
Mechanisms of DNA Damage Recognition by UDG and PARP1 in the Nucleosome

 , , , and
, , , and
Abstract
1. Introduction
2. Methods
2.1. Docking
2.2. Molecular Dynamics
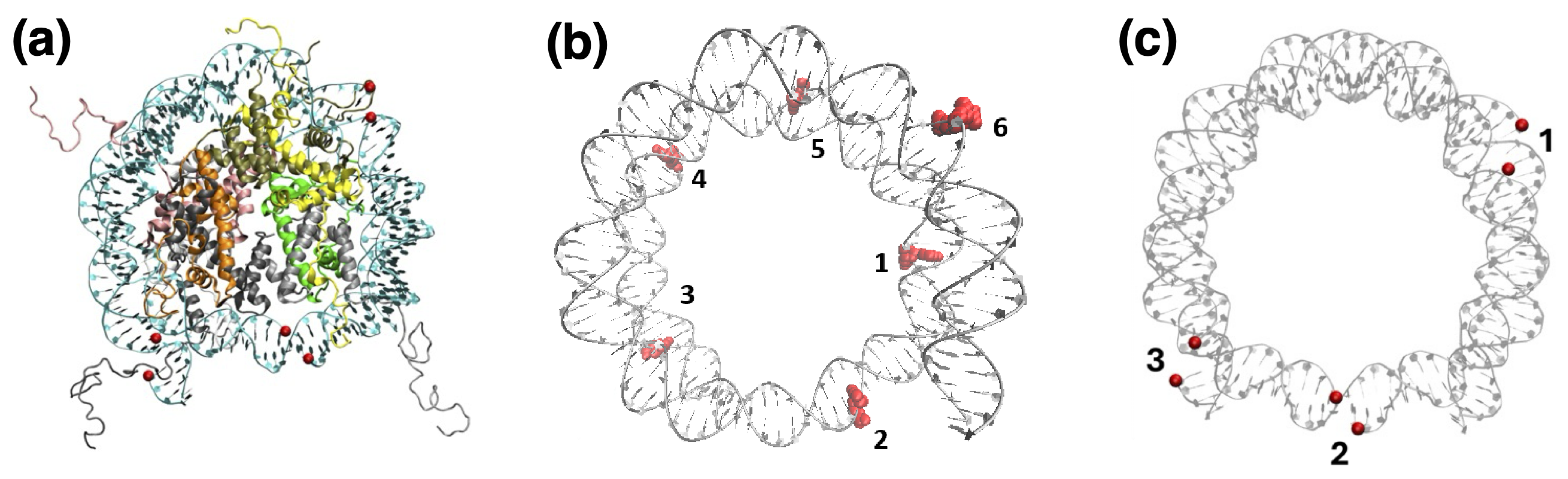
2.3. Nucleosome
3. Results: Uracil and UDG
3.1. Structures of UDG and Flipped-Out Nucleotides
3.2. Nucleosome Accessibility and UDG Positioning
3.3. Structural Dynamics of UDG Interactions in the Nucleosome
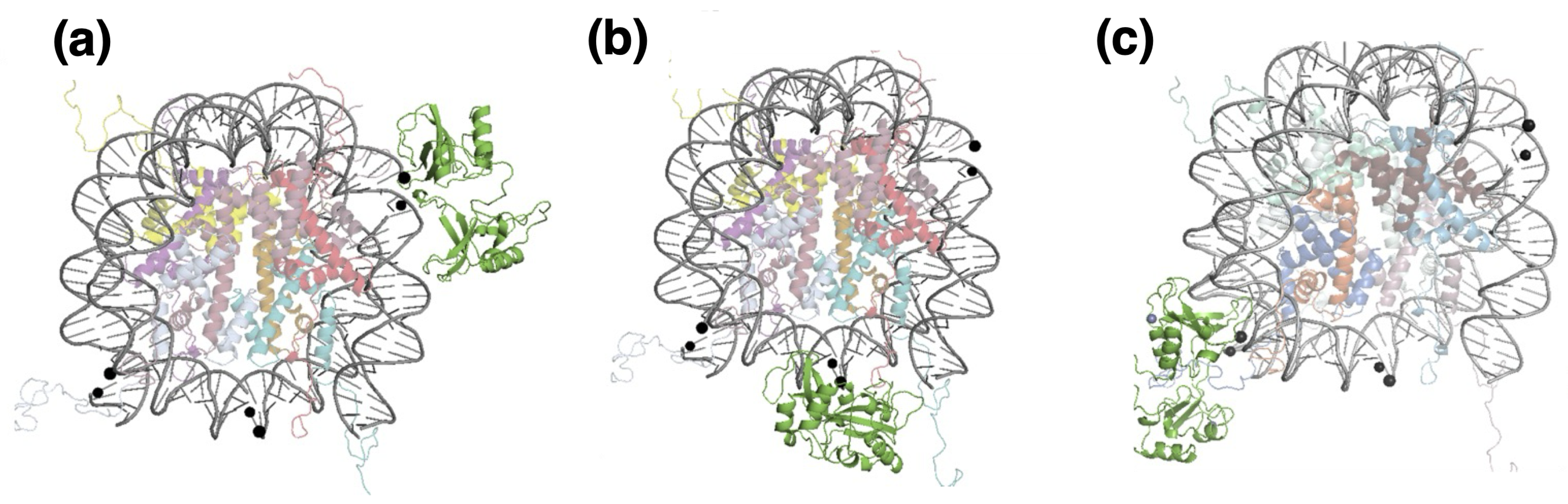
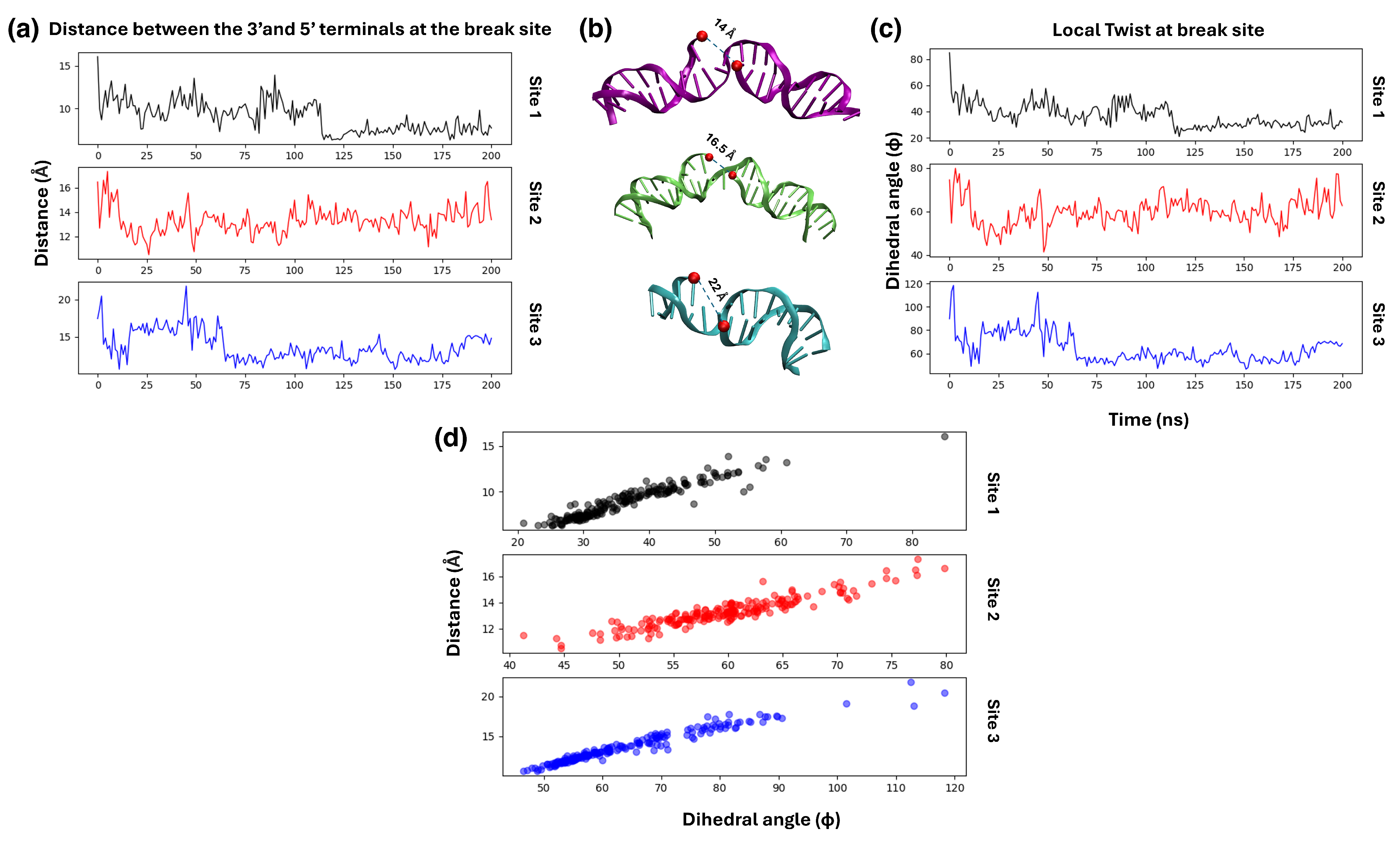
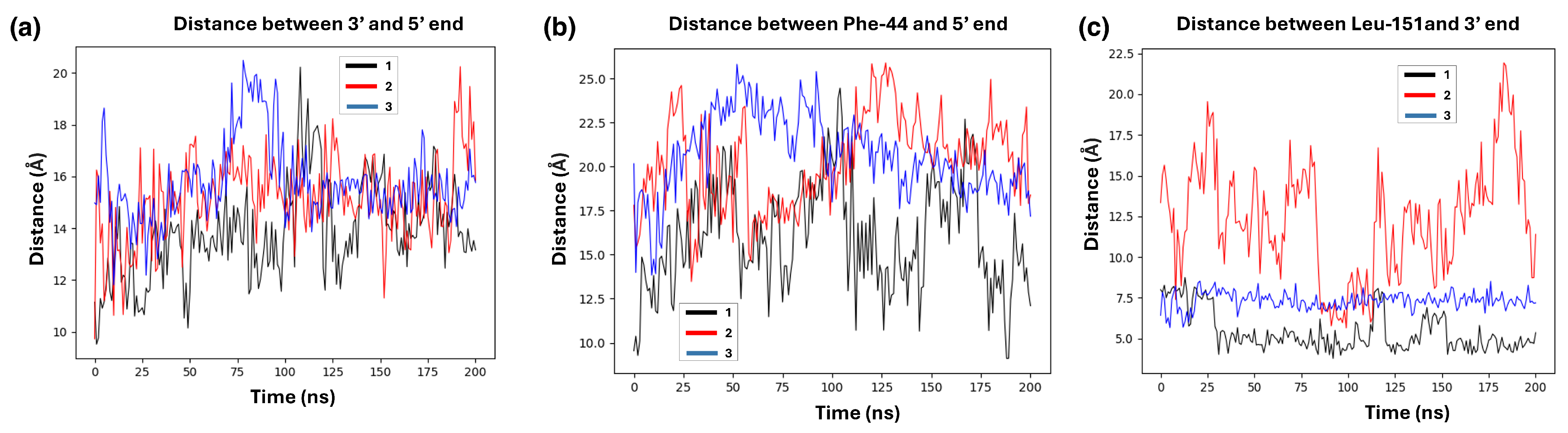
4. Results: Single-Strand Breaks and PARP1
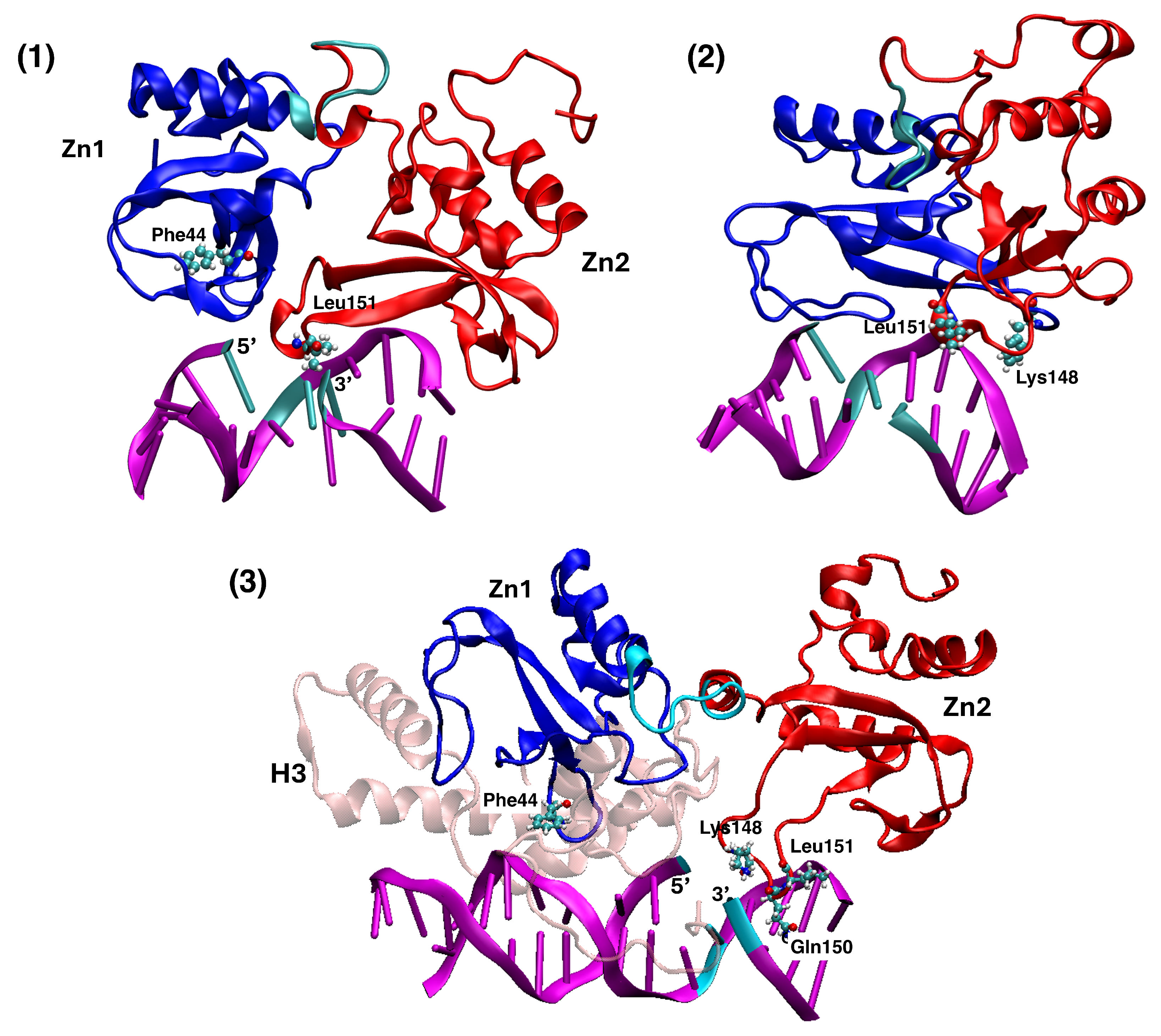
4.1. Structures of PARP1 and Single-Strand Breaks
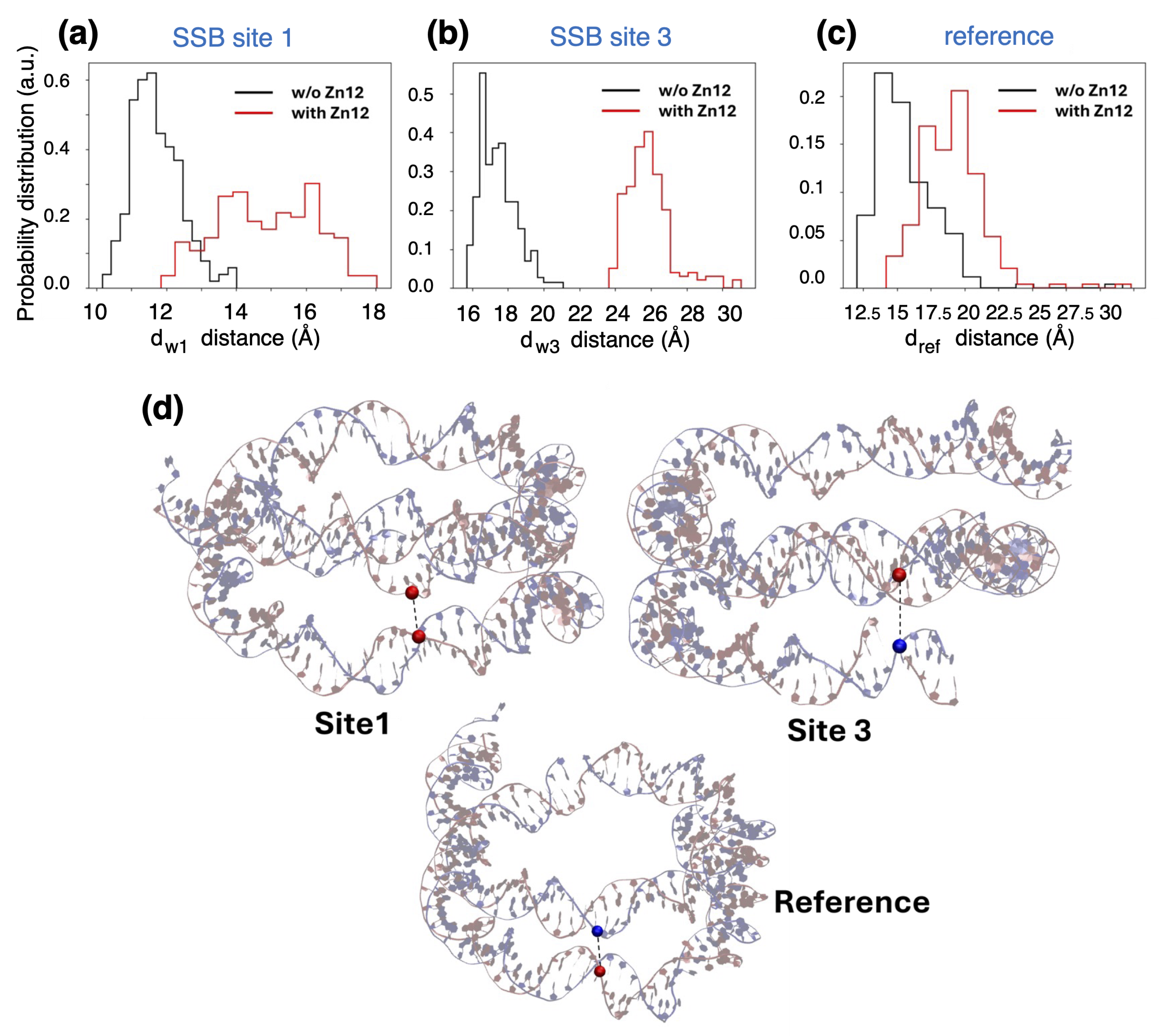
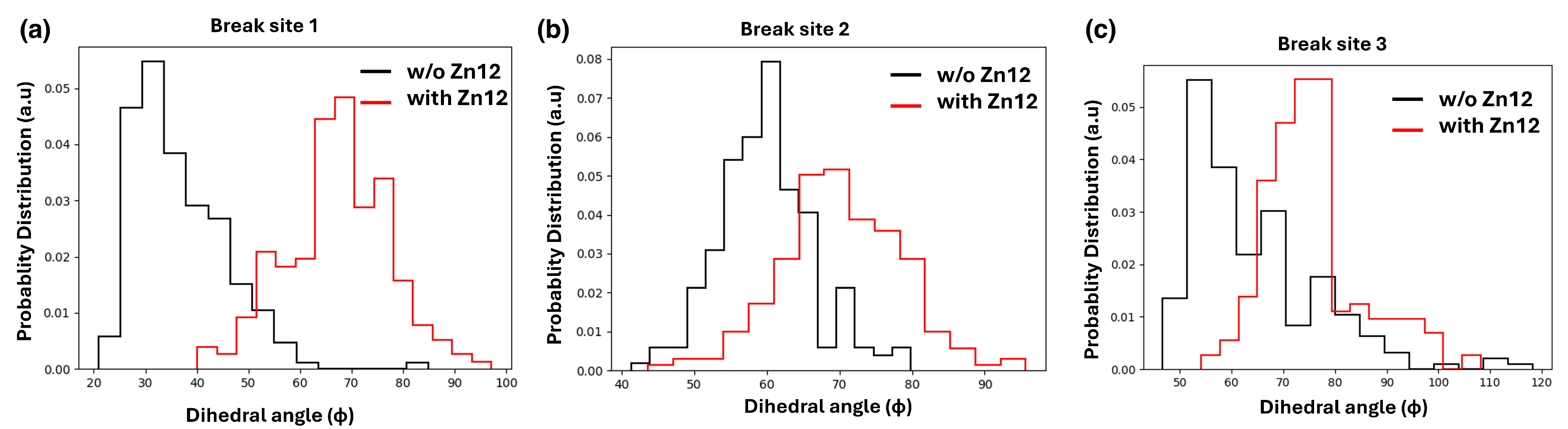
4.2. Analysing the Zn-Finger Domain Contacts with SSB
4.3. Nucleosome Reorganization by PARP1 and the Role of Histone Tails
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DNA | deoxyribonucleic acid |
| SSB | single-strand break |
| DSB | double-strand break |
| NCP | nucleosome core particle |
| MD | molecular dynamics |
| UDG | uracil DNA glycosylase |
| PARP | poly-ADP-ribose polymerase |
| DDR | DNA damage repair |
| BER | base-excision repair |
| SSBR | single-strand break repair |
| SHL | superhelical symmetry axis |
| PDB | protein data bank |
| RMSD | root-mean-squared displacement |
| RMSF | root-mean-squared fluctuation |
| FRET | Forster resonance energy-transfer |
| NMR | nuclear magnetic resonance |
References
- Krokan, H.E.; Drabløs, F.; Slupphaug, G. Uracil in DNA—Occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [PubMed]
- Von Sonntag, C. Recent Trends in Radiation Chemistry; Wishart, J.F., Ed.; World Scientific: Singapore, 2010; p. 543. [Google Scholar]
- Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; pp. 379–390. [Google Scholar]
- Porro, M.L.T.; Greenberg, M.M. Double-Strand Breaks from a Radical Commonly Produced by DNA-Damaging Agents. Chem. Res. Toxycology 2015, 28, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Weinfeld, M.; Soderling, K.J. 32P-postlabeling detection of radiation-induced DNA damage: Identification and estimation of thymine glycols and phosphoglycolate termini. Biochemistry 1999, 30, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Akopiants, K.; Mohapatra, S.; Lin, P.S.; Valerie, K.; Ramsden, D.A.; Lees-Miller, S.P.; Povirk, L.F. Tyrosyl-DNA phosphodiesterase and the repair of 3’-phosphoglycolate-terminated DNA double-strand breaks. DNA Repair 2009, 8, 901–911. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kacmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Branze, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Robertson, A.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef]
- Friedman, J.I.; Stivers, J.T. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry 2010, 49, 4957–4967. [Google Scholar] [CrossRef]
- Bonnet, I.; Biebricher, A.; Porté, P.L.; Loverdo, C.; Bénichou, O.; Voituriez, R.; Escudé, C.; Wende, W.; Pingoud, A.; Desbiolles, P. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008, 36, 4118–4127. [Google Scholar] [CrossRef] [PubMed]
- Monico, C.; Capitanio, M.; Belcastro, G.; Vanzi, F.; Pavone, F.S. Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level. Int. J. Mol. Sci. 2013, 14, 3961–3992. [Google Scholar] [CrossRef] [PubMed]
- Kamagata, K.; Itoh, Y.; Subekti, D.R.G. How p53 Molecules Solve the Target DNA Search Problem: A Review. Int. J. Mol. Sci. 2020, 21, 1031. [Google Scholar] [CrossRef] [PubMed]
- Cleri, F.; Landuzzi, F.; Blossey, R. Mechanical evolution of DNA double-strand breaks in the nucleosome. PLoS Comput. Biol. 2018, 14, e1006224. [Google Scholar] [CrossRef]
- Cleri, F.; Giordano, S.; Blossey, R. Nucleosome Array Deformation in Chromatin is Sustained by Bending, Twisting and Kinking of Linker DNA. J. Mol. Biol. 2023, 435, 168263. [Google Scholar] [CrossRef]
- Sarma, P.A.; Abbadie, C.; Cleri, F. Cooperative dynamics of PARP1 zinc-finger domains in the detection of DNA single-strand breaks. Sci. Rep. 2024, 14, 23257. [Google Scholar] [CrossRef]
- Cao, C.; Jiang, Y.L.; Stivers, J.T.; Song, F. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 2004, 11, 1230–1236. [Google Scholar] [CrossRef]
- Yin, Y.; Yang, L.; Zheng, G.; Zhao, X.S. Dynamics of spontaneous flipping of a mismatched base in DNA duplex. Proc. Natl. Acad. Sci. USA 2014, 111, 8043–8048. [Google Scholar] [CrossRef]
- Sarangi, M.K.; Zvoda, V.; Holte, M.N.; Becker, N.A.; Peters, J.P.; Maher, L.J.; Ansari, A. Evidence for a bind-then-bend mechanism for architectural DNA binding protein yNhp6A. Nucleic Acids Res. 2019, 47, 2781–2791. [Google Scholar] [CrossRef]
- Panigrahi, A.; Vemuri, H.; Aggarwal, M.; Pitta, K.; Krishnan, M. Sequence specificity, energetics and mechanism of mismatch recognition by DNA damage sensing protein Rad4/XPC. Nucleic Acids Res. 2020, 48, 2246–2257. [Google Scholar] [CrossRef]
- Yoshua, S.B.; Watson, G.D.; Howard, J.A.L.; Velasco-Berrelleza, V.; Leake, M.C.; Noy, A. Integration host factor bends and bridges DNA in a multiplicity of binding modes with varying specificity. Nucleic Acids Res. 2021, 49, 8684–8698. [Google Scholar] [PubMed]
- Baral, S.; Chakraborty, S.; Steinbach, P.J.; Paul, D.; Min, J.H.; Ansari, A. Evidence for intrinsic DNA dynamics and deformability in damage sensing by the Rad4/XPC nucleotide excision repair complex. Nucleic Acids Res. 2025, 53, gkae1290. [Google Scholar]
- Cole, H.A.; Tabor-Godwin, J.M.; Hayes, J.J. Uracil DNA Glycosylase Activity on Nucleosomal DNA Depends on Rotational Orientation of Targets. J. Biol. Chem. 2010, 285, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Stahley, M.R.; Xu, J.; Friedman, J.I.; Sun, Y.; McKnight, J.N.; Gray, J.J.; Bowman, G.D.; Stivers, J.T. Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry 2012, 51, 6028–6038. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Smerdon, M.J. The Structural Location of DNA Lesions in Nucleosome Core Particles Determines Accessibility by Base Excision Repair Enzymes. J. Biol. Chem. 2013, 288, 13863–13875. [Google Scholar] [PubMed]
- Olmon, E.D.; Delaney, S. Differential Ability of Five DNA Glycosylases to Recognize and Repair Damage on Nucleosomal DNA. ACS Chem. Biol. 2017, 12, 692–701. [Google Scholar]
- Tarantino, M.E.; Dow, B.J.; Drohat, A.C.; Delaney, S. Nucleosomes and the three glycosylases: High, medium, and low levels of excision by the uracil DNA glycosylase superfamily. DNA Repair 2018, 72, 56–63. [Google Scholar] [CrossRef]
- Sultanov, D.; Gerasimova, N.; Kudryashova, K.; Maluchenko, N.; Kotova, E.; Langelier, M.F.; Pascal, J.; Kirpichnikov, M.; Feofanov, A.; Studitsky, V. Unfolding of core nucleosomes by PARP1 revealed by spFRET microscopy. AIMS Genet. 2017, 4, 021–031. [Google Scholar]
- Maluchenko, N.V.; Nilov, D.K.; Pushkarev, S.V.; Kotova, E.Y.; Gerasimova, N.S.; Kirpichnikov, M.P.; Langelier, M.F.; Pascal, J.M.; Akhtar, M.S.; Feofanov, A.V.; et al. Mechanisms of Nucleosome Reorganization by PARP1. Int. J. Mol. Sci. 2021, 22, 12127. [Google Scholar] [CrossRef]
- Saravanan, V.; Raouraoua, N.; Brysbaert, G.; Giordano, S.; Lensink, M.F.; Cleri, F.; Blossey, R. The ‘very moment’ when UDG recognizes a flipped-out uracil base in dsDNA. Sci. Rep. 2025, 15, 7993. [Google Scholar] [CrossRef]
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef]
- Sefer, A.; Kallis, E.; Eilert, T.; Röcker, C.; Kolesnikova, O.; Neuhaus, D.; Eustermann, S.; Michaelis, J. Structural dynamics of DNA strand break sensing by PARP-1 at a single-molecule level. Nat. Commun. 2022, 13, 6569. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical and/or biophysical information. J. Am. Chem. Soc. 2006, 125, 1731–1737. [Google Scholar] [CrossRef]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2015, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 9, 988996. [Google Scholar] [CrossRef]
- Rodríguez-Lumbreras, L.A.; Jiménez-García, B.; Giménez-Santamarina, S.; Fernández-Recio, J. pyDockDNA: A new web server for energy-based protein-DNA docking and scoring. Front. Mol. Biosci. 2022, 9, 988996. [Google Scholar] [CrossRef]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF-Chimera, a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Berendsen, H.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgrading and Validation of the AMBER Force Field for Histidine and Cysteine Zinc(II)-Binding Residues in Sites with Four Protein Ligands. J. Chem. Inf. Model. 2019, 59, 3803–3816. [Google Scholar] [CrossRef] [PubMed]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force-field for DNA simulations. Nat. Meth. 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Love, O.; Galindo-Murillo, R.; Zgarbová, M.; Sponer, J.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA - 2023 Edition. J. Chem. Theory Comput. 2023, 19, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Wang, H.; Xiong, L.; Cramer, P. Structures and implications of TBP-nucleosome complexes. Proc. Natl. Acad. Sci. USA 2021, 118, e2108859118. [Google Scholar]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Lowary, P.; Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef]
- Stivers, J.T. Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry 1998, 38, 952–963. [Google Scholar]
- Parikh, S.S.; Putnam, C.D.; Tainer, J. Lessons learned from structural results on uracil-DNA glycosylase. Mutat. Res. Repair 2000, 460, 183–199. [Google Scholar]
- Parikh, S.; Walcher, G.; Jones, G.; Slupphaug, G.; Krokan, H.; Blackburn, G.; Tainer, J. Uracil-DNA glycosylase-DNA substrate and product structures: Conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl. Acad. Sci. USA 2000, 97, 5083–5088. [Google Scholar] [CrossRef]
- Parikh, S.; Mol, C.; Slupphaug, G.; Bharati, S.; Krokan, H.; Tainer, J. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998, 17, 5214–5226. [Google Scholar] [CrossRef] [PubMed]
- Earl, C.; Bagnéris, C.; Zeman, K.; Cole, A.; Barrett, T.; Savva, R. A structurally conserved motif in gamma-herpesvirus uracil-DNA glycosylases elicits duplex nucleotide-flipping. Nucleic Acids Res. 2018, 46, 4286–4300. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Grollman, A.P. The DNA trackwalkers: Principles of lesion search and recognition by DNA glycosylases. Mutat. Res. Mol. Mech. Mutagen. 2005, 57, 24–54. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D.; Wallace, S.S.; Pederson, D.S. Rules of Engagement for Base Excision Repair in Chromatin. J. Cell Physiol. 2013, 228, 258–266. [Google Scholar] [CrossRef]
- Assenza, S.; Pérez, R. Accurate Sequence-Dependent Coarse-Grained Model for Conformational and Elastic Properties of Double-Stranded DNA. J. Chem. Theory Comput. 2022, 18, 3239–3256. [Google Scholar] [CrossRef]
- Clark, N.J.; Kramer, M.; Muthurajan, U.M.; Luger, K. Alternative modes of binding of poly-(ADP-ribose) polymerase-1 to free DNA and nucleosomes. J. Biol. Chem. 2012, 287, 32430–32439. [Google Scholar] [CrossRef]
- Sharma, D.; De Falco, L.; Padavattan, S.; Rao, C.; Geifman-Shochat, S.; Liu, C.F.; Davey, C.A. PARP1 exhibits enhanced association and catalytic efficiency with γH2A.X-nucleosome. Nat. Commun. 2019, 10, 5751. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Ngo, T.T.M.; Ha, T. Nucleosomes undergo slow spontaneous gaping. Nucleic Acids Res. 2015, 43, 3964–3971. [Google Scholar] [CrossRef]
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)–Dependent Regulation of DNA Repair by the Chromatin Remodeling Enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Mechetin, G.V.; Nevinsky, G.A. Uracil-DNA glycosylase: Structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 685, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Porecha, R.H.; Stivers, J.T. Uracil-DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc. Natl. Acad. Sci. USA 2008, 105, 10791–10796. [Google Scholar] [CrossRef]
- Hedglin, M.; O’Brien, P.J. Hopping enables a DNA repair glycosylase to search both strands and bypass a bound protein. ACS Chem. Biol. 2010, 5, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef]
- Muthurajan, U.M.; Hepler, M.R.; Hieb, A.R.; Clark, N.J.; Kramer, M.; Yao, T.; Luger, K. Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc. Natl. Acad. Sci. USA 2014, 111, 12752–12757. [Google Scholar] [CrossRef]
- Weaver, T.M.; Hoitsma, N.M.; Spencer, J.J.; Gakhar, L.; Schnicker, N.J.; Freudenthal, B.D. Structural basis for APE1 processing DNA damage in the nucleosome. Nat. Commun. 2022, 13, 5390. [Google Scholar] [CrossRef]
- Widom, J. Equilibrium and dynamic nucleosome stability. Methods Mol. Biol. 1999, 119, 61–77. [Google Scholar]
- Schiessel, H.; Widom, J.; Bruinsma, R.F.; Gelbart, W.M. Polymer reptation and nucleosome repositioning. Phys. Rev. Lett. 2001, 86, 4414–4417. [Google Scholar] [CrossRef]
- Becker, P.B. Nucleosome sliding: Facts and fiction. EMBO J. 2002, 21, 4749–4753. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SHL Position | 5′-Tetramer | 3′-Tetramer | Histone Tail | Accessibility | Chain-Nucl. | Substitution | H-Bonds | Hydrophob. | Salt Bridges | (kcal/mol) | Interface Area (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| +1.5 | TTTT | ACCG | yes | hard | I–15 | T | 1 / 1 | 2 / 2 | 0 / 0 | —13.5/—1.1 | 706 |
| ***** | ** | U | 2 / 2 | 0 / 0 | 0 / 0 | —10.5/—1.3 | 783 | ||||
| 0 (dyad) | GCTG | CCCC | close | easy | I–3 | T | 8 / 4 | 1 / 1 | 1 / 0 | —6.4/—2.6 | 502 |
| ** | **** | U | 4 / 4 | 2 / 1 | 3 / 1 | —9.8/—3.9 | 725 | ||||
| +0.5 | GTCC | CCGC | no | hard | I–5 | T | 0 / 0 | 0 / 0 | 0 / 0 | —3.7/—1.3 | 495 |
| ** | * | U | 1 / 0 | 0 / 0 | 0 / 0 | —5.0/—1.3 | 514 | ||||
| +6.5 | TATA | ACAT | no | medium | I–64 | T | 3 / 2 | 0 / 0 | 1 / 0 | —11.8/—2.1 | 632 |
| *** | *** | U | 1 / 1 | 1 / 1 | 1 / 0 | —12.2/—4.2 | 648 | ||||
| +4.5 | CTCC | GGCA | yes | easy | I–46 | T | 5 / 4 | 2 / 2 | 1 / 1 | —4.9/—3.1 | 773 |
| **** | ** | U | 7 / 4 | 1 / 1 | 2 / 1 | —13.7/—2.4 | 1061 | ||||
| +3 | TTAC | CCCT | no | easy | I–34 | T | 4 / 1 | 0 / 0 | 0 / 0 | —3.2/—3.8 | 622 |
| *** | **** | U | 8 / 6 | 1 / 1 | 0 / 0 | —7.1/—3.1 | 687 | ||||
| —5 | GGCT | CGGC | no | medium | J–53 | T | 9 / 5 | 3 / 1 | 1 / 0 | —10.1/—4.8 | 649 |
| ** | * | U | 3 / 2 | 1 / 1 | 2 / 1 | —11.2/—4.1 | 751 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghediri, S.; Sarma, P.A.P.; Saravanan, V.; Abbadie, C.; Blossey, R.; Cleri, F. Mechanisms of DNA Damage Recognition by UDG and PARP1 in the Nucleosome. Biomolecules 2025, 15, 649. https://doi.org/10.3390/biom15050649
Ghediri S, Sarma PAP, Saravanan V, Abbadie C, Blossey R, Cleri F. Mechanisms of DNA Damage Recognition by UDG and PARP1 in the Nucleosome. Biomolecules. 2025; 15(5):649. https://doi.org/10.3390/biom15050649
Chicago/Turabian StyleGhediri, Safwen, Parvathy A. P. Sarma, Vinnarasi Saravanan, Corinne Abbadie, Ralf Blossey, and Fabrizio Cleri. 2025. "Mechanisms of DNA Damage Recognition by UDG and PARP1 in the Nucleosome" Biomolecules 15, no. 5: 649. https://doi.org/10.3390/biom15050649
APA StyleGhediri, S., Sarma, P. A. P., Saravanan, V., Abbadie, C., Blossey, R., & Cleri, F. (2025). Mechanisms of DNA Damage Recognition by UDG and PARP1 in the Nucleosome. Biomolecules, 15(5), 649. https://doi.org/10.3390/biom15050649