLV1H12 and BLV5B8 [
18]. Both antibodies exhibit a unique β-stalk and knob structure stabilized by eight β-sheet hydrogen bonds (bovine-specific) [
19]. The β-stalk connects to the antibody scaffold via a β-ring at its base. The descending chain contains alternating aromatic motifs (typically YXYXY), with BLV1H12 featuring “YTYNY” and BLV5B8 featuring “HSYEF” [
20]. These motifs form stacked steps, enhancing β-sheet stability.
In conventional antibodies, the CDR typically serves as the contact point for binding to antigens. However, in antibodies containing a bovine ultra-long CDR H3, the CDR H1 and H2 fragments mainly play a role in supporting and stabilizing the knob structure and are not directly involved in antigen binding [
21]. In most cases, only the CDR H3 in bovine ultra-long CDR H3 antibodies is used to bind antigens. Overall, the bovine immune system produces a unique CDR H3 sequence, which folds to form beta ribbon stalks and a knob structure. Changes in the number and binding mode of cysteines allow bovine antibodies to exhibit unique functions in terms of antigen recognition [
22]. Previous studies have shown that antibodies with a unique bovine ultra-long CDR H3 can be used as broadly neutralizing antibodies (bnAbs) to neutralize HIV and treat HIV infection [
23]. This indicates that they may also possess similar advantages against bovine pathogens such as rotavirus and foot-and-mouth disease virus. Although research on ultra-long CDR H3 antibodies has mainly focused on human diseases like HIV and cancer, their application to bovine pathogens could fill a crucial gap in the fields of veterinary medicine and agricultural biotechnology, bringing hope for the reduction in economic losses in the livestock industry.
A phage display library is a diversified phage clone population in which every clone contains a random foreign DNA insert and, hence, presents a different molecule on its surface [
24]. The key advantage of phage display libraries is the possibility to test a vast number of phages in every round due to the high transformation efficiency, allowing for the identification of the most promising binders [
25]. Meanwhile, phage display technology is also suitable for screening small-molecule antibody fragments, such as single-chain fragment variables (scFvs) and single-domain antibodies (sdAbs, also called nanobodies).
In our previous study, we prepared and characterized single-domain antibodies by immunizing Bactrian camels and constructing a phage display antibody library. These single-domain antibodies can be developed as a bispecific nanobody to target tumor cells or as a virus-specific binder for the detection and purification of viruses [
26,
27,
28]. Based on our previous work, the development of bovine ultra-long CDR H3 antibodies using phage display technology appears feasible. Bovine pathogen-specific ultra-long CDR H3 antibodies may be a good candidate for the treatment and prevention of diseases since they are derived from cattle, and there is no immune reaction when applied to cattle. This could provide an effective approach for developing passive immunotherapeutics for bovine infectious diseases. This study aimed to utilize phage display antibody library technology to generate ultra-long CDR H3 antibodies that specifically target BRV, evaluate the binding and neutralizing activities of the antibodies as well as their therapeutic efficacy, and demonstrate that ultra-long CDR H3 antibodies could serve as effective agents for the prevention and treatment of bovine rotavirus infections.
2. Materials and Methods
2.1. Bovine Immunization
A five-month-old Holstein male calf was obtained from the dairy farm in the suburbs of Hohhot, Inner Mongolia Autonomous Region, and used for immunization. The calf was injected with 3 mL of the group A BRV NMG strain (concentration: 10
−6.13/100 μL (TCID
50); GenBank accession number MN807286.1; provided by Professor Weiguang Zhou from the College of Veterinary Medicine, Inner Mongolia Agricultural University, Hohhot, China [
29]). After inactivation, it was mixed with an equal volume of sterilized Montanide ISA206 (SEPPIC, Colombes, France) adjuvant. A 5 mL volume of blood was collected from the jugular vein of the calf and allowed to stand until it naturally coagulated. Then, the serum was collected and stored at −20 °C and used as a negative control at a dilution of 1:1000. The calf was immunized 4 times in a 2-week interval. One week after each immunization, blood was collected from the jugular vein and placed at 4 °C to separate the serum for the ELISAs to evaluate the antibody titers. One week after the fourth immunization, 50 mL of blood was collected from the jugular vein of the calf, which was used to isolate peripheral blood mononuclear cells (PBMCs). The calf was housed and fed in the Laboratory Animal Center, College of Veterinary Medicine, Inner Mongolia Agricultural University, with free access to food and water. All experimental procedures were carried out in accordance with the agency and the national guidelines and regulations and were approved by the Experimental Animal Use and Care Committee of Inner Mongolia Agricultural University (approval number: NND2022023).
2.2. Construction and Screening of Phage Display Library
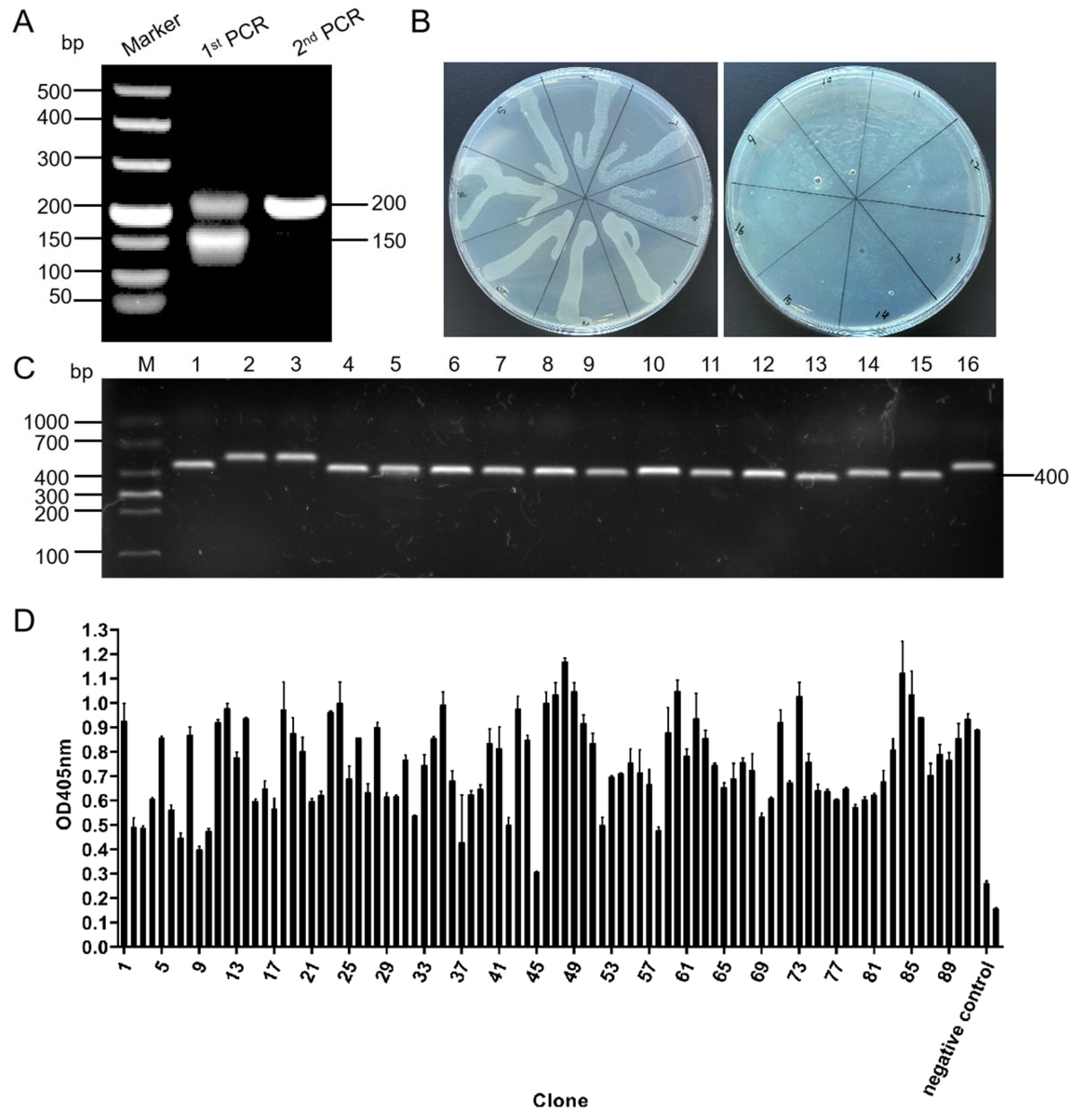
Calf PBMCs were isolated from the whole blood using bovine blood lymphocyte separation kits (Tianjin Haoyang Biological Products Technology Co., Ltd., Tianjin, China). The total RNA was extracted from the PBMCs using TRIzol reagent (Ambion, Austin, TX, USA), and RT-PCR amplification was performed using the One Step PrimeScript™ RT-PCR (Takara Bio, Shiga, Japan) reaction kit. The bovine ultra-long CDR H3 antibody fragment was amplified using nested PCR and the method used in the study by Joyce C and others, with some modifications [
30]. All the primers used in this study are listed in
Table 1. During the first round of PCR, primers P1 and P2, which anneal to the end of framework-3 and the head of framework-4 of the bovine immunoglobulin heavy chain, respectively, were used to amplify the fragment encompassing the complete bovine immunoglobulin heavy chain CDR H3 region. The PCR products were electrophoresed on agarose gel. The band containing the bovine ultra-long CDR H3 sequence was recovered by cutting it, extracting it from the gel, and using it as a template for the second round of PCR. The bovine ultra-long CDR H3 gene was amplified using the primer sets R311-R326. The PCR products were digested with the restriction enzymes
Nco I and
Not I (Takara Bio, Shiga, Japan) and ligated into the pMECS plasmid vector (a gift from Professor Serge Muyldermans, Vrije Universitaire Brussel, Belgium) with T4 ligase (Takara Bio, Shiga, Japan). The ligation product was transformed into
E. coli TG1 competent cells (GE Healthcare, Chicago, IL, USA) to construct the antibody library. The capacity of the antibody library was estimated by calculating the number of colony-forming units on the 2×YT-AG plate containing ampicillin and glucose by diluting the antibody library in a 10-fold dilution series. The transformation efficiency and ultra-long CDR H3 gene insertion rates were assessed using PCR and the sequencing primers MP57 and GIII for the pMECS plasmid vectors.
The antibody library was added to 200 mL of 2×YT medium, and the library was cultured in a shaker at 37 °C, 250 r/min for 2 h. The M13K07 helper phage was added (NBbiolab, Chengdu, China), and the culture was incubated at 37 °C for 1 h to allow for infection and rescue. The cultured bacteria solution was centrifuged at 4000× g for 10 min at 4 °C, the supernatant was discarded, and the bacteria pellet was resuspended with 2×YT-AK medium containing ampicillin and kanamycin and grown at 37 °C, 220 r/min overnight. The next day, the culture medium was centrifuged at 4 °C and 7197× g for 15 min; 20% PEG8000 was added to the supernatant, which was then centrifuged at 7197× g at 4 °C for 25 min to precipitate the phages. The pellet was resuspended in PBS to obtain the recombinant phage library.
The phage library was added to 5 mL immunotubes coated with BRV at the TCID50 (10−6.13/100 μL), incubated for 1 h at 37 °C, and then the bound phages were washed with 2 mL of PBS. The bound recombinant phage was used to infect E. coli TG1 (OD600nm = 0.6), and the M13K07 helper phage was added for rescue. The phages were harvested, purified, and used for a new round of enrichment. The E. coli TG1 infected with the bound phages from the final enrichment were grown on 2×YT-AG plates. Ninety-two clones were randomly picked from the 2×YT-AG plate, added to 2×YT-AG liquid medium, and incubated at 37 °C and 250 r/min overnight. The M13K07 helper phage was added and allowed to infect for 1 h before being centrifuged at 14,000 r/min for 5 min. The bacteria pellets were resuspended in 2×YT-AK medium and incubated at 37 °C and 250 r/min overnight. The medium was centrifuged at 14,000 r/min for 5 min; 20% PEG8000 was added to the supernatant, and the mixture was centrifuged at 7197× g for 30 min. The recombinant phage was introduced to an ELISA plate pre-coated with BRV at the TCID50 (10−6.13/100 L) and incubated for 2 h at room temperature to facilitate binding. The M13K07 helper phage was used as a negative control, and PBS buffer was used as a blank control. The anti-M13 bacteriophage secondary antibody (AlpSdAbs VHH, Chengdu, China) was diluted to a concentration of 1:5000 and added to each well and allowed to bind for 1 h at room temperature. A TMB solution was added for color development, and the OD405nm value was measured using a microplate reader. The absorbance of the experimental group/negative control ≥2.1 was regarded as a positive result.
2.3. Expression and Purification of the Bovine Ultra-Long CDR H3
The selected recombinant phages from the phage ELISA carrying the bovine ultra-long CDR H3 gene were ligated to the expression vector and transformed for expression, purification, and identification. The plasmids of the positive clones were isolated from the phage ELISA and digested with the restriction enzymes Nco I and Not I. The digested bovine ultra-long CDR H3 gene fragments were ligated into the pET-22b (+) plasmid using T4 DNA ligase. The ligation products were transformed into E. coli BL21 (DE3) competent cells (Sangon Biotech, Shanghai, China). The expression of recombinant ultra-long CDR H3 antibodies was induced by incubating the cells with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Solarbio Life Sciences, Beijing, China) for 12 h, and then the cells were collected and sonicated. The bacterial lysates were centrifuged, and the precipitates and supernatants were collected separately and analyzed using SDS-PAGE. Ni-NTA Sefinose™ Resin (Sangon Biotech, Shanghai, China) was used to purify the expressed recombinant ultra-long CDR H3 antibodies, and the recombinant protein was renatured with a gradient urea buffer from 8 M to 2 M and finally dialyzed in PBS after purification and analyzed using SDS-PAGE.
2.4. Binding Activity and Specificity of the Bovine Ultra-Long CDR H3 Antibodies
In order to determine the binding activity, the purified bovine ultra-long CDR H3 clones were diluted and added as the primary antibody to a 96-well plate coated with BRV at the TCID50 (10−5.41/100 μL). At the same time, the sonicated E. coli BL21 (DE3) lysate was used to transform the empty pET-22b (+) vector and used as the negative control, while PBS buffer was used as the blank control. The HRP-conjugated 6*His-tag mouse monoclonal antibody (Proteintech Group, Wuhan, China) was used as the secondary antibody at a concentration of 1:10,000. A TMB solution was added for color development, and the OD450nm value was measured using a microplate reader.
2.5. Immunofluorescence
MA-104 African green monkey fetal kidney cells were added to a 6-well cell culture dish at a concentration of 106 cells/well and grown at 37 °C and 5% CO2 for 24 h. The cell culture medium was discarded, and 2 mL of DMEM medium was added to each well. The BRV was diluted to the TCID50 (10−5.41/100 μL), and 2 mL of virus diluent was added to each well, and DMEM was added to the control group. The cells were cultured in an incubator for 2 h. The medium was discarded, and 1 mL of pre-cooled ice methanol was added to each well for 20 min at room temperature to fix the cells. The fixative was aspirated, and the cells were rinsed with DPBS 3 times for 5 min each time. A 2 mL volume of a 5% bovine serum albumin (BSA) solution was added to each well, and the dish was incubated at room temperature for 1 h before 2 mL of the recombinant ultra-long CDR H3 working solution (100 μg/mL) was added to each well. Then, the dish was incubated overnight in a refrigerator. The next day, the liquid was discarded, and the cells were rinsed with Dulbecco’s Phosphate-Buffered Saline (DPBS) 3 times for 5 min each. The CoraLite®488-conjugated 6*His His-Tag Mouse Monoclonal antibody (Proteintech Group, Wuhan, China) was used as the secondary antibody at a concentration of 1:500; 2 mL of the working solution of the secondary antibody was added to each well, and the dish was incubated at room temperature for 1 h, and rinsed with PBST 3 times for 5 min each time. A 200 μL volume of a ready-to-use 4′, 6-diamidino-2-phenylindole (DAPI) (Beyotime Biotechnology, Shanghai, China) working solution was added to each well, and the dish was incubated at room temperature for 4 min, and rinsed with PBST 3 times for 5 min each time. The fluorescence signal was detected using a laser scanning confocal microscope (ZEISS LSM-800, Oberkochen, Germany).
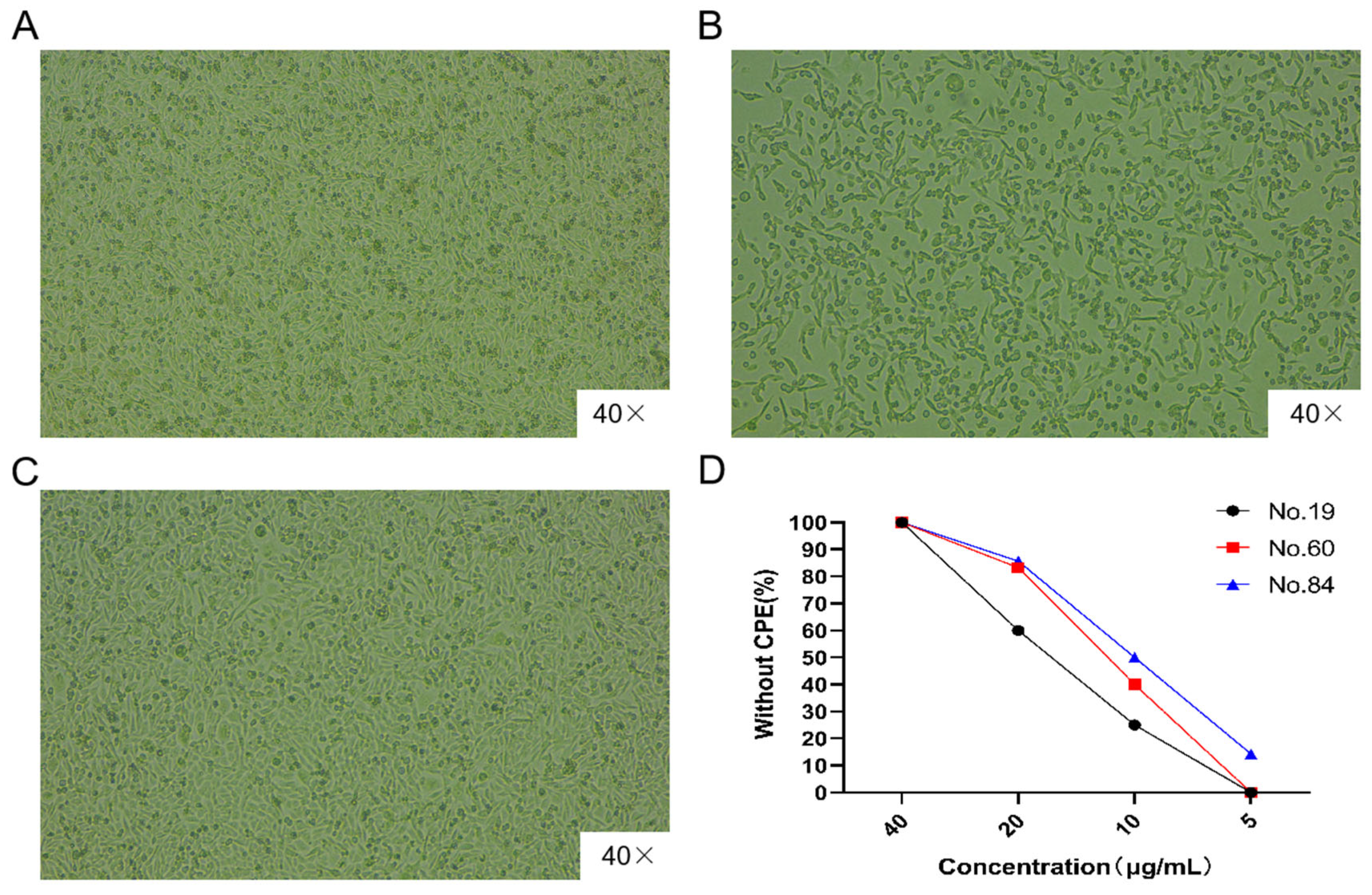
2.6. Neutralizing Activity of the Bovine Ultra-Long CDR H3
MA-104 cells were incubated at a density of 8000 cells/well in a 96-well cell culture dish at 37 °C and 5% CO2 for 24 h. BRV was diluted to the TCID50 (10−5.41/100 μL), and 100 μL of virus diluent was mixed with an equal volume of purified recombinant ultra-long CDR H3 clone 84 to final concentrations of 5, 10, 20, and 40 μg/mL. The antibody and virus mixtures were incubated at 37 °C and 5% CO2 for 1 h and then added to the 96-well cell culture dish and incubated for 3–5 days to observe the CPEs on the cells. At the same time, PBS + BRV was used as the positive infection control, and MA-104 cells were used as the untreated, uninfected blank group.
2.7. The Suckling Rat Challenge Model
2.7.1. BRV Challenge Test in Suckling Rats
We planned to utilize the suckling rat as a model for BRV infection to verify the effect of the recombinant ultra-long CDR H3 antibodies following similar methodologies to studies using animal models for viral infection research [
31,
32]. Female Wistar rats at five months of age were obtained from the Inner Mongolia Medical University. To avoid the effects of biological differences between the dams and their respective pups, the pups were shuffled before all experiments, and the litter size was adjusted to five pups per dam. The control and inoculated rats were housed separately in the Laboratory Animal Center of the College of Veterinary Medicine, Inner Mongolia Agricultural University, with free access to food and water. Five-day-old Wistar rats were weighed and divided into two groups: the BRV and PBS groups, with five rats in each group. BRV was orally administered to the BRV group at a dose of 100 μL/g (10
−5.41/100 μL (TCID
50)) according to the weight of the rats at 0 h. The rats in the PBS group were also orally administered with PBS at a dose of 100 μL per gram of body weight. The body weight of each rat and the occurrence of diarrhea in each group were monitored from 0 h to 156 h. The rats in the BRV and PBS groups were euthanized, and the spleen and small intestine were sampled at 24, 48, 84, 132, and 156 h.
2.7.2. PCR Detection of Virus
The virus genome RNA was extracted from the spleen and small intestine of the rats using the Viral Genome DNA/RNA Extraction Kit (Spin Column Type) (TIANGEN, Beijing, China). The primers BRV-F and BRV-R (
Table 1) were used to amplify the BRV nonstructural protein 5 (
NSP5) gene [
33] using the One Step PrimeScript™ RT-PCR Reaction Kit (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions. The PCR products were analyzed using electrophoresis on a 2% agarose gel and purified to recover the products. These were ligated into the pMD19-T vector to construct the recombinant plasmid pMD19-T-NSP5.
2.7.3. RT-qPCR
The recombinant plasmid pMD19-T-NSP5 was diluted ten-fold from 10
10 copies to 10
1 copies and used as a template along with the primers BRV-F and BRV-R and BRV-Probe [
33] to perform RT-qPCR using the One Step PrimeScript™ RT-PCR reaction kit (Perfect Real Time) (Takara Bio, Shiga, Japan). The reaction system consisted of the following: 12.5 μL of 2×One Step RT-PCR Buffer III, 0.5 μL of ExTaq HS, 0.5
L of PrimeScript RT Enzyme Mix II, 1 μL of BRV-Probe (10
M), 1.0 μL each of the BRV-F/BRV-R primers (10 μM), and 2.0 μL of template, which was brought to a final volume of 25.0 μL using nuclease-free water (ddH
2O). The RT-qPCR reaction conditions were as follows: initial reverse transcription at 42 °C for 5 min, followed by PCR activation at 95 °C for 10 s, and 45 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 30 s. After the reaction, a standard curve was plotted using Excel.
The total RNA from spleen and small intestine samples from the suckling rats was extracted and used as a template, along with the primers BRV-F and BRV-R and BRV-Probe [
33], to perform RT-qPCR using the same reaction as above to obtain the Cq value and perform an absolute quantitative analysis.
2.7.4. Histopathological Assessment of Rat Intestine Slices
Intestinal samples obtained from the small intestines of the rats in each group were placed in tissue embedding cassettes and immersed in a 4% paraformaldehyde (PFA) solution in a screw-cap bottle and incubated at room temperature for 24 h before rinsing with water for 8 h. The tissue samples underwent a stepwise dehydration process using graded ethanol concentrations (70%, 80%, 90%, 95%, 100%, and 100%), followed by treatment with xylene. Subsequently, the samples were infiltrated with soft wax and hard wax, each for a duration of 50 min. The tissues were cut into 5 μm thick sections with a microtome (Kedi KD-202A rotary microtome, Hangzhou, China), and the sections were floated in a water bath at 60 °C until they became smooth on the water surface before they were carefully lifted onto a glass slide (Citotest Labware Manufacturing, Jiangning, China). The glass slides were dried on a heater until the sections had fully spread out. The sections were stained with HE (Beyotime Biotechnology, Shanghai, China) and observed using an optical microscope (NIKON ECLIPSE Ts2, Tokyo, Japan) to check for intestinal epithelial cell damage, inflammation, and vacuolization in the villi.
2.8. Calves Challenge Model
2.8.1. BRV Challenge Test and Treatment of Calves
The twelve 5-day-old Holstein male calves that were used for the BRV challenge were obtained from a dairy farm in the suburbs of Hohhot city, Inner Mongolia Autonomous Region, China, and raised in the Laboratory Animal Center of the College of Veterinary Medicine, Inner Mongolia Agricultural University. Twelve Holstein male calves were randomly divided into three groups: a BRV group (calves No. 1–4), an antibody group (calves No. 5–8.), and a PBS group (calves No. 9–12), with four calves in each group. They were fed 2 L of calf milk replacer (Inner Mongolia Knight Dairy Group Co., Ltd., Hohhot, China) twice a day.
The calves in the BRV and antibody groups were orally administered 10 mL of BRV (10
−5.41/100 μL (TCID
50)). The calves in the PBS group were orally administered 10 mL of PBS. After administration, the calves were observed at different time points, and the clinical symptoms were recorded. The evaluation of clinical symptoms included measuring the rectal temperature, mental state, feeding status, and occurrences of diarrhea. Fecal samples from the calves in each group were regularly collected, and the fecal score was recorded according to the scoring system shown in
Supplementary Table S1. The BRV-infected calves in the antibody group received intravenous injection of the recombinant bovine ultra-long CDR H3 clones 60 and 84 at a dose of 1 mg/kg antibody protein filtered into 500 mL of physiological saline, once a day for a total of six days. The calves that died during the experimental procedure were dissected and sampled immediately after death. Except for the animals that died during the experiment, the animals in the PBS, BRV, and antibody groups in the other experiments were all euthanized at the end of the experimental period. This process was carried out in accordance with the guidelines and regulations of the relevant institutions and the state and was approved by the Experimental Animal Use and Care Committee of Inner Mongolia Agricultural University.
2.8.2. RT-qPCR Detection of Virus in Calf Fecal Samples
A total of 0.2 g of the collected fecal sample was placed in 1 mL of PBS, frozen and thawed three times, filtered, and centrifuged at 12,000 r/min for 1 min to collect the supernatant. The virus genomic RNA was extracted using a virus genomic DNA/RNA extraction kit (TIANGEN, Beijing, China). The virus genomic RNA was used as a template to perform RT-qPCR (the reaction system was the same as that of the RT-qPCR system used for the Wistar rat samples) to obtain the Cq value and perform an absolute quantitative analysis. One calf from each group was randomly selected, and its RT-qPCR product was run on a 2% agarose gel.
2.8.3. Stability of Anti-BRV Recombinant Bovine Ultra-Long CDR H3 Antibodies in Bovine Serum
The stability of the recombinant antibodies was assessed by incubating them with bovine serum at varying durations to monitor their binding activity to BRV using ELISAs. Briefly, 7.5 μg of the recombinant bovine ultra-long CDR H3 antibody was incubated with bovine serum at 37 °C. The incubated samples were harvested at 3, 6, 12, 24, and 48 h and frozen at −20 °C. The 0 h samples were immediately frozen at −20 °C after the addition of serum and served as the control group. The samples at each time point were used as primary antibodies and were added to 96-well Stripwell™ microplates precoated with 100 μL of BRV (10−5.9/100 μL (TCID50)). The secondary antibody was an HRP-conjugated 6*His-tagged mouse monoclonal antibody (Proteintech Group, Wuhan, China) at a concentration of 1:10,000. After adding the TMB substrate, the plate was read at OD450 nm using a microplate reader.
2.8.4. Histopathological Assessment of Calf Intestine Slices
The histopathological assessment of the calves intestine slice was the same as the steps for the Wistar rat samples.
2.9. Statistical Analysis
GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used for the statistical analysis. Each experiment was repeated independently three times, and the measurement data are expressed as the mean ± SD. Two-way ANOVA was used to analyze the differences between multiple groups. * p values < 0.05 were considered to be statistically significant.
4. Discussion
Calf diarrhea is a major problem affecting the development of the cattle industry. The causes of calf diarrhea are complicated and are mainly divided into infectious and non-infectious factors. Non-infectious factors are usually attributed to the level of feeding and management. At present, large pastures provide better control over calf diarrhea caused by non-infectious factors [
35]. As for infectious factors, a variety of pathogens have been identified, including rotavirus and
E. coli [
36]. Calves are highly susceptible to BRV. After being infected, the main symptom is diarrhea, accompanied by an elevated body temperature, depression, anorexia, increased salivation, abdominal pain, and diarrhea. The excreta are watery and contain mucus and blood. It can also cause various complications in the body. Moreover, calves will excrete the virus for life after infection. In severe cases, it can lead to the death of the calves [
37].
Bovine ultra-long CDR H3 is the smallest antibody fragment known to be capable of binding to antigens. Compared with typical antibodies, bovine ultra-long CDR H3 antibodies have a longer complementarity-determining region, which provides a larger contact surface for antigen–antibody interactions. It can specifically recognize and bind to relatively concealed antigenic sites, whereas the Fab of traditional antibodies and scFv typically can only recognize the surface-exposed sites of the antigens. This characteristic enables bovine ultra-long CDR H3 antibodies to possess a broad and strong antigen-binding ability even in the absence of light chains. Besides the above advantages, bovine-derived ultra-long CDR H3 antibodies have a smaller structure compared to other antibodies. They can better enter and fuse with the body through the knob region. After separation, they behave like a peptide rich in disulfide bonds, and their molecular weight is approximately one-third that of a single-domain antibody. Such antibodies have a relatively strong antigen recognition ability and a relatively high affinity. Due to the high diversity and strong affinity of the bovine ultra-long CDR H3 structure [
38], it has great potential in the development of new antibodies. Since the light chain of the bovine ultra-long CDR H3 does not participate in antigen binding and is only used to maintain the antibody structure, only the mispairing problem of the heavy chain needs to be addressed when constructing bispecific antibodies. Researchers developed a bispecific antibody targeting EGFR/NKp30 based on the above principle, providing a new direction for the construction of bispecific antibodies in the future [
39]. Given the conformational characteristics and size (>50 aa) of the ultra-long CDR H3 region in bovine antibodies, this feature can be utilized to develop small-molecule antibodies [
40].
The reason for the formation of the bovine ultra-long CDR H3 is due to the limited number of antibody gene compositions in cows compared with mice and humans, as well as other vertebrates. There are only 12 functional
VH, 23
DH, and 4
JH gene fragments in cows compared to humans, who have 36–49
VH, 23
DH, and 6
JH gene fragments. In terms of diversity, theoretically, humans have as many as 6700 possible VDJ combinations, compared with just over 1000 for bovines. This makes the diversity in cows much lower, so cows develop ultra-long CDR H3 antibodies to maximize their antibody diversity [
41].
The current management of BRV-induced calf diarrhea predominantly relies on antibiotic therapy. However, escalating antimicrobial resistance and regulatory restrictions on antibiotic usage have heightened treatment risks, while the absence of specific antibody-based therapeutics underscores the urgent need for targeted anti-BRV immunotherapeutics as a promising preventive strategy. In this context, bovine ultra-long CDR H3 antibodies and chicken egg yolk IgY antibodies exhibit complementary advantages in passive immunotherapy for calves. The structural configuration of bovine ultra-long CDR H3 facilitates tissue penetration, enabling deep infiltration into target tissues for therapeutic action [
14]. Phage display technology allows for the efficient screening of high-affinity CDR H3 variants, supporting the rapid development of pathogen-specific therapeutic molecules against bovine pathogens. Nevertheless, the current research remains constrained to in vitro screening, preliminary in vivo models, and delivery optimization, with insufficient exploration of neutralization mechanisms in complex physiological environments and a notable absence of long-term efficacy data [
30]. In contrast, chicken IgY antibodies demonstrated utility in clinical applications and scalable production. In studies by Vega C and others, oral IgY administration to a BRV infection model was found to effectively reduce the diarrhea duration, viral shedding, and clinical manifestations including fever, dehydration, and anorexia [
30]. Produced through the mass immunization of hens, spray-dried IgY from egg yolk exhibits enhanced stability and significantly lower production costs compared to bovine ultra-long CDR H3 antibodies.
Bovine ultra-long CDR H3 antibodies hold substantial promise as novel therapeutics owing to their low immunogenicity and precise targeting capabilities, although their in vivo mechanisms of action warrant further elucidation. Chicken IgY antibodies, with their demonstrated clinical efficacy and cost-effectiveness, may serve as adjuncts to colostrum replacement strategies, although they will require careful consideration of the potential systemic immunosuppression risks. These two antibody platforms demonstrate complementary technical and applicational profiles, with the optimal approach contingent upon the therapeutic objectives, economic constraints, and operational environment.
In this study, the results showed that after challenge with BRV, the suckling rats did not exhibit the typical symptoms such as diarrhea or weight loss. Further analysis of the RT-qPCR data indicated that no BRV infection was detected in the small intestines and internal organs of the suckling rats. Although previous studies have successfully used suckling mice as a BRV challenge model, our results indicate that this was not the case in this study. This difference may be due to the use of different virus strains and the pathogenic properties of the virus. In view of this, we ultimately chose to directly use calves as the BRV challenge animals. The experimental results showed that the calves exhibited relatively obvious disease symptoms.
In this study, the treatment regimen involved intravenously injecting recombinant antibodies daily (1 mg/kg, once a day for 6 consecutive days). Despite the inherent instability and degradation of antibodies in bovine serum, continuous supplementation with exogenous antibodies through daily dosing ensured the maintenance of effective antibody concentrations in vivo, thus mitigating the problem of insufficient serum stability and compensating for the short half-life of the antibodies. This guaranteed that the antibody levels remained above the virus neutralization threshold during the critical period of virus replication. As can be seen from the experimental results, after the fourth day, compared with the BRV-infected calves, the antibody-treated calves showed a gradual alleviation of their diarrhea symptoms, a restoration of appetite, and a significant reduction in their viral load, as detected by RT-qPCR. These results indicate that although the serum half-life of a single antibody dose is limited, daily antibody supplementation can continuously inhibit virus replication, reduce intestinal lesions, and ultimately achieve a lasting protective effect within 6 days.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}