
Modulating the CXCR2 Signaling Axis Using Engineered Chemokine Fusion Proteins to Disrupt Myeloid Cell Infiltration in Pancreatic Cancer
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Subcloning
2.3. Protein Expression and Purification
2.4. Western Blotting
2.5. Migration Assays
2.6. Thermodynamic Stability Assay
2.7. Aggregation Assay
2.8. Production of Lentiviral Particles and Infection of K562 Cells
2.9. Flow Cytometry
2.10. MDSC Isolation from Mouse Spleens
2.11. CyTOF
3. Results
3.1. CXCR2 Ligands Are the Most Abundant Factors Secreted by PDAC Cells
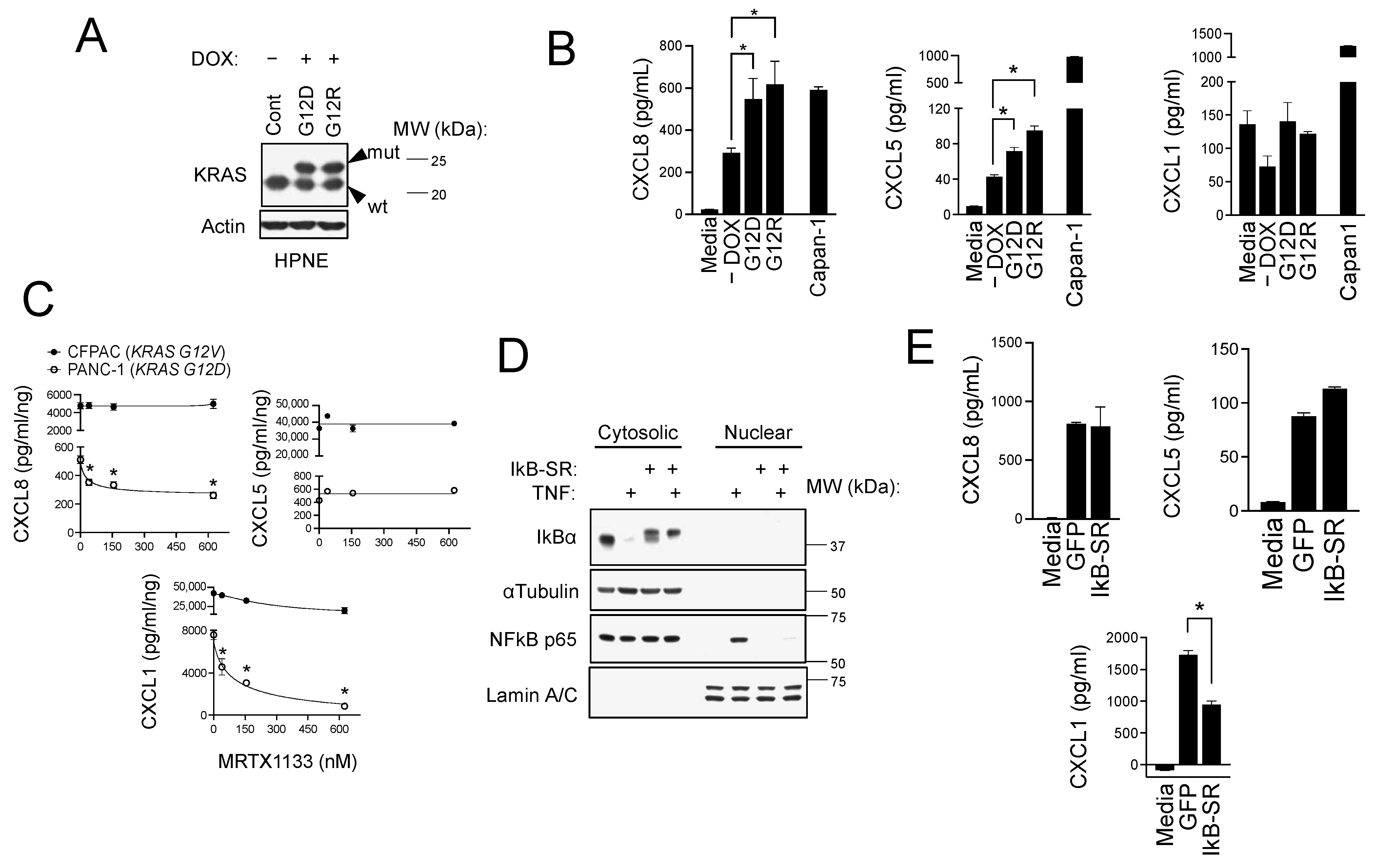
3.2. CXCR2 Ligand Production in PDAC Cells Is Induced by Mutant KRAS and NFκB Signaling
3.3. CXCR2 Is Part of the Immunophenotype of Myeloid-Derived Suppressor Cells (MDSCs) in Mice with PDAC
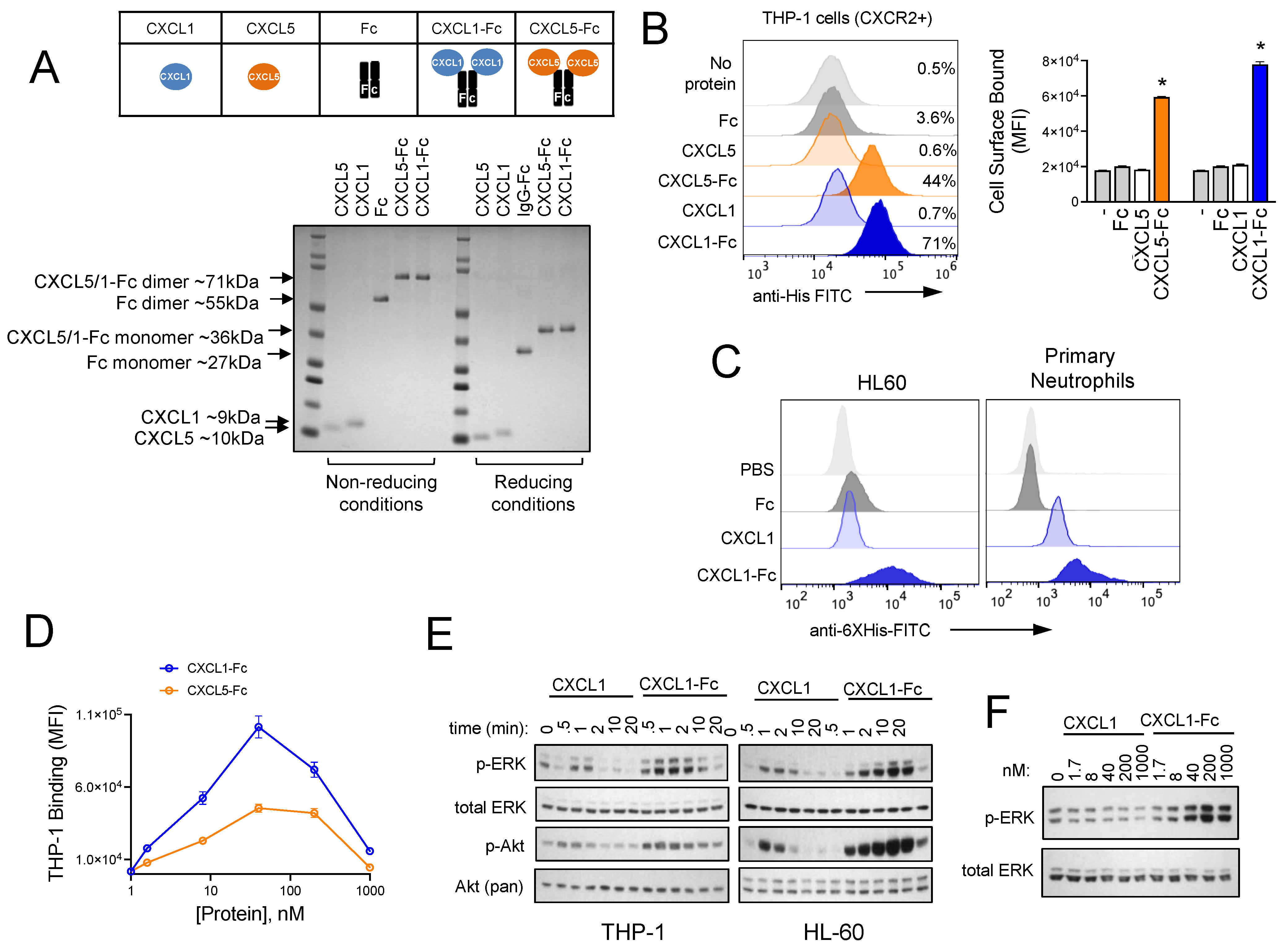
3.4. Design and Characterization of CXCR2 Ligand Fusion Proteins
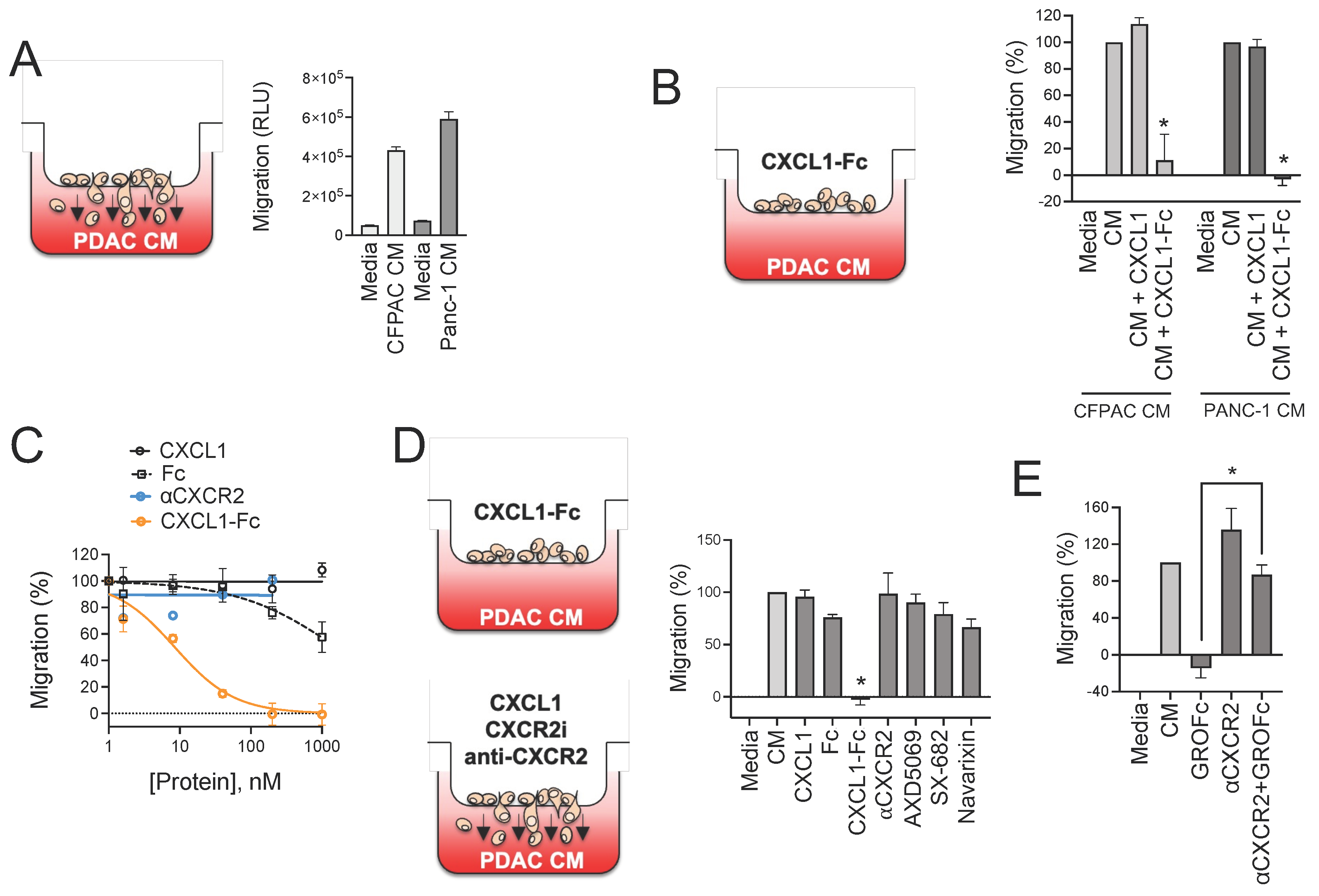
3.5. CXCR2 Ligand Fusion Protein Disrupts Chemotaxis of Myeloid Cells
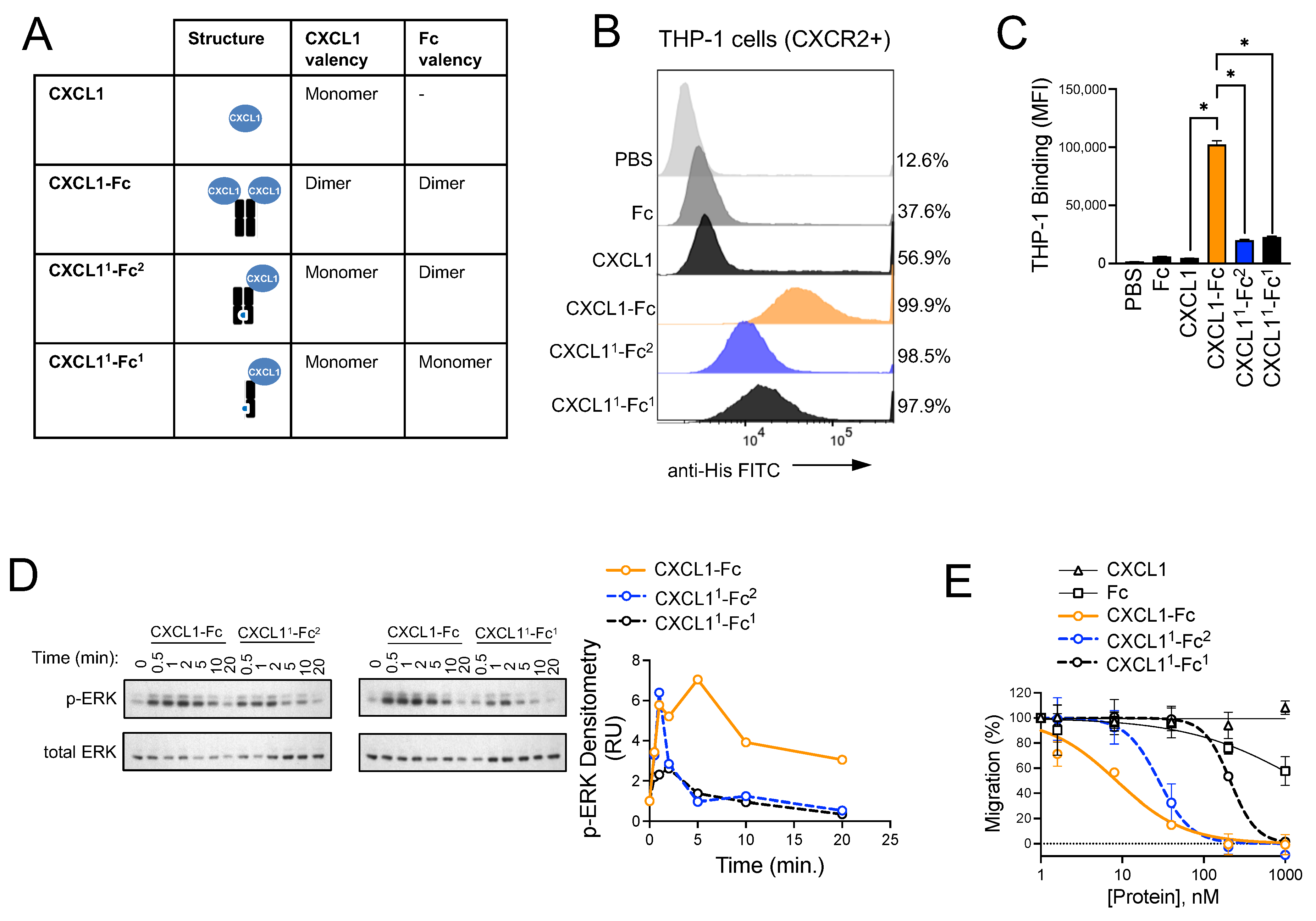
3.6. Structural Investigation of CXCL1-Fc
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Society, A.C. Cancer Facts & Figures 2024; American Cancer Society: Atlanta, GA, USA, 2024. [Google Scholar]
- Murphy, K.J.; Chambers, C.R.; Herrmann, D.; Timpson, P.; Pereira, B.A. Dynamic Stromal Alterations Influence Tumor-Stroma Crosstalk to Promote Pancreatic Cancer and Treatment Resistance. Cancers 2021, 13, 3481. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Increased Levels of Granulocytic Myeloid-Derived Suppressor Cells in Peripheral Blood and Tumour Tissue of Pancreatic Cancer Patients. J. Immunol. Res. 2014, 2014, 879897. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS–Induced Expression of ICAM-1 in Pancreatic Acinar Cells Causes Attraction of Macrophages to Expedite the Formation of Precancerous Lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases During the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Forget, M.-A.; Lucas, F.A.S.; Alvarez, H.A.; Haymaker, C.; Chattopadhyay, C.; Kim, S.-H.; Ekmekcioglu, S.; Grimm, E.A.; et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016, 6, 35848. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.-J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.-W.; et al. Defective Localization With Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10, 496. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal—Redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol. Immunother. 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Sharma, V.; Sachdeva, N.; Gupta, V.; Nada, R.; Jacob, J.; Sahni, D.; Aggarwal, A. IL-6 is associated with expansion of myeloid-derived suppressor cells and enhanced immunosuppression in pancreatic adenocarcinoma patients. Scand. J. Immunol. 2021, 94, e13107. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.; Shi, M.; Chen, Y.; Yu, D.; Zhao, C.; Gu, Y.; Yang, B.; Guo, S.; Ding, G.; et al. CD13hi Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. OncoImmunology 2017, 6, e1258504. [Google Scholar] [CrossRef]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.-J.; Zabaleta, J.; Ochoa, A.C. l-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Y.; Zhao, K.; Yin, K.; Wang, S. Function of reactive oxygen species in myeloid-derived suppressor cells. Front. Immunol. 2023, 14, 1226443. [Google Scholar] [CrossRef]
- Hanson, E.M.; Clements, V.K.; Sinha, P.; Ilkovitch, D.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Down-Regulate L-Selectin Expression on CD4+ and CD8+ T Cells. J. Immunol. 2009, 183, 937–944. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-β1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-Derived Suppressor Cells Promote Cross-Tolerance in B-Cell Lymphoma by Expanding Regulatory T Cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef]
- Beury, D.W.; Parker, K.H.; Nyandjo, M.; Sinha, P.; Carter, K.A.; Ostrand-Rosenberg, S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukoc. Biol. 2014, 96, 1109–1118. [Google Scholar] [CrossRef]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef]
- Wang, F. The Signaling Mechanisms Underlying Cell Polarity and Chemotaxis. Cold Spring Harb. Perspect. Biol. 2009, 1, a002980. [Google Scholar] [CrossRef]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; Van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef]
- Unverdorben, F.; Richter, F.; Hutt, M.; Seifert, O.; Malinge, P.; Fischer, N.; Kontermann, R.E. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. mAbs 2016, 8, 120–128. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Giordano, G.; Febbraro, A.; Venditti, M.; Campidoglio, S.; Olivieri, N.; Raieta, K.; Parcesepe, P.; Imbriani, G.C.; Remo, A.; Pancione, M. Targeting Angiogenesis and Tumor Microenvironment in Metastatic Colorectal Cancer: Role of Aflibercept. Gastroenterol. Res. Pract. 2014, 2014, 526178. [Google Scholar] [CrossRef] [PubMed]
- Shirmohammadi, E.; Ebrahimi, S.-E.S.; Farshchi, A.; Salimi, M. The efficacy of etanercept as anti-breast cancer treatment is attenuated by residing macrophages. BMC Cancer 2020, 20, 836. [Google Scholar] [CrossRef]
- Tan, J.K.; Aphale, A.; Malaviya, R.; Sun, Y.; Gottlieb, A.B. Mechanisms of Action of Etanercept in Psoriasis. J. Investig. Dermatol. Symp. Proc. 2007, 12, 38–45. [Google Scholar] [CrossRef]
- Nowicka, M.; Krieg, C.; Crowell, H.L.; Weber, L.M.; Hartmann, F.J.; Guglietta, S.; Becher, B.; Levesque, M.P.; Robinson, M.D. CyTOF workflow: Differential discovery in high-throughput high-dimensional cytometry datasets. F1000Research 2017, 6, 748. [Google Scholar] [CrossRef]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. Author Correction: High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 1773–1775. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Velez-Delgado, A.; Donahue, K.L.; Brown, K.L.; Du, W.; Irizarry-Negron, V.; Menjivar, R.E.; Lasse Opsahl, E.L.; Steele, N.G.; The, S.; Lazarus, J.; et al. Extrinsic KRAS Signaling Shapes the Pancreatic Microenvironment Through Fibroblast Reprogramming. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1673–1699. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Loukopoulos, P.; Kanetaka, K.; Takamura, M.; Shibata, T.; Sakamoto, M.; Hirohashi, S. Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas 2004, 29, 193–203. [Google Scholar] [CrossRef]
- Moore, P.S.; Sipos, B.; Orlandini, S.; Sorio, C.; Real, F.X.; Lemoine, N.R.; Gress, T.; Bassi, C.; Kloppel, G.; Kalthoff, H.; et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001, 439, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat. Rev. Immunol. 2002, 2, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Fujioka, S.; Sclabas, G.M.; Schmidt, C.; Niu, J.; Frederick, W.A.; Dong, Q.G.; Abbruzzese, J.L.; Evans, D.B.; Baker, C.; Chiao, P.J. Inhibition of constitutive NF-kappa B activity by I kappa B alpha M suppresses tumorigenesis. Oncogene 2003, 22, 1365–1370. [Google Scholar] [CrossRef]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Leabman, M.K.; Meng, Y.G.; Kelley, R.F.; Deforge, L.E.; Cowan, K.J.; Iyer, S. Effects of altered FcγR binding on antibody pharmacokinetics in cynomolgus monkeys. mAbs 2013, 5, 896–903. [Google Scholar] [CrossRef]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef]
- Sano, M.; Ijichi, H.; Takahashi, R.; Miyabayashi, K.; Fujiwara, H.; Yamada, T.; Kato, H.; Nakatsuka, T.; Tanaka, Y.; Tateishi, K.; et al. Blocking CXCLs-CXCR2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis 2019, 8, 8. [Google Scholar] [CrossRef]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, D.R.; Molczyk, C.; Purohit, A.; Saxena, S.; Sturgeon, R.; Dave, B.J.; Kumar, S.; Batra, S.K.; Singh, R.K. Small molecule antagonist of CXCR2 and CXCR1 inhibits tumor growth, angiogenesis, and metastasis in pancreatic cancer. Cancer Lett. 2023, 563, 216185. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, C.; Vilgelm, A.E.; Chen, S.C.; Ayers, G.D.; Johnson, C.A.; Richmond, A. Targeted Deletion of CXCR2 in Myeloid Cells Alters the Tumor Immune Environment to Improve Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Jazayeri, J.A.; Carroll, G.J. Fc-based cytokines: Prospects for engineering superior therapeutics. BioDrugs 2008, 22, 11–26. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Caronni, N.; La Terza, F.; Vittoria, F.M.; Barbiera, G.; Mezzanzanica, L.; Cuzzola, V.; Barresi, S.; Pellegatta, M.; Canevazzi, P.; Dunsmore, G.; et al. IL-1β+ macrophages fuel pathogenic inflammation in pancreatic cancer. Nature 2023, 623, 415–422. [Google Scholar] [CrossRef]
- Sitaru, S.; Budke, A.; Bertini, R.; Sperandio, M. Therapeutic inhibition of CXCR1/2: Where do we stand? Intern. Emerg. Med. 2023, 18, 1647–1664. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kuang, W.; Wang, D.; Yuan, K.; Yang, P. Expanding role of CXCR2 and therapeutic potential of CXCR2 antagonists in inflammatory diseases and cancers. Eur. J. Med. Chem. 2023, 250, 115175. [Google Scholar] [CrossRef] [PubMed]
- Lazaar, A.L.; Miller, B.E.; Donald, A.C.; Keeley, T.; Ambery, C.; Russell, J.; Watz, H.; Tal-Singer, R.; Bardin, P.; Bremner, P.; et al. CXCR2 antagonist for patients with chronic obstructive pulmonary disease with chronic mucus hypersecretion: A phase 2b trial. Respir. Res. 2020, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Geva, R.; Chung, H.C.; Lemech, C.; Miller, W.H.; Hansen, A.R.; Lee, J.-S.; Tsai, F.; Solomon, B.J.; Kim, T.M.; et al. CXCR2 antagonist navarixin in combination with pembrolizumab in select advanced solid tumors: A phase 2 randomized trial. Investig. New Drugs 2024, 42, 145–159. [Google Scholar] [CrossRef]
- Gu, H.; Deng, W.; Zheng, Z.; Wu, K.; Sun, F. CCL2 produced by pancreatic ductal adenocarcinoma is essential for the accumulation and activation of monocytic myeloid-derived suppressor cells. Immun. Inflamm. Dis. 2021, 9, 1686–1695. [Google Scholar] [CrossRef]
- Korbecki, J.; Kupnicka, P.; Chlubek, M.; Goracy, J.; Gutowska, I.; Baranowska-Bosiacka, I. CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int. J. Mol. Sci. 2022, 23, 2168. [Google Scholar] [CrossRef]
- Jin, T.; Xu, X.; Hereld, D. Chemotaxis, chemokine receptors and human disease. Cytokine 2008, 44, 1–8. [Google Scholar] [CrossRef]
- Devalaraja, R.M.; Nanney, L.B.; Qian, Q.; Du, J.; Yu, Y.; Devalaraja, M.N.; Richmond, A. Delayed Wound Healing in CXCR2 Knockout Mice. J. Investig. Dermatol. 2000, 115, 234–244. [Google Scholar] [CrossRef]
- Öhnstedt, E.; Vågesjö, E.; Fasth, A.; Lofton Tomenius, H.; Dahg, P.; Jönsson, S.; Tyagi, N.; Åström, M.; Myktybekova, Z.; Ringstad, L.; et al. Engineered bacteria to accelerate wound healing: An adaptive, randomised, double-blind, placebo-controlled, first-in-human phase 1 trial. eClinicalMedicine 2023, 60, 102014. [Google Scholar] [CrossRef]
- Yang, H.; Liu, L.; Xu, F. The promises and challenges of fusion constructs in protein biochemistry and enzymology. Appl. Microbiol. Biotechnol. 2016, 100, 8273–8281. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christopher, B.N.; Golick, L.; Basar, A.; Reyes, L.; Robinson, R.M.; Angerstein, A.O.; Krieg, C.; Hobbs, G.A.; Guttridge, D.C.; O’Bryan, J.P.; et al. Modulating the CXCR2 Signaling Axis Using Engineered Chemokine Fusion Proteins to Disrupt Myeloid Cell Infiltration in Pancreatic Cancer. Biomolecules 2025, 15, 645. https://doi.org/10.3390/biom15050645
Christopher BN, Golick L, Basar A, Reyes L, Robinson RM, Angerstein AO, Krieg C, Hobbs GA, Guttridge DC, O’Bryan JP, et al. Modulating the CXCR2 Signaling Axis Using Engineered Chemokine Fusion Proteins to Disrupt Myeloid Cell Infiltration in Pancreatic Cancer. Biomolecules. 2025; 15(5):645. https://doi.org/10.3390/biom15050645
Chicago/Turabian StyleChristopher, Benjamin N., Lena Golick, Ashton Basar, Leticia Reyes, Reeder M. Robinson, Aaron O. Angerstein, Carsten Krieg, G. Aaron Hobbs, Denis C. Guttridge, John P. O’Bryan, and et al. 2025. "Modulating the CXCR2 Signaling Axis Using Engineered Chemokine Fusion Proteins to Disrupt Myeloid Cell Infiltration in Pancreatic Cancer" Biomolecules 15, no. 5: 645. https://doi.org/10.3390/biom15050645
APA StyleChristopher, B. N., Golick, L., Basar, A., Reyes, L., Robinson, R. M., Angerstein, A. O., Krieg, C., Hobbs, G. A., Guttridge, D. C., O’Bryan, J. P., & Dolloff, N. G. (2025). Modulating the CXCR2 Signaling Axis Using Engineered Chemokine Fusion Proteins to Disrupt Myeloid Cell Infiltration in Pancreatic Cancer. Biomolecules, 15(5), 645. https://doi.org/10.3390/biom15050645