
Inflammasome-Mediated Neuroinflammation: A Key Driver in Alzheimer’s Disease Pathogenesis

 ,
,  and
and 
Abstract
1. Introduction
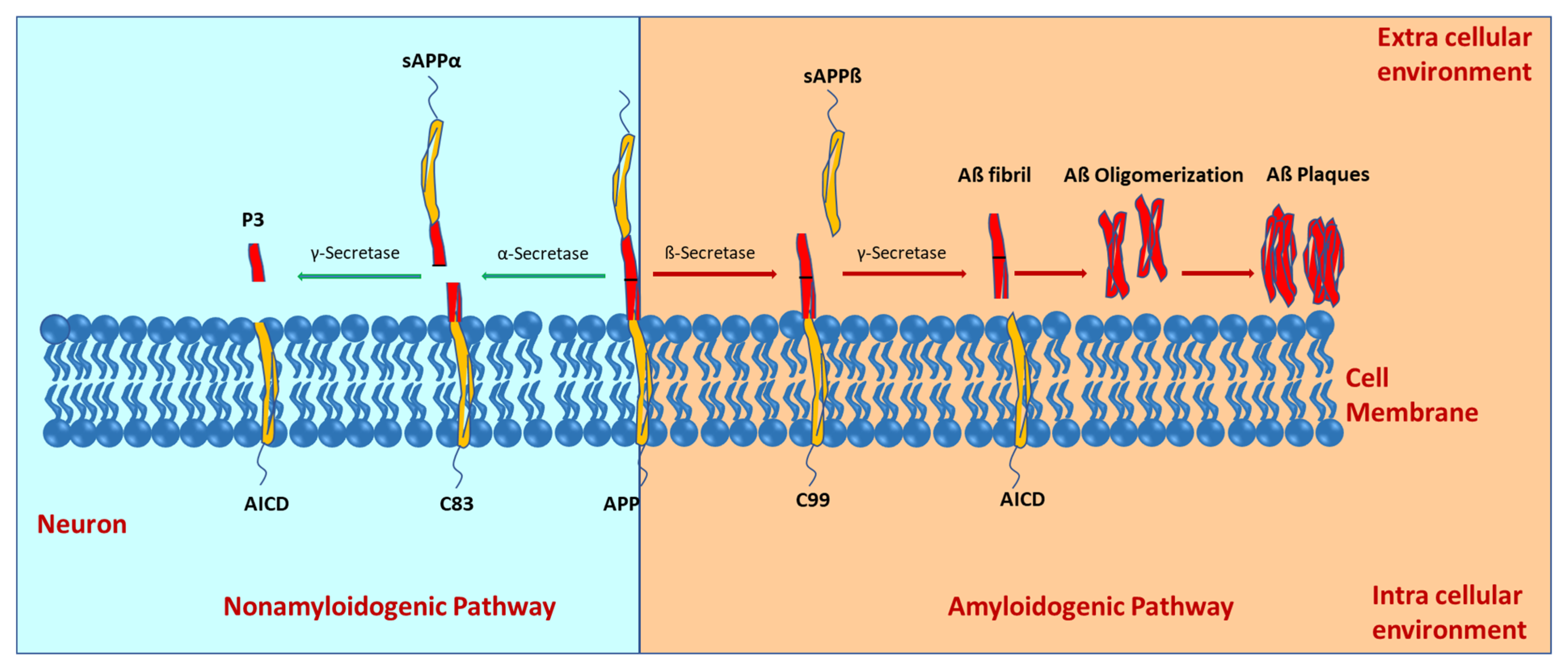
2. Molecular Mechanisms of Alzheimer’s Disease Development
3. Systemic Inflammation and Microglial Dynamics in AD Progression
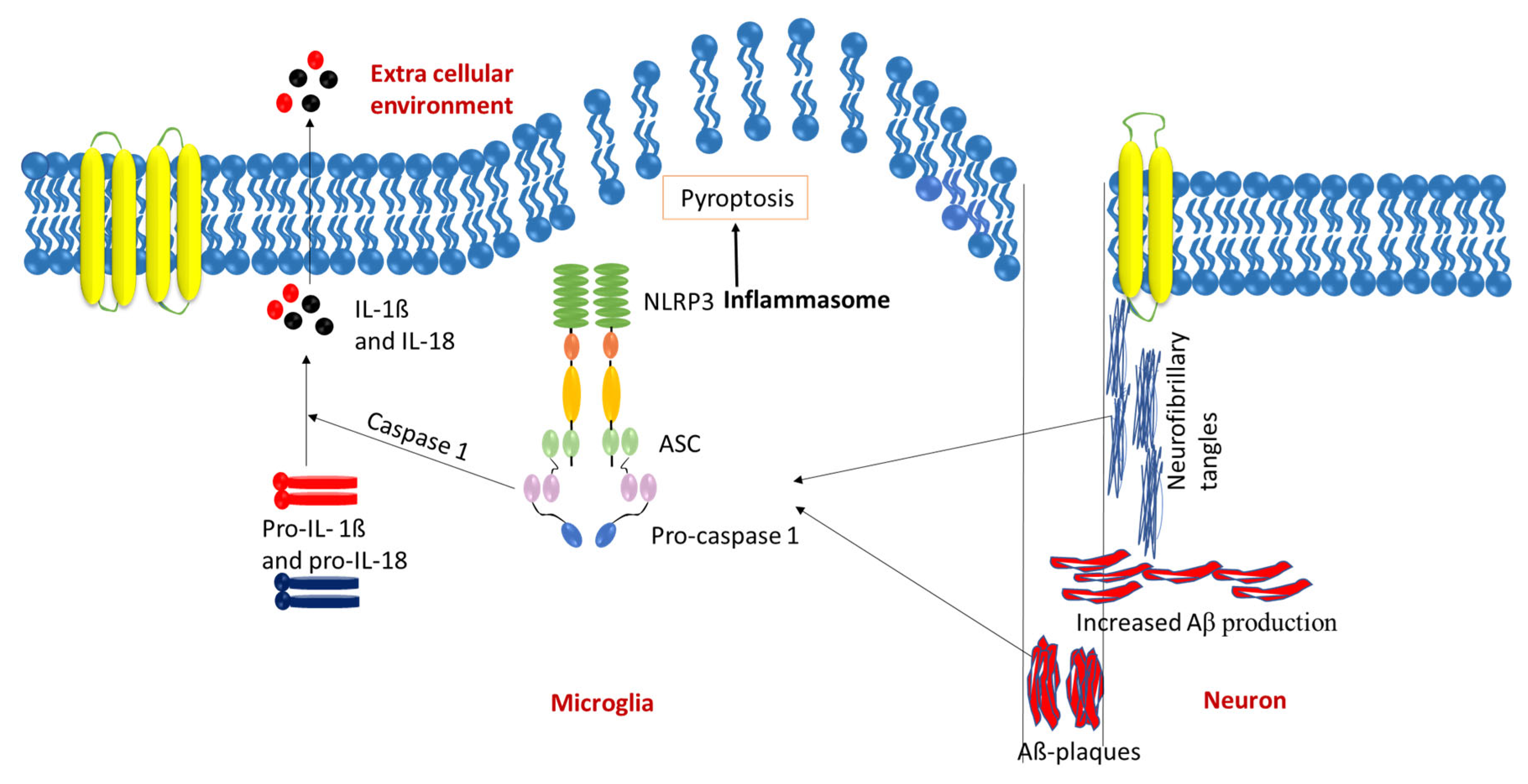
4. NLRP3 Inflammasome in AD Pathology
5. Therapeutic Molecules Targeting Inflammasomes in Alzheimer’s Disease

{kind=link}
{kind=link}
| Inhibitor | Class | Mechanism of Action | AD-Specific Effects/Findings |
|---|---|---|---|
| Thioredoxin-1 [52] | Antioxidant protein | Inhibits NLRP1-mediated pyroptosis, linked to NLRP3 activity | Reduces neuronal death and neuroinflammation in AD models |
| Methylchamaejasmin [53] | Biflavonoid (natural) | Suppresses NLRP3 activation, reducing inflammatory response | Anti-inflammatory effects in cell models of neuroinflammation relevant to AD |
| Colchicine (SBN-284) [54] | Colchicine hybrid | Inhibits cholinesterase and suppresses NLRP3 activation | Promising candidate for addressing cholinergic dysfunction and neuroinflammation in AD |
| Acacetin [60] | Flavonoid (natural) | Blocks NLRP3 via MAPK/NF-κB pathway, reduces oxidative stress | Decreases inflammation in AD-relevant models |
| Echinatin [61] | Flavonoid (natural) | Inhibits NLRP3 activation, reducing neuroinflammation | Protects neuronal health in AD models |
| Ghrelin [62] | Hormone | Upregulates autophagy, inhibits NLRP3 activation | Improves cognitive function in AD models by reducing inflammation |
| Beta-hydroxybutyrate (BHB) [63,64] | Ketone body | Reduces NLRP3 activity, modulates neuroinflammation | Neuroprotective in AD models, influences metabolic pathways |
| Gardenia jasminoides extract [55] | Natural compound | Inhibits NLRP3 activation | Attenuates memory impairment in AD models |
| Gastrodin [56] | Natural compound | Suppresses NLRP3 activation | Provides neuroprotection and alleviates neuropathic pain in AD models |
| Oridonin [72,73] | Natural compound (diterpenoid) | Covalently inhibits NLRP3, blocking NEK7 interaction and inflammasome activation | Reduces synaptic loss, neuroinflammation, and cognitive deficits in AD models |
| Salvianolic acid B [57] | Natural compound | Modulates macrophage polarization (M1 to M2), reduces NLRP3 activation | Anti-inflammatory effects in AD models |
| Vitenegu acid [58] | Natural compound | Specifically inhibits NLRP3 oligomerization | Potential candidate to suppress inflammasome activation in AD |
| Epigallocatechin-3-gallate (EGCG) [59] | Polyphenol (natural) | Inhibits NLRP3 via ROS/TXNIP/NLRP3 pathway, reduces oxidative stress | Neuroprotective effects in AD models by reducing inflammation and oxidative stress |
| Resveratrol [74] | Polyphenol (natural) | Modulates NLRP3 and neuroinflammatory pathways, antioxidative | Reduces inflammation and activates protective mechanisms in AD models |
| PCSK9 [66] | Protein target | Reduces NLRP3 activation when knocked out | Decreases Aβ deposition and neuroinflammation in AD mouse models |
| NT-0796 [67] | Small molecule | Blocks NLRP3 oligomerization, preventing inflammasome assembly and cytokine release | Reduces neuroinflammation in preclinical AD models, brain-penetrant with therapeutic promise |
| Tranilast [70,71] | Small molecule | Inhibits NLRP3 by binding the NACHT domain, reducing inflammasome activation | Alleviates neuroinflammation and cognitive deficits in AD mouse models |
| Glibenclamide [61] | Sulfonylurea | Inhibits NLRP3 activation via ATP-sensitive potassium channels (K_ATP) | Inhibits NLRP3 and promotes autophagy in AD |
| BPBA [65] | Synthetic hybrid | Inhibits Aβ aggregation and NLRP3 activation (benzothiazole + o-aminobenzoic acid) | Reduces Aβ oligomers and IL-1β levels and improves cognition in AD mouse models |
| Nicotinamide mononucleotide (NMN) [69] | Synbiotic | Enhances mitochondrial function, reduces NLRP3 activation | Improves cognition and reduces neuroinflammation in preclinical AD models |
| Extracellular vesicles (EVs) [75] | Nanotechnology (hiPSC-derived) | Modulates microglial activation, reduces NLRP3 activity | Preserves cognitive function by altering microglial inflammatory profile in AD models |
| Adiponectin (gene therapy) [76] | Gene therapy | Increases adiponectin expression, reduces NLRP3 activation | Improves cognitive outcomes and reduces neuroinflammation in AD models |
6. Inflammasome Inhibitors in Clinical Trials
| Inhibitor | Class | Mechanism of Action | AD-Specific Effects/Findings |
|---|---|---|---|
| Canakinumab [84] | Monoclonal antibody | Neutralizes IL-1β, a key NLRP3 inflammasome product, reducing neuroinflammation | Completed Phase II trial (NCT04795466) for cognition in MCI/mild AD [84] |
| Anakinra [82,83] | Recombinant protein | Blocks IL-1β receptor, inhibiting NLRP3-driven inflammatory signaling | Preclinical reduction in IL-1β signaling in APP/PS1 AD models; no active AD trials, tested in other conditions [82,83] |
| Sargramostim [87] | Recombinant protein | Stimulates GM-CSF, suppresses proinflammatory cytokines, enhances microglial Aβ phagocytosis | Completed Phase II trial (NCT01409915), ongoing Phase II (NCT04902703) for mild/moderate AD; reduces inflammation [87] |
| Dapansutrile [77,78] (OLT1177) | Small molecule | Selectively inhibits NLRP3, reducing inflammasome activation and cytokine release | Completed Phase II trial for gout (NCT02104050); reduces IL-1β, microglial activation, and Aβ plaques in APP/PS1 AD models; no AD trials [77,78] |
| VX-765 (Belnacasan) [79,80,81,82] | Small molecule | Inhibits caspase-1, blocking NLRP3-mediated IL-1β and IL-18 activation, BBB-penetrant | Completed Phase II trial for epilepsy (e.g., NCT01048255); reduces IL-1β, Aβ deposition, and improves cognition in AD models; no AD trials [79,80,81] |
| Lenalidomide [85,86] | Thalidomide derivative | Reduces proinflammatory cytokines (TNF-α, IL-1β, IL-6, IL-12) linked to inflammasomes | Ongoing MCLENA-1 Phase II trial (NCT04032626) for aMCI due to AD; aims to reduce neuroinflammation and cognitive decline [85,86] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villegas, S.; Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef] [PubMed]
- Husemann, J.; Loike, J.D.; Anankov, R.; Febbraio, M.; Silverstein, S.C. Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia 2002, 40, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Cescato, M.; Zhu, Y.Y.J.; Le Corre, L.; Py, B.F.; Georgin-Lavialle, S.; Rodero, M.P. Implication of the LRR Domain in the Regulation and Activation of the NLRP3 Inflammasome. Cells 2024, 13, 1365. [Google Scholar] [CrossRef]
- McManus, R.M.; Latz, E. NLRP3 inflammasome signalling in Alzheimer’s disease. Neuropharmacology 2024, 252, 109941. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, Y.; Zhang, Q.; Liu, H.; Liu, Y. The Potential Roles of Exosomes Carrying APP and Tau Cleavage Products in Alzheimer’s Disease. J. Clin. Med. 2023, 12, 1883. [Google Scholar] [CrossRef]
- Suh, Y.-H.; Checler, F. Amyloid Precursor Protein, Presenilins, and α-Synuclein: Molecular Pathogenesis and Pharmacological Applications in Alzheimer’s Disease. Pharmacol. Rev. 2002, 54, 469–525. [Google Scholar] [CrossRef]
- Hur, J.-Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Azargoonjahromi, A. The duality of amyloid-β: Its role in normal and Alzheimer’s disease states. Mol. Brain 2024, 17, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Kabir, T.; Rahman, S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef]
- Schoonhoven, D.N.; Coomans, E.M.; Millán, A.P.; van Nifterick, A.M.; Visser, D.; Ossenkoppele, R.; Tuncel, H.; van der Flier, W.M.; Golla, S.S.V.; Scheltens, P.; et al. Tau protein spreads through functionally connected neurons in Alzheimer’s disease: A combined MEG/PET study. Brain 2023, 146, 4040–4054. [Google Scholar] [CrossRef]
- Guan, P.-P.; Cao, L.-L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Li, B.; Shi, J.; Xu, J.; Yuan, M. Targeting Mitochondrial Dysfunction in Neurodegenerative Diseases: Expanding the Therapeutic Approaches by Plant-Derived Natural Products. Pharmaceuticals 2023, 16, 277. [Google Scholar] [CrossRef]
- Cheignon, C.; Collin, F.; Sabater, L.; Hureau, C. Oxidative Damages on the Alzheimer’s Related-Aβ Peptide Alters Its Ability to Assemble. Antioxidants 2023, 12, 472. [Google Scholar] [CrossRef]
- Denechaud, M.; Geurs, S.; Comptdaer, T.; Bégard, S.; Garcia-Núñez, A.; Pechereau, L.-A.; Bouillet, T.; Vermeiren, Y.; De Deyn, P.P.; Perbet, R.; et al. Tau promotes oxidative stress-associated cycling neurons in S phase as a pro-survival mechanism: Possible implication for Alzheimer’s disease. Prog. Neurobiol. 2022, 223, 102386. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Early-onset Alzheimer Disease and Its Variants. Contin. Lifelong Learn. Neurol. 2019, 25, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Aloi, M.S.; Prater, K.E.; Sánchez, R.E.A.; Beck, A.; Pathan, J.L.; Davidson, S.; Wilson, A.; Keene, C.D.; de la Iglesia, H.; Jayadev, S.; et al. Microglia specific deletion of miR-155 in Alzheimer’s disease mouse models reduces amyloid-β pathology but causes hyperexcitability and seizures. J. Neuroinflammation 2023, 20, 60. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Heath, L.; Preuss, C.; Jayadev, S.; Garden, G.A.; Greenwood, A.K.; Sieberts, S.K.; De Jager, P.L.; Ertekin-Taner, N.; Carter, G.W.; et al. Molecular estimation of neurodegeneration pseudotime in older brains. Nat. Commun. 2020, 11, 5781. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, A.B.; Hennessy, E.; Murray, C.L.; Nazmi, A.; Delaney, H.J.; Healy, D.; Fagan, S.G.; Rooney, M.; Stewart, E.; Lewis, A.; et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer’s disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimer’s Dement. 2021, 17, 1735–1755. [Google Scholar] [CrossRef]
- He, X.-F.; Xu, J.-H.; Li, G.; Li, M.-Y.; Li, L.-L.; Pei, Z.; Zhang, L.-Y.; Hu, X.-Q. Correction to: NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2022, 13, 489. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2019, 155, 650–661. [Google Scholar] [CrossRef]
- Nakanishi, A.; Kaneko, N.; Takeda, H.; Sawasaki, T.; Morikawa, S.; Zhou, W.; Kurata, M.; Yamamoto, T.; Akbar, S.M.F.; Zako, T.; et al. Amyloid β directly interacts with NLRP3 to initiate inflammasome activation: Identification of an intrinsic NLRP3 ligand in a cell-free system. Inflamm. Regen. 2018, 38, 27. [Google Scholar] [CrossRef]
- Hwang, C.J.; Choi, D.-Y.; Park, M.H.; Hong, J.T. NF-κB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol. Disord.–Drug Targets 2019, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Atluri, V.S.R.; Tiwari, S.; Rodriguez, M.; Kaushik, A.; Yndart, A.; Kolishetti, N.; Yatham, M.; Nair, M. Inhibition of Amyloid-Beta Production, Associated Neuroinflammation, and Histone Deacetylase 2-Mediated Epigenetic Modifications Prevent Neuropathology in Alzheimer’s Disease in vitro Model. Front. Aging Neurosci. 2020, 11, 342. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Liu, J.; Yao, Z.; Xiao, Y.; Zhang, X.; Zhang, Y.; Xu, J. NF-κB-Induced Upregulation of miR-146a-5p Promoted Hippocampal Neuronal Oxidative Stress and Pyroptosis via TIGAR in a Model of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 653881. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Milner, M.T.; Maddugoda, M.; Götz, J.; Burgener, S.S.; Schroder, K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr. Opin. Immunol. 2021, 68, 116–124. [Google Scholar] [CrossRef]
- Van Zeller, M.; Dias, D.M.; Sebastião, A.M.; Valente, C.A. NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 939–961. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Mirioglu, S.; Uludag, O.; Hurdogan, O.; Kumru, G.; Berke, I.; Doumas, S.A.; Frangou, E.; Gul, A. AA Amyloidosis: A Contemporary View. Curr. Rheumatol. Rep. 2024, 26, 248–259. [Google Scholar] [CrossRef]
- Losa, M.; Emmenegger, M.; De Rossi, P.; Schürch, P.M.; Serdiuk, T.; Pengo, N.; Capron, D.; Bieli, D.; Bargenda, N.; Rupp, N.J.; et al. The ASC inflammasome adapter governs SAA-derived protein aggregation in inflammatory amyloidosis. EMBO Mol. Med. 2024, 16, 2024–2042. [Google Scholar] [CrossRef]
- Ye, L.; Hu, M.; Mao, R.; Tan, Y.; Sun, M.; Jia, J.; Xu, S.; Liu, Y.; Zhu, X.; Xu, Y.; et al. Conditional knockout of AIM2 in microglia ameliorates synaptic plasticity and spatial memory deficits in a mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2023, 30. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; Zaheer, A. Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 2017, 60, 1143–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gai, Y.; Liu, Y.; Meng, D.; Zeng, Y.; Luo, Y.; Zhang, H.; Wang, Z.; Yang, M.; Li, Y.; et al. Tau induces inflammasome activation and microgliosis through acetylating NLRP3. Clin. Transl. Med. 2024, 14, e1623. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Li, Y.; Liu, X.; Liang, W.; Li, Y.; Wu, L.; Wang, Z.; Song, W. FUBP3 mediates the amyloid-β-induced neuronal NLRP3 expression. Neural Regen. Res. 2024, 20, 2068–2083. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Hu, D.; Sun, X.; Fujioka, H.; Lundberg, K.; Chan, E.R.; Wang, Q.; Xu, R.; Flanagan, M.E.; et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 2020, 6, eabb8680. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Hoyle, C.; Pointer, A.; Lawrence, C.; Pickering-Brown, S.; Brough, D.; Ryan, S. C9orf72 dipeptides activate the NLRP3 inflammasome. Brain Commun. 2024, 6, fcae282. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Zhao, N.; Geng, F.; Zhu, M.; Ren, Q. Synergism of ApoE4 and systemic infectious burden is mediated by the APOE-NLRP3 axis in Alzheimer’s disease. Psychiatry Clin. Neurosci. 2024, 78, 517–526. [Google Scholar] [CrossRef]
- Yu, Y.; Lv, J.; Ma, D.; Han, Y.; Zhang, Y.; Wang, S.; Wang, Z. Microglial ApoD-induced NLRC4 inflammasome activation promotes Alzheimer’s disease progression. Anim. Model. Exp. Med. 2024. [Google Scholar] [CrossRef]
- Kumar, S.; Budhathoki, S.; Oliveira, C.B.; Kahle, A.D.; Calhan, O.Y.; Lukens, J.R.; Deppmann, C.D. Role of the caspase-8/RIPK3 axis in Alzheimer’s disease pathogenesis and Aβ-induced NLRP3 inflammasome activation. J. Clin. Investig. 2023, 8, e157433. [Google Scholar] [CrossRef]
- Daily, K.P.; Badr, A.; Eltobgy, M.; Estfanous, S.; Whitham, O.; Tan, M.H.; Carafice, C.; Krause, K.; McNamara, A.; Hamilton, K.; et al. DNA hypomethylation promotes the expression of CASPASE-4 which exacerbates inflammation and amyloid-β deposition in Alzheimer’s disease. Alzheimer’s Res. Ther. 2024, 16, 29. [Google Scholar] [CrossRef]
- Flores, J.; Fillion, M.-L.; LeBlanc, A.C. Caspase-1 inhibition improves cognition without significantly altering amyloid and inflammation in aged Alzheimer disease mice. Cell Death Dis. 2022, 13, 864. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, H.; Sun, L.; Xu, Y.; Zeng, X. Thioredoxin-1 Protects Neurons Through Inhibiting NLRP1-Mediated Neuronal Pyroptosis in Models of Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 9723–9734. [Google Scholar] [CrossRef] [PubMed]
- Owona, B.A.; Mary, A.; Messi, A.N.; Ravichandran, K.A.; Mbing, J.N.; Pegnyemb, E.; Moundipa, P.F.; Heneka, M.T. Biflavonoid Methylchamaejasmin and Khaya grandifoliola Extract Inhibit NLRP3 Inflammasome in THP-1 Cell Model of Neuroinflammation. Mol. Neurobiol. 2024, 62, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.N.; Nuthakki, V.K.; Sharma, A.; Malik, S.; Tabassum, M.; Kumar, R.; Choudhary, S.; Iqbal, F.; Tufail, Z.; Mondhe, D.M.; et al. Synthesis and Biological Evaluation of Colchicine─Aryl/Alkyl Amine Hybrids as Potential Noncytotoxic Cholinesterase Inhibitors: Identification of SBN-284 as a Dual Inhibitor of Cholinesterases and NLRP3 Inflammasome. ACS Chem. Neurosci. 2024, 15, 2779–2794. [Google Scholar] [CrossRef]
- Wang, Y.; Gong, Q.; Pan, H.; Wang, X.; Yan, C. Gardenia jasminoides J. Ellis extract attenuates memory impairment in rats with Alzheimer’s disease by suppressing NLRP3 inflammasome. Brain Res. 2023, 1824, 148687. [Google Scholar] [CrossRef]
- Jin, J.; Kang, D.H.; Lee, G.H.; Kim, W.M.; Choi, J.I. Intrathecal gastrodin alleviates allodynia in a rat spinal nerve ligation model through NLRP3 inflammasome inhibition. BMC Complement. Med. Ther. 2024, 24, 213. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Liu, X.; Zhang, J.; Zhang, Y.; Wen, Y.; Yang, G. Salvianolic acid B exerts protective effects against Aβ-induced neuroinflammation through the inhibition of NLRP3 inflammasome activation and switching of M1/M2 polarization. Tissue Cell 2023, 85, 102260. [Google Scholar] [CrossRef]
- Di, Q.; Zhao, X.; Lin, J.; Li, X.; Li, X.; Tang, H.; Zhang, R.; Xiao, W.; Chen, W. A new acid isolated from V. negundo L. inhibits NLRP3 inflammasome activation and protects against inflammatory diseases. Front. Immunol. 2023, 14, 1174463. [Google Scholar] [CrossRef]
- Xiao, Y.; Yang, C.; Si, N.; Chu, T.; Yu, J.; Yuan, X.; Chen, X.-T. Epigallocatechin-3-gallate Inhibits LPS/AβO-induced Neuroinflammation in BV2 Cells through Regulating the ROS/TXNIP/NLRP3 Pathway. J. Neuroimmune Pharmacol. 2024, 19, 31. [Google Scholar] [CrossRef]
- Bu, J.; Mahan, Y.; Zhang, S.; Wu, X.; Zhang, X.; Zhou, L.; Zhang, Y. Acacetin inhibits inflammation by blocking MAPK/NF-κB pathways and NLRP3 inflammasome activation. Front. Pharmacol. 2024, 15, 1286546. [Google Scholar] [CrossRef]
- Kim, E.Y.; Im, J.H.; Han, J.; Cho, W.-J. Structure-based design and synthesis of sulfonylureas as novel NLRP3 inhibitors for Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2024, 99, 129622. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, L. Ghrelin inhibits NLRP3 inflammasome activation by upregulating autophagy to improve Alzheimer’s disease. Vitr. Cell. Dev. Biol.–Anim. 2023, 59, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Nomura, M.; Murad, N.F.; Madhavan, S.S.; Mu, W.-C.; Eap, B.; Garcia, T.Y.; Aguirre, C.G.; Verdin, E.; Ellerby, L.; et al. Ketogenic Diet Reduces Age-Induced Chronic Neuroinflammation in Mice. Aging Biol. 2024, 2, 20240038. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, L.; Shang, Y.; Zhu, Z.; Jin, S.; Guo, Z.; Wang, X. Concurrent suppression of Aβ aggregation and NLRP3 inflammasome activation for treating Alzheimer’s disease. Chem. Sci. 2022, 13, 2971–2980. [Google Scholar] [CrossRef]
- Vilella, A.; Bodria, M.; Papotti, B.; Zanotti, I.; Zimetti, F.; Remaggi, G.; Elviri, L.; Potì, F.; Ferri, N.; Lupo, M.G.; et al. PCSK9 ablation attenuates Aβ pathology, neuroinflammation and cognitive dysfunctions in 5XFAD mice. Brain, Behav. Immun. 2023, 115, 517–534. [Google Scholar] [CrossRef]
- Harrison, D.; Billinton, A.; Bock, M.G.; Doedens, J.R.; Gabel, C.A.; Holloway, M.K.; Porter, R.A.; Reader, V.; Scanlon, J.; Schooley, K.; et al. Discovery of Clinical Candidate NT-0796, a Brain-Penetrant and Highly Potent NLRP3 Inflammasome Inhibitor for Neuroinflammatory Disorders. J. Med. Chem. 2023, 66, 14897–14911. [Google Scholar] [CrossRef]
- Saresella, M.; Zoia, C.P.; La Rosa, F.; Bazzini, C.; Sala, G.; Grassenis, E.; Marventano, I.; Hernis, A.; Piancone, F.; Conti, E.; et al. Glibenclamide-Loaded Engineered Nanovectors (GNVs) Modulate Autophagy and NLRP3-Inflammasome Activation. Pharmaceuticals 2023, 16, 1725. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Xu, H.; Liu, X.; He, Y.; Gu, J. NMN Synbiotics: A Multifaceted Therapeutic Approach for Alzheimer’s Disease. Neurochem. Res. 2024, 49, 2888–2896. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP 3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Thapak, P.; Bishnoi, M.; Sharma, S.S. Tranilast, a Transient Receptor Potential Vanilloid 2 Channel (TRPV2) Inhibitor Attenuates Amyloid β-Induced Cognitive Impairment: Possible Mechanisms. NeuroMolecular Med. 2021, 24, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, L.; Yang, H.; Li, C.; Hui, Z.; Xu, Y.; Zhu, X. Oridonin Attenuates Synaptic Loss and Cognitive Deficits in an Aβ1–42-Induced Mouse Model of Alzheimer’s Disease. PLoS ONE 2016, 11, e0151397. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Bartra, C.; Yuan, Y.; Vuraić, K.; Valdés-Quiroz, H.; Garcia-Baucells, P.; Slevin, M.; Pastorello, Y.; Suñol, C.; Sanfeliu, C. Resveratrol Activates Antioxidant Protective Mechanisms in Cellular Models of Alzheimer’s Disease Inflammation. Antioxidants 2024, 13, 177. [Google Scholar] [CrossRef]
- Madhu, L.N.; Kodali, M.; Upadhya, R.; Rao, S.; Shuai, B.; Somayaji, Y.; Attaluri, S.; Kirmani, M.; Gupta, S.; Maness, N.; et al. Intranasally Administered EVs from hiPSC-Derived NSCs Alter the Transcriptomic Profile of Activated Microglia and Conserve Brain Function in an Alz-heimer’s Model 2024. Journal of extracellular vesicles 2024, 13, e12519. [Google Scholar] [CrossRef]
- Ng, R.C.-L.; Jian, M.; Ma, O.K.-F.; Xiang, A.W.; Bunting, M.; Kwan, J.S.-C.; Wong, C.W.-K.; Yick, L.-W.; Chung, S.K.; Lam, K.S.-L.; et al. Liver-specific adiponectin gene therapy suppresses microglial NLRP3-inflammasome activation for treating Alzheimer’s disease. J. Neuroinflammation 2024, 21, 77. [Google Scholar] [CrossRef]
- Klück, V.A.; Jansen, T.L.T.; Janssen, M.; Comarniceanu, A.; Efdé, M.; Tengesdal, I.W.; Schraa, K.; Cleophas, M.C.P.; Scribner, C.L.; Skouras, D.B.; et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020, 2, e270–e280. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. 2020, 117, 32145–32154. [Google Scholar] [CrossRef]
- Study of VX-765 in Subjects with Treatment-Resistant Partial Epilepsy. Available online: https://ctv.veeva.com/study/study-of-vx-765-in-subjects-with-treatment-resistant-partial-epilepsy (accessed on 27 March 2025).
- Ravizza, T.; Lucas, S.; Balosso, S.; Bernardino, L.; Ku, G.; Noé, F.; Malva, J.; Randle, J.C.R.; Allan, S.; Vezzani, A. Inactivation of Caspase-1 in Rodent Brain: A Novel Anticonvulsive Strategy. Epilepsia 2006, 47, 1160–1168. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef]
- Green, E.A.; Metz, D.; Galinsky, R.; Atkinson, R.; Skuza, E.M.; Clark, M.; Gunn, A.J.; Kirkpatrick, C.M.; Hunt, R.W.; Berger, P.J.; et al. Anakinra Pilot–a clinical trial to demonstrate safety, feasibility and pharmacokinetics of interleukin 1 receptor antagonist in preterm infants. Front. Immunol. 2022, 13, 1022104. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Alfaro, M.; Cuadrado-Tejedor, M.; Lasarte-Cia, A.; Navarro, F.; Vivas, I.; Espelosin, M.; Cartas-Cejudo, P.; Fernández-Irigoyen, J.; Santamaría, E.; et al. Improvement of cognitive function in wild-type and Alzheimer’s disease mouse models by the immunomodulatory properties of menthol inhalation or by depletion of T regulatory cells. Front. Immunol. 2023, 14, 1130044. [Google Scholar] [CrossRef] [PubMed]
- Melchiorri, D.; Merlo, S.; Micallef, B.; Borg, J.-J.; Dráfi, F. Alzheimer’s disease and neuroinflammation: Will new drugs in clinical trials pave the way to a multi-target therapy? Front. Pharmacol. 2023, 14, 1196413. [Google Scholar] [CrossRef]
- Martiniani, R.; Di Loreto, V.; Di Sano, C.; Lombardo, A.; Liberati, A.M. Biological Activity of Lenalidomide and Its Underlying Therapeutic Effects in Multiple Myeloma. Adv. Hematol. 2012, 2012, 842945. [Google Scholar] [CrossRef]
- Decourt, B.; Wilson, J.; Ritter, A.; Dardis, C.; DiFilippo, F.P.; Zhuang, X.; Cordes, D.; Lee, G.; Fulkerson, N.D.; Rose, T.S.; et al. MCLENA-1: A Phase II Clinical Trial for the Assessment of Safety, Tolerability, and Efficacy of Lenalidomide in Patients with Mild Cognitive Impairment Due to Alzheimer’s Disease. Open Access J. Clin. Trials 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Potter, H.; Woodcock, J.H.; Boyd, T.D.; Coughlan, C.M.; O’Shaughnessy, J.R.; Borges, M.T.; Thaker, A.A.; Raj, B.A.; Adamszuk, K.; Scott, D.; et al. Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer’s disease. Alzheimer’s Dement. 2021, 7, e12158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGroarty, J.; Salinas, S.; Evans, H.; Jimenez, B.; Tran, V.; Kadavakollu, S.; Vashist, A.; Atluri, V. Inflammasome-Mediated Neuroinflammation: A Key Driver in Alzheimer’s Disease Pathogenesis. Biomolecules 2025, 15, 676. https://doi.org/10.3390/biom15050676
McGroarty J, Salinas S, Evans H, Jimenez B, Tran V, Kadavakollu S, Vashist A, Atluri V. Inflammasome-Mediated Neuroinflammation: A Key Driver in Alzheimer’s Disease Pathogenesis. Biomolecules. 2025; 15(5):676. https://doi.org/10.3390/biom15050676
Chicago/Turabian StyleMcGroarty, Julie, Shelbi Salinas, Hayden Evans, Bryan Jimenez, Vincent Tran, Samuel Kadavakollu, Arti Vashist, and Venkata Atluri. 2025. "Inflammasome-Mediated Neuroinflammation: A Key Driver in Alzheimer’s Disease Pathogenesis" Biomolecules 15, no. 5: 676. https://doi.org/10.3390/biom15050676
APA StyleMcGroarty, J., Salinas, S., Evans, H., Jimenez, B., Tran, V., Kadavakollu, S., Vashist, A., & Atluri, V. (2025). Inflammasome-Mediated Neuroinflammation: A Key Driver in Alzheimer’s Disease Pathogenesis. Biomolecules, 15(5), 676. https://doi.org/10.3390/biom15050676