

Structural Biology in the AlphaFold Era: How Far Is Artificial Intelligence from Deciphering the Protein Folding Code?




Abstract
1. The Central Role of Proteins in Life and Their Extraordinary Chemical and Structural Complexity
2. The Protein Folding Code and Its Formulation(s)
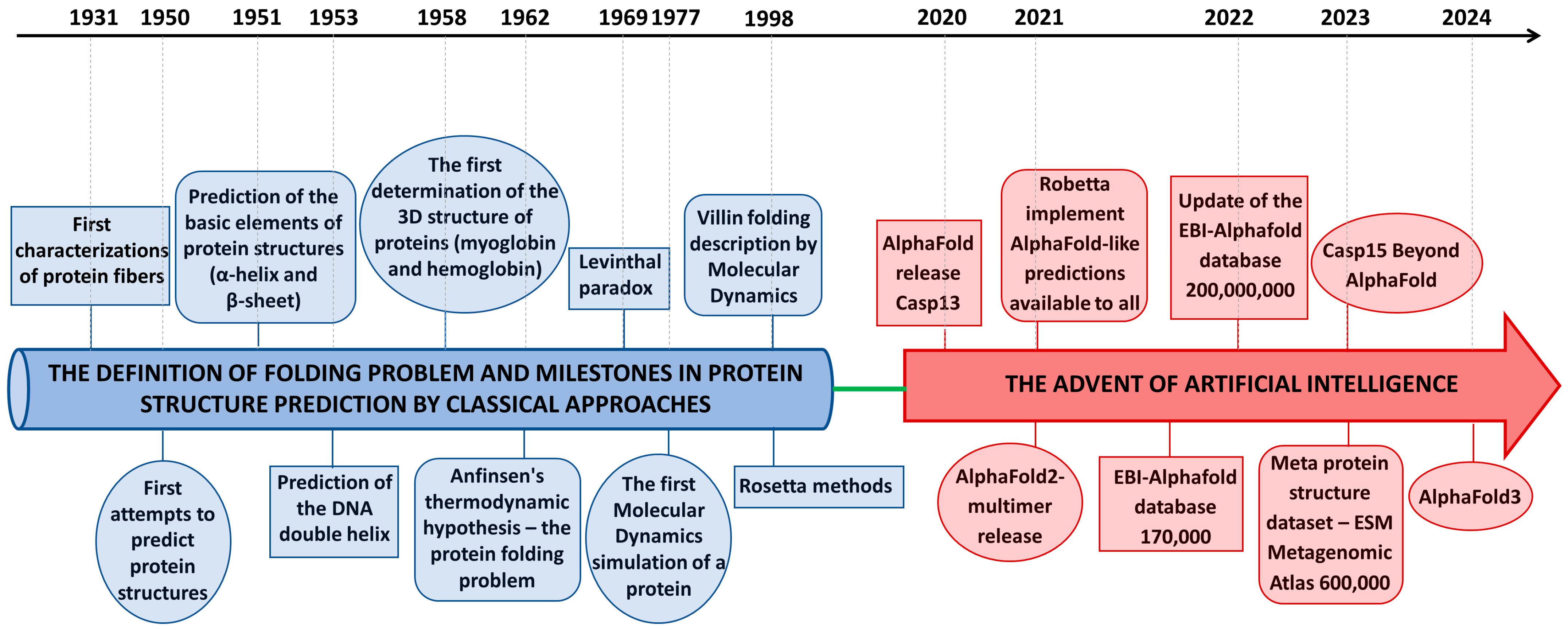
3. From Ab Initio to Empirical Approaches
4. The Advent of Machine Learning Techniques: The AlphaFold Revolution
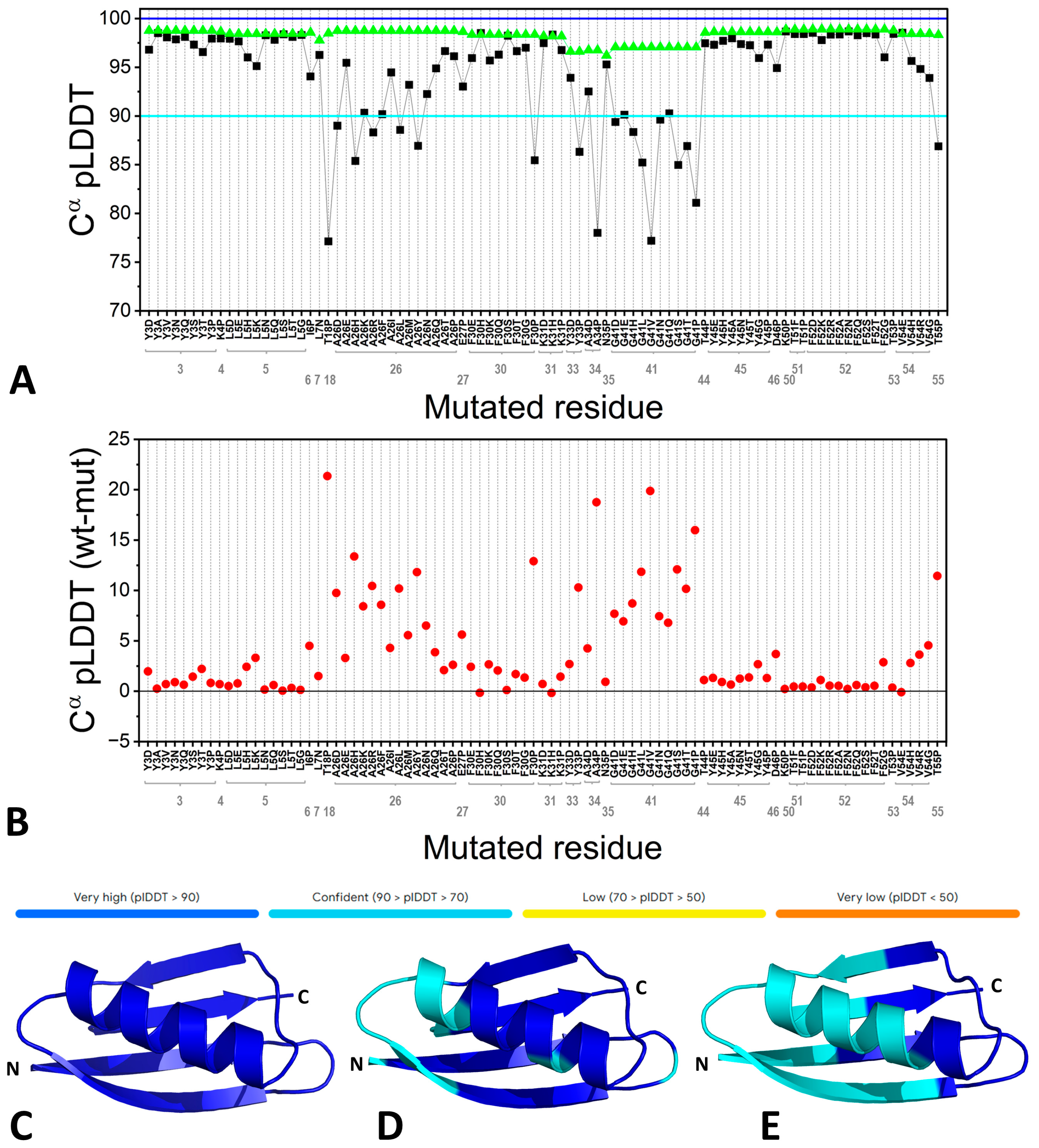
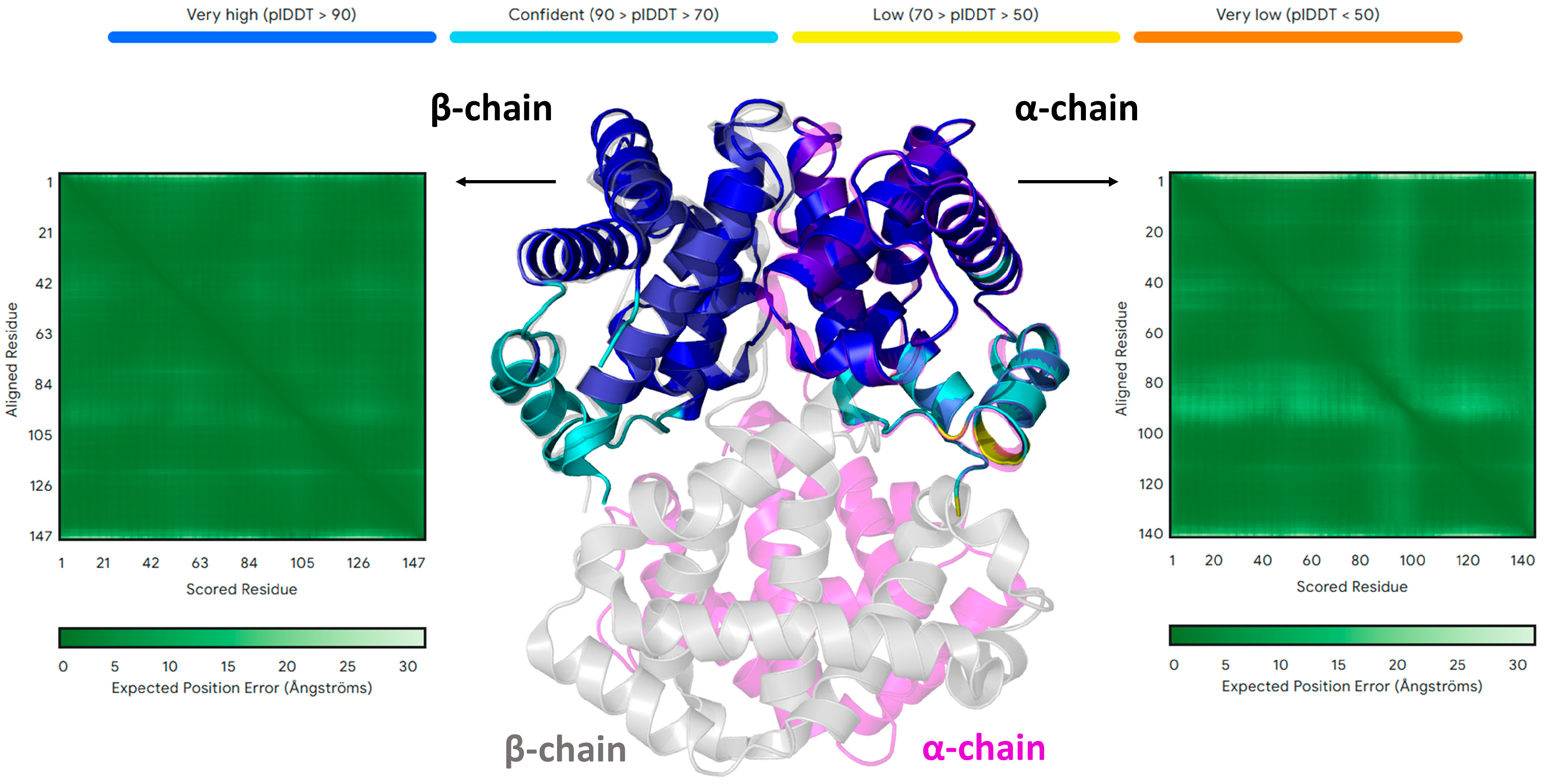
5. AlphaFold and the Folding Problem
6. Conclusions and Perspectives for Structural Biology and Beyond
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, R.; Zhang, Y. Toward the Solution of the Protein Structure Prediction Problem. J. Biol. Chem. 2021, 297, 100870. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Burley, S.K. Protein Data Bank (PDB): Fifty-Three Years Young and Having a Transformative Impact on Science and Society. Quart. Rev. Biophys. 2025, 58, e9. [Google Scholar] [CrossRef] [PubMed]
- Levinthal, C. Are There Pathways for Protein Folding? J. Chim. Phys. 1968, 65, 44–45. [Google Scholar] [CrossRef]
- Zwanzig, R.; Szabo, A.; Bagchi, B. Levinthal’s Paradox. Proc. Natl. Acad. Sci. USA 1992, 89, 20–22. [Google Scholar] [CrossRef]
- Anfinsen, C.B.; Haber, E.; Sela, M.; White, F.H. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. USA 1961, 47, 1309–1314. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles That Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Anfinsen, C.B.; Scheraga, H.A. Experimental and Theoretical Aspects of Protein Folding. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1975; Volume 29, pp. 205–300. ISBN 978-0-12-034229-7. [Google Scholar]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef]
- Rumbley, J.; Hoang, L.; Mayne, L.; Englander, S.W. An Amino Acid Code for Protein Folding. Proc. Natl. Acad. Sci. USA 2001, 98, 105–112. [Google Scholar] [CrossRef]
- Wrinch, D.M. The Cyclol Hypothesis and the “Globular” Proteins. Proc. R. Soc. Lond. A 1937, 161, 505–524. [Google Scholar] [CrossRef]
- Bragg, W.L.; Kendrew, J.C.; Perutz, M.F. Polypeptide Chain Configurations in Crystalline Proteins. Proc. R. Soc. Lond. A 1950, 203, 321–357. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A.C.T. Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Giegé, R. A Historical Perspective on Protein Crystallization from 1840 to the Present Day. FEBS J. 2013, 280, 6456–6497. [Google Scholar] [CrossRef]
- Pauling, L.; Corey, R.B.; Branson, H.R. The Structure of Proteins: Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. Configurations of Polypeptide Chains With Favored Orientations Around Single Bonds: Two New Pleated Sheets. Proc. Natl. Acad. Sci. USA 1951, 37, 729–740. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Kartha, G. Structure of Collagen. Nature 1955, 176, 593–595. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F.H.C. The Structure of Collagen. Nature 1955, 176, 915–916. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Moult, J.; Pedersen, J.T.; Judson, R.; Fidelis, K. A Large-scale Experiment to Assess Protein Structure Prediction Methods. Proteins 1995, 23, ii–iv. [Google Scholar] [CrossRef]
- Das, R.; Baker, D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Progress and Challenges in Protein Structure Prediction. Curr. Opin. Struct. Biol. 2008, 18, 342–348. [Google Scholar] [CrossRef]
- Ginalski, K. Comparative Modeling for Protein Structure Prediction. Curr. Opin. Struct. Biol. 2006, 16, 172–177. [Google Scholar] [CrossRef]
- Rawlings, C.J.; Fox, J.P. Artificial Intelligence in Molecular Biology: A Review and Assessment. Phil. Trans. R. Soc. Lond. B 1994, 344, 353–363. [Google Scholar] [CrossRef]
- Sternberg, M.J.; King, R.D.; Lewis, R.A.; Muggleton, S. Application of Machine Learning to Structural Molecular Biology. Phil. Trans. R. Soc. Lond. B 1994, 344, 365–371. [Google Scholar] [CrossRef]
- Rost, B. PHD: Predicting One-Dimensional Protein Structure by Profile-Based Neural Networks. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1996; Volume 266, pp. 525–539. ISBN 978-0-12-182167-8. [Google Scholar]
- Pirovano, W.; Heringa, J. Protein Secondary Structure Prediction. In Data Mining Techniques for the Life Sciences; Carugo, O., Eisenhaber, F., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 609, pp. 327–348. ISBN 978-1-60327-240-7. [Google Scholar]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein Structure Prediction Using Multiple Deep Neural Networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins 2019, 87, 1141–1148. [Google Scholar] [CrossRef]
- Callaway, E. ‘It Will Change Everything’: DeepMind’s AI Makes Gigantic Leap in Solving Protein Structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef]
- Service, R.F. ‘The Game Has Changed.’ AI Triumphs at Protein Folding. Science 2020, 370, 1144–1145. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and Improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer 2021. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Genc, A.G.; McGuffin, L.J. Beyond AlphaFold2: The Impact of AI for the Further Improvement of Protein Structure Prediction. In Prediction of Protein Secondary Structure; Kloczkowski, A., Kurgan, L., Faraggi, E., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2025; Volume 2867, pp. 121–139. ISBN 978-1-07-164195-8. [Google Scholar]
- Liu, J.; Guo, Z.; Wu, T.; Roy, R.S.; Chen, C.; Cheng, J. Improving AlphaFold2-Based Protein Tertiary Structure Prediction with MULTICOM in CASP15. Commun. Chem. 2023, 6, 188. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Kwon, D. RNA Function Follows Form—Why Is It so Hard to Predict? Nature 2025, 639, 1106–1108. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Jumper, J.; Hassabis, D. Protein Structure Predictions to Atomic Accuracy with AlphaFold. Nat. Methods 2022, 19, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Baker, D. Deep Learning and Protein Structure Modeling. Nat. Methods 2022, 19, 13–14. [Google Scholar] [CrossRef]
- Barrio-Hernandez, I.; Yeo, J.; Jänes, J.; Mirdita, M.; Gilchrist, C.L.M.; Wein, T.; Varadi, M.; Velankar, S.; Beltrao, P.; Steinegger, M. Clustering Predicted Structures at the Scale of the Known Protein Universe. Nature 2023, 622, 637–645. [Google Scholar] [CrossRef]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A Structural Biology Community Assessment of AlphaFold2 Applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef]
- Burke, D.F.; Bryant, P.; Barrio-Hernandez, I.; Memon, D.; Pozzati, G.; Shenoy, A.; Zhu, W.; Dunham, A.S.; Albanese, P.; Keller, A.; et al. Towards a Structurally Resolved Human Protein Interaction Network. Nat. Struct. Mol. Biol. 2023, 30, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Al-Janabi, A. Has DeepMind’s AlphaFold Solved the Protein Folding Problem? BioTechniques 2022, 72, 73–76. [Google Scholar] [CrossRef]
- Ahdritz, G.; Bouatta, N.; Floristean, C.; Kadyan, S.; Xia, Q.; Gerecke, W.; O’Donnell, T.J.; Berenberg, D.; Fisk, I.; Zanichelli, N.; et al. OpenFold: Retraining AlphaFold2 Yields New Insights into Its Learning Mechanisms and Capacity for Generalization. Nat. Methods 2024, 21, 1514–1524. [Google Scholar] [CrossRef]
- Wang, L.; Wen, Z.; Liu, S.-W.; Zhang, L.; Finley, C.; Lee, H.-J.; Fan, H.-J.S. Overview of AlphaFold2 and Breakthroughs in Overcoming Its Limitations. Comput. Biol. Med. 2024, 176, 108620. [Google Scholar] [CrossRef]
- Nisthal, A.; Wang, C.Y.; Ary, M.L.; Mayo, S.L. Protein Stability Engineering Insights Revealed by Domain-Wide Comprehensive Mutagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16367–16377. [Google Scholar] [CrossRef] [PubMed]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate Proteome-Wide Missense Variant Effect Prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.W.; Walter, J.C. Predictomes, a Classifier-Curated Database of AlphaFold-Modeled Protein-Protein Interactions. Mol. Cell 2025, 85, 1216–1232.e5. [Google Scholar] [CrossRef]
- Esposito, L.; Balasco, N.; Smaldone, G.; Berisio, R.; Ruggiero, A.; Vitagliano, L. AlphaFold-Predicted Structures of KCTD Proteins Unravel Previously Undetected Relationships among the Members of the Family. Biomolecules 2021, 11, 1862. [Google Scholar] [CrossRef]
- Esposito, L.; Balasco, N.; Vitagliano, L. Alphafold Predictions Provide Insights into the Structural Features of the Functional Oligomers of All Members of the KCTD Family. Int. J. Mol. Sci. 2022, 23, 13346. [Google Scholar] [CrossRef]
- Balasco, N.; Esposito, L.; Smaldone, G.; Salvatore, M.; Vitagliano, L. A Comprehensive Analysis of the Structural Recognition between KCTD Proteins and Cullin 3. Int. J. Mol. Sci. 2024, 25, 1881. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Liebschner, D.; Croll, T.I.; Williams, C.J.; McCoy, A.J.; Poon, B.K.; Afonine, P.V.; Oeffner, R.D.; Richardson, J.S.; Read, R.J.; et al. AlphaFold Predictions Are Valuable Hypotheses and Accelerate but Do Not Replace Experimental Structure Determination. Nat. Methods 2024, 21, 110–116. [Google Scholar] [CrossRef]
- Read, R.J.; Baker, E.N.; Bond, C.S.; Garman, E.F.; Van Raaij, M.J. AlphaFold and the Future of Structural Biology. Acta Crystallogr. D Struct. Biol. 2023, 79, 556–558. [Google Scholar] [CrossRef]
- Bijak, V.; Szczygiel, M.; Lenkiewicz, J.; Gucwa, M.; Cooper, D.R.; Murzyn, K.; Minor, W. The Current Role and Evolution of X-Ray Crystallography in Drug Discovery and Development. Expert. Opin. Drug Discov. 2023, 18, 1221–1230. [Google Scholar] [CrossRef]
- Bai, X.; McMullan, G.; Scheres, S.H.W. How Cryo-EM Is Revolutionizing Structural Biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The Revolution Will Not Be Crystallized: A New Method Sweeps through Structural Biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Markwick, P.R.L.; Malliavin, T.; Nilges, M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput. Biol. 2008, 4, e1000168. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.S.; Hacking, I. The Structure of Scientific Revolutions, 4th ed.; University of Chicago Press: Chicago, IL, USA, 2012; ISBN 978-0-226-45811-3. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Residue | pLDDT | Mutation | pLDDT in the Single-Point Mutants | pLDDT in the Hexa-Mutant | pLDDT in the Octa-Mutant |
|---|---|---|---|---|---|
| Y3 | 98.76 | Y3A | 98.51 | 90.01 | 89.11 |
| L5 | 98.43 | L5D | 97.93 | 92.94 | 90.24 |
| A26 | 98.75 | A26P | 96.13 | 89.03 | 76.83 |
| F30 | 98.35 | F30G | 97.00 | 90.71 | 86.31 |
| G41 | 97.06 | G41P | 81.09 | - | 93.96 |
| Y45 | 98.62 | Y45G | 95.94 | 92.23 | 86.23 |
| F52 | 98.89 | F52G | 96.02 | 93.10 | 88.60 |
| V54 | 98.44 | V54G | 93.90 | - | 92.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balasco, N.; Esposito, L.; Vitagliano, L. Structural Biology in the AlphaFold Era: How Far Is Artificial Intelligence from Deciphering the Protein Folding Code? Biomolecules 2025, 15, 674. https://doi.org/10.3390/biom15050674
Balasco N, Esposito L, Vitagliano L. Structural Biology in the AlphaFold Era: How Far Is Artificial Intelligence from Deciphering the Protein Folding Code? Biomolecules. 2025; 15(5):674. https://doi.org/10.3390/biom15050674
Chicago/Turabian StyleBalasco, Nicole, Luciana Esposito, and Luigi Vitagliano. 2025. "Structural Biology in the AlphaFold Era: How Far Is Artificial Intelligence from Deciphering the Protein Folding Code?" Biomolecules 15, no. 5: 674. https://doi.org/10.3390/biom15050674
APA StyleBalasco, N., Esposito, L., & Vitagliano, L. (2025). Structural Biology in the AlphaFold Era: How Far Is Artificial Intelligence from Deciphering the Protein Folding Code? Biomolecules, 15(5), 674. https://doi.org/10.3390/biom15050674