
Pharmacological Modulation of the Unfolded Protein Response as a Therapeutic Approach in Cutaneous T-Cell Lymphoma
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Culture
2.3. Flow Cytometry
2.4. Western Blot
2.5. Rt-qPCR
2.6. Cell Viability Assays
2.7. Production of HSPA5, HSPA6, and the Substrate-Binding Domain of HSPA5 and HSPA6
2.8. Fluorescence Polarization Assay
2.9. Determining Effect of CTCL Secreted Factors on Naïve T-Cell Activation via IFN-γ ELISA
2.10. Cytokine Bead Array
3. Results
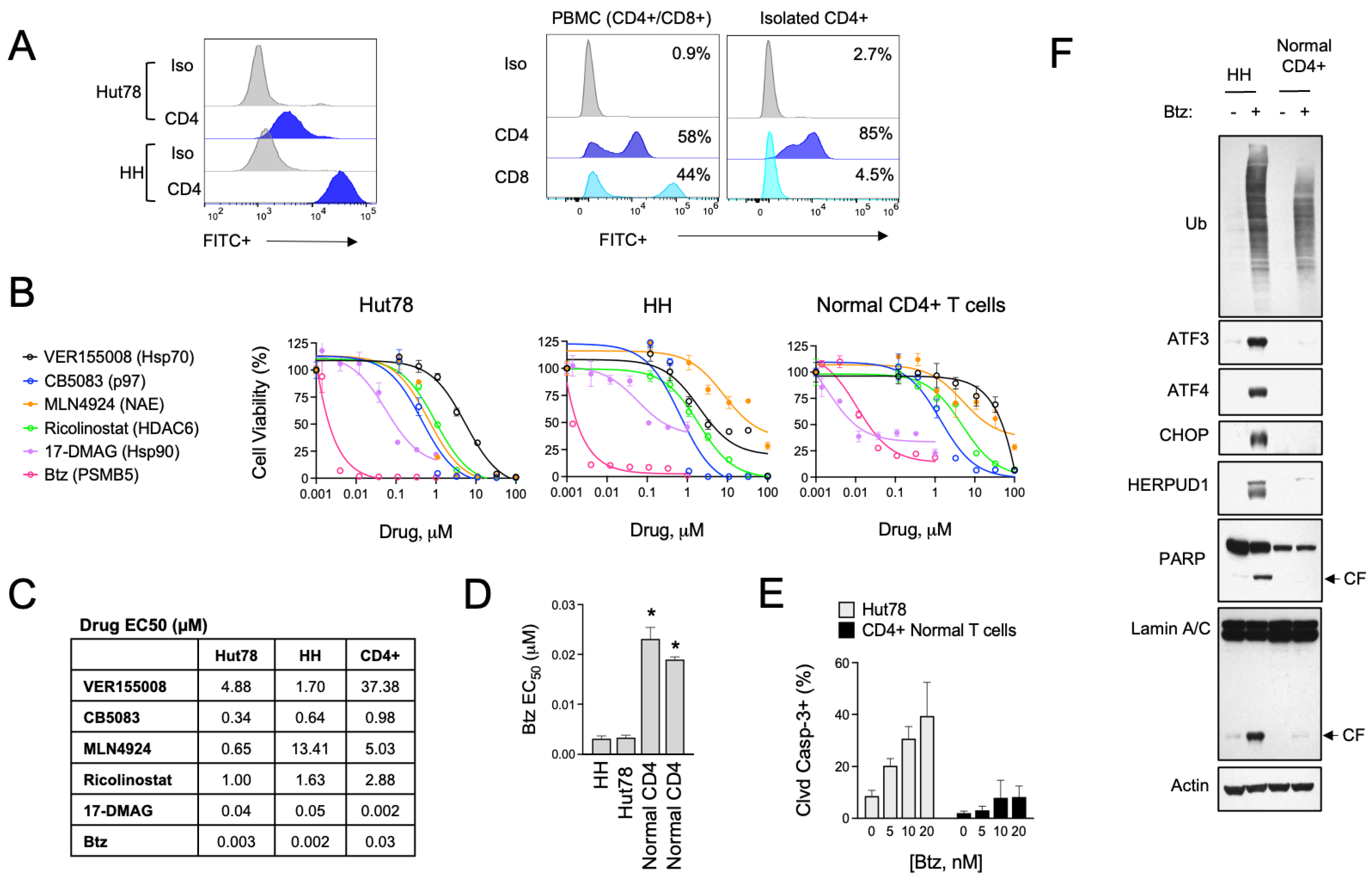
3.1. Divergent ER Stress Response Signaling in CTCL and Normal CD4+ T Cells
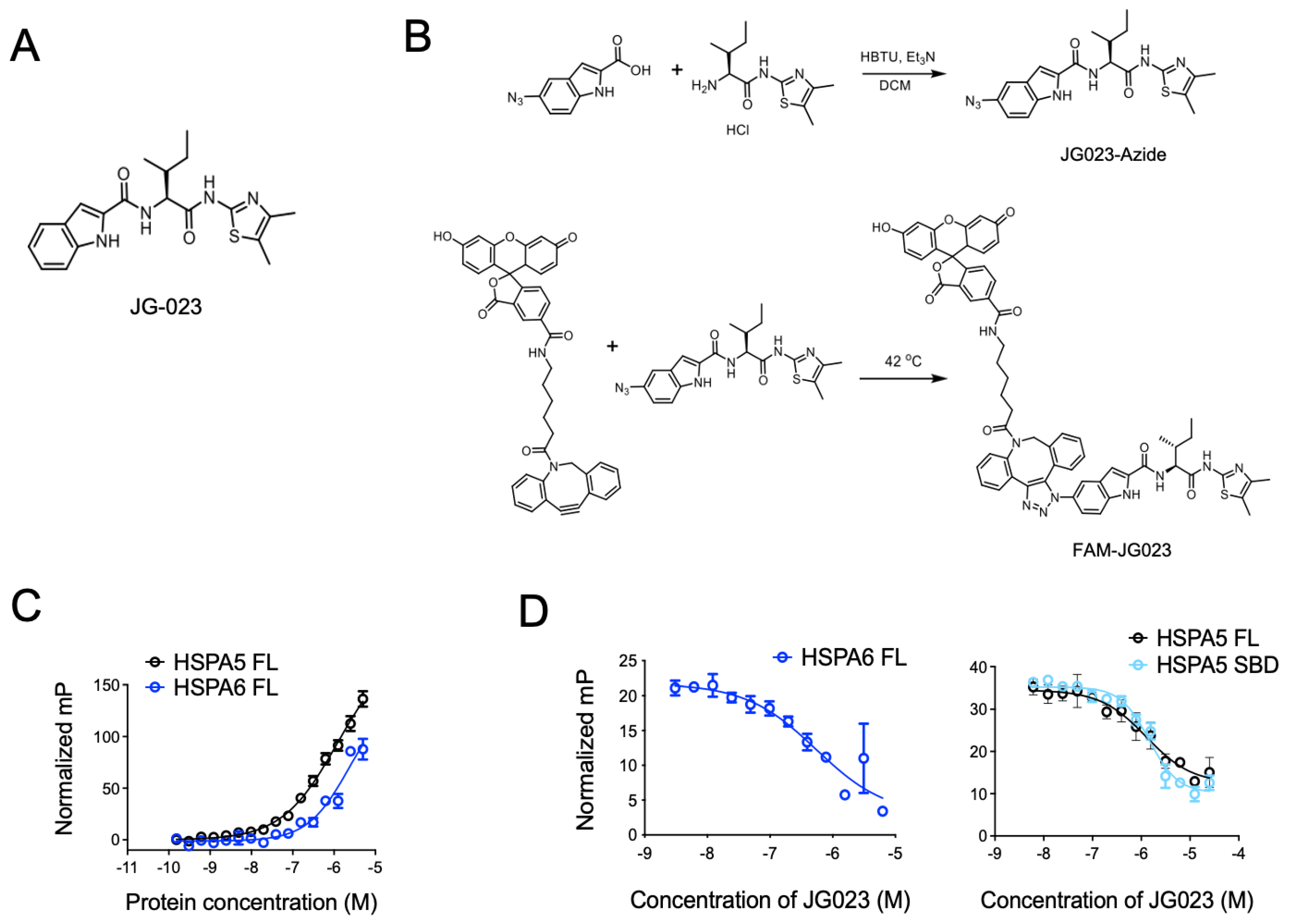
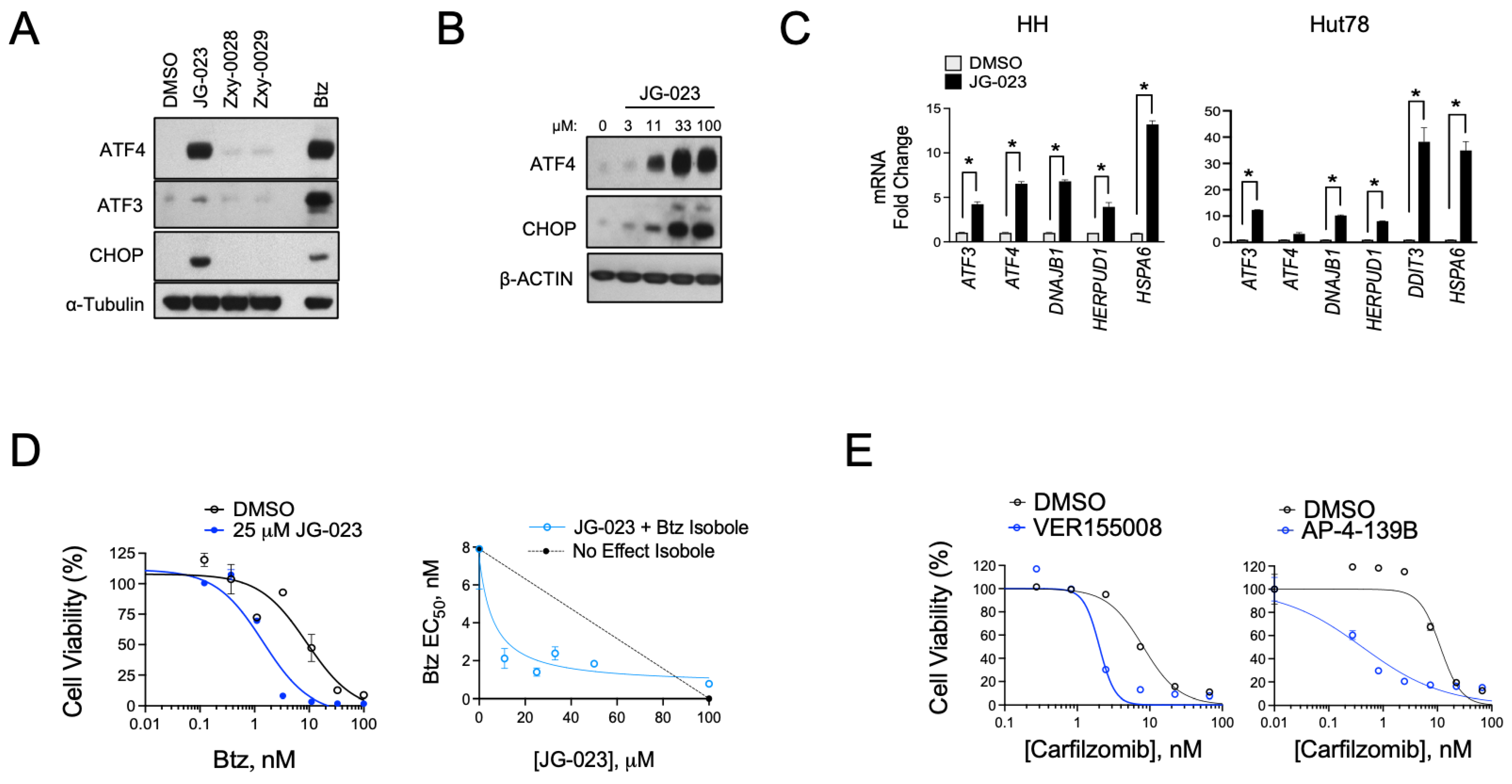
3.2. HSPA5/6 Play a Dominant Role in the ER Stress Response in CTCL Cells
3.3. HSPA5/6-Specific Inhibition Enhances ER Stress-Induced Death of CTCL Cells
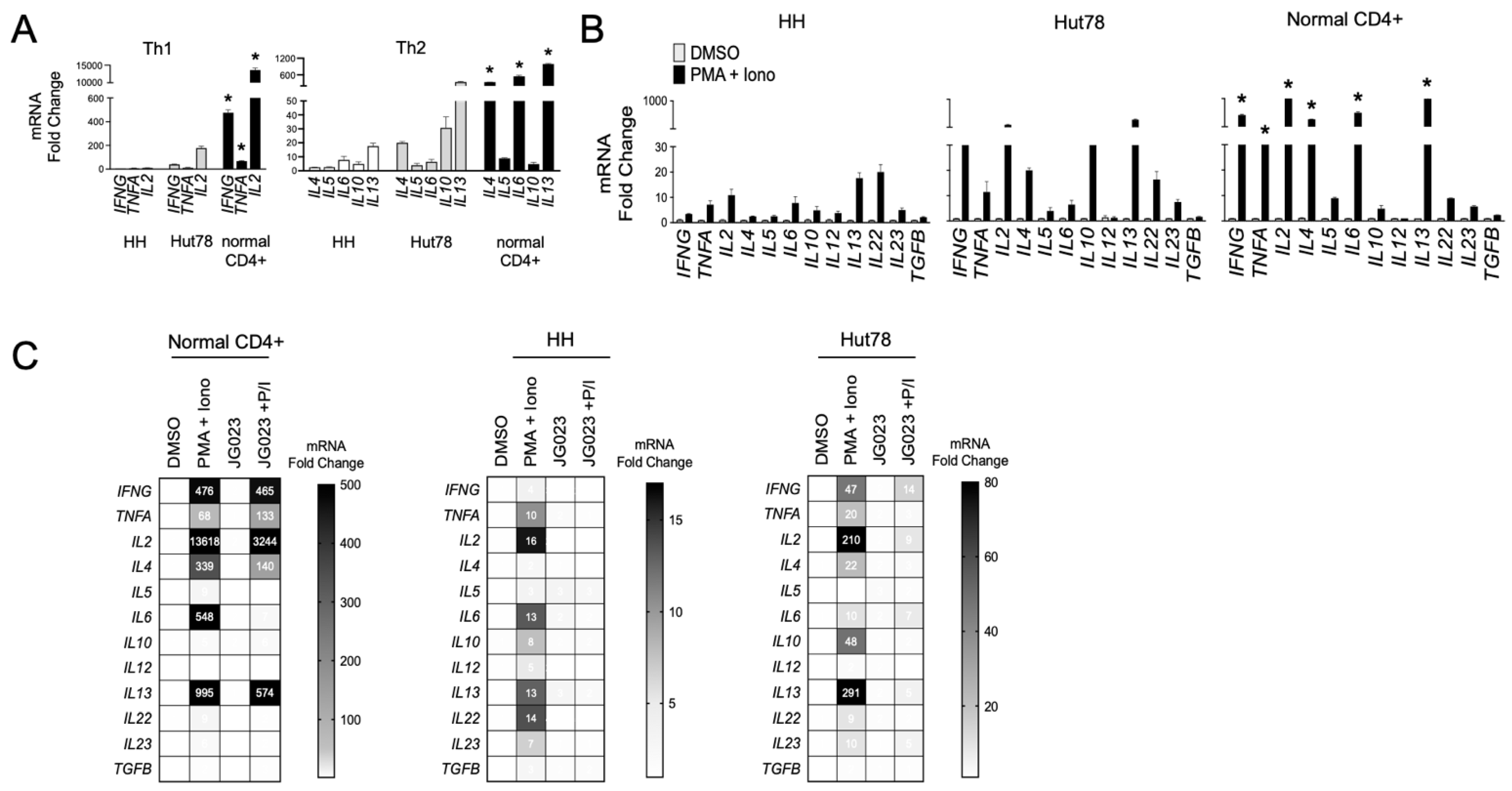
3.4. HSPA5/6 Inhibition Suppresses Th2 Cytokine Production by CTCL Cells
3.5. CTCL-Secreted Factors Suppress Normal T-Cell Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olsen, E.; Vonderheid, E.; Pimpinelli, N.; Willemze, R.; Kim, Y.; Knobler, R.; Zackheim, H.; Duvic, M.; Estrach, T.; Lamberg, S.; et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007, 110, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Alpdogan, O.; Kartan, S.; Johnson, W.; Sokol, K.; Porcu, P. Systemic therapy of cutaneous T-cell lymphoma (CTCL). Chin. Clin. Oncol. 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Liu, H.L.; Mraz-Gernhard, S.; Varghese, A.; Hoppe, R.T. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: Clinical prognostic factors and risk for disease progression. Arch. Dermatol. 2003, 139, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Scarisbrick, J.J.; Bagot, M.; Ortiz-Romero, P.L. The changing therapeutic landscape, burden of disease, and unmet needs in patients with cutaneous T-cell lymphoma. Br. J. Haematol. 2021, 192, 683–696. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.H.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sezary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic landscape of cutaneous T cell lymphoma. Nat. Genet. 2015, 47, 1011–1019. [Google Scholar] [CrossRef]
- da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef]
- McGirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, X.; Hwang, S.T.; Liu, J. The Role of Tumor Microenvironment in Mycosis Fungoides and Sezary Syndrome. Ann. Dermatol. 2021, 33, 487–496. [Google Scholar] [CrossRef]
- Patil, K.; Kuttikrishnan, S.; Khan, A.Q.; Ahmad, F.; Alam, M.; Buddenkotte, J.; Ahmad, A.; Steinhoff, M.; Uddin, S. Molecular pathogenesis of Cutaneous T cell Lymphoma: Role of chemokines, cytokines, and dysregulated signaling pathways. Semin. Cancer Biol. 2022, 86, 382–399. [Google Scholar] [CrossRef]
- Miyashiro, D.; Souza, B.d.C.e.; Torrealba, M.P.; Manfrere, K.C.G.; Sato, M.N.; Sanches, J.A. The Role of Tumor Microenvironment in the Pathogenesis of Sezary Syndrome. Int. J. Mol. Sci. 2022, 23, 936. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jin, S.; Hu, S.; Li, R.; Pan, H.; Liu, Y.; Lai, P.; Xu, D.; Sun, J.; Liu, Z.; et al. Single-cell transcriptomics links malignant T cells to the tumor immune landscape in cutaneous T cell lymphoma. Nat. Commun. 2022, 13, 1158. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: A biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Vowels, B.R.; Lessin, S.R.; Cassin, M.; Jaworsky, C.; Benoit, B.; Wolfe, J.T.; Rook, A.H. Th2 cytokine mRNA expression in skin in cutaneous T-cell lymphoma. J. Investig. Dermatol. 1994, 103, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Rook, A.H.; Vowels, B.R.; Jaworsky, C.; Singh, A.; Lessin, S.R. The immunopathogenesis of cutaneous T-cell lymphoma. Abnormal cytokine production by Sezary T cells. Arch. Dermatol. 1993, 129, 486–489. [Google Scholar] [CrossRef]
- Kural, Y.B.; Su, O.; Onsun, N.; Uras, A.R. Atopy, IgE and eosinophilic cationic protein concentration, specific IgE positivity, eosinophil count in cutaneous T Cell lymphoma. Int. J. Dermatol. 2010, 49, 390–395. [Google Scholar] [CrossRef]
- Spellberg, B.; Edwards, J.E., Jr. Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Walker, J.A.; McKenzie, A.N.J. T(H)2 cell development and function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef]
- Axelrod, P.I.; Lorber, B.; Vonderheid, E.C. Infections complicating mycosis fungoides and Sezary syndrome. JAMA 1992, 267, 1354–1358. [Google Scholar] [CrossRef]
- Nguyen, V.; Huggins, R.H.; Lertsburapa, T.; Bauer, K.; Rademaker, A.; Gerami, P.; Guitart, J. Cutaneous T-cell lymphoma and Staphylococcus aureus colonization. J. Am. Acad. Dermatol. 2008, 59, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sezary syndrome. Br. J. Dermatol. 2008, 159, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Willerslev-Olsen, A.; Krejsgaard, T.; Lindahl, L.M.; Bonefeld, C.M.; Wasik, M.A.; Koralov, S.B.; Geisler, C.; Kilian, M.; Iversen, L.; Woetmann, A.; et al. Bacterial toxins fuel disease progression in cutaneous T-cell lymphoma. Toxins 2013, 5, 1402–1421. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Todd, D.J.; McHeyzer-Williams, L.J.; Kowal, C.; Lee, A.-H.; Volpe, B.T.; Diamond, B.; McHeyzer-Williams, M.G.; Glimcher, L.H. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 2009, 206, 2151–2159. [Google Scholar] [CrossRef]
- Henriksson, J.; Chen, X.; Gomes, T.; Ullah, U.; Meyer, K.B.; Miragaia, R.; Duddy, G.; Pramanik, J.; Yusa, K.; Lahesmaa, R.; et al. Genome-Wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk Between Activation and Differentiation. Cell 2019, 176, 882–896.e18. [Google Scholar] [CrossRef]
- Kemp, K.L.; Lin, Z.; Zhao, F.; Gao, B.; Song, J.; Zhang, K.; Fang, D. The serine-threonine kinase inositol-requiring enzyme 1alpha (IRE1alpha) promotes IL-4 production in T helper cells. J. Biol. Chem. 2013, 288, 33272–33282. [Google Scholar] [CrossRef]
- Ambrose, A.J.; Sivinski, J.; Zerio, C.J.; Zhu, X.; Godek, J.; Kumirov, V.K.; Brujas, T.C.; Garcia, J.T.; Annadurai, A.; Schmidlin, C.J.; et al. Discovery and Development of a Selective Inhibitor of the ER Resident Chaperone Grp78. J. Med. Chem. 2023, 66, 677–694. [Google Scholar] [CrossRef]
- Duncan, R.M.; Reyes, L.; Moats, K.; Robinson, R.M.; Murphy, S.A.; Kaur, B.; Stessman, H.A.; Dolloff, N.G. ATF3 Coordinates Antitumor Synergy Between Epigenetic Drugs and Protein Disulfide Isomerase Inhibitors. Cancer Res. 2020, 80, 3279–3291. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.M.; Basar, A.P.; Reyes, L.; Duncan, R.M.; Li, H.; Dolloff, N.G. PDI inhibitor LTI6426 enhances panobinostat efficacy in preclinical models of multiple myeloma. Cancer Chemother. Pharmacol. 2022, 89, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Klock, H.E.; Lesley, S.A. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 2009, 498, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Boumsell, L.; Bernard, A.; Reinherz, E.L.; Nadler, L.M.; Ritz, J.; Coppin, H.; Richard, Y.; Dubertret, L.; Valensi, F.; Degos, L.; et al. Surface antigens on malignant Sezary and T-CLL cells correspond to those of mature T cells. Blood 1981, 57, 526–530. [Google Scholar] [CrossRef]
- Rook, A.H.; Gottlieb, S.L.; Wolfe, J.T.; Vowels, B.R.; Sood, S.S.; Niu, Z.; Lessin, S.R.; Fox, F.E. Pathogenesis of cutaneous T-cell lymphoma: Implications for the use of recombinant cytokines and photopheresis. Clin. Exp. Immunol. 1997, 107 (Suppl. S1), 16–20. [Google Scholar]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2019, 33, 1011–1022. [Google Scholar] [CrossRef]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Strobel, E.D.; Hyman, S.L.; Reitz, A.B.; Dolloff, N.G. Tuning isoform selectivity and bortezomib sensitivity with a new class of alkenyl indene PDI inhibitor. Eur. J. Med. Chem. 2020, 186, 111906. [Google Scholar] [CrossRef]
- Brocchieri, L.; de Macario, E.C.; Macario, A.J. hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol. Biol. 2008, 8, 19. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.R.; Lee, A.S. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, T.; Leung, J.C.; Leu, J.I.-J.; Basu, S.; Poli, A.N.R.; Parris, J.L.; Indeglia, A.; Martynyuk, T.; Good, M.; Gnanapradeepan, K.; et al. A Novel Inhibitor of HSP70 Induces Mitochondrial Toxicity and Immune Cell Recruitment in Tumors. Cancer Res. 2020, 80, 5270–5281. [Google Scholar] [CrossRef] [PubMed]
- Asadullah, K.; Docke, W.D.; Haeussler, A.; Sterry, W.; Volk, H.D. Progression of mycosis fungoides is associated with increasing cutaneous expression of interleukin-10 mRNA. J. Investig. Dermatol. 1996, 107, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Miyagaki, T.; Sugaya, M.; Suga, H.; Kamata, M.; Ohmatsu, H.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. IL-22, but not IL-17, dominant environment in cutaneous T-cell lymphoma. Clin. Cancer Res. 2011, 17, 7529–7538. [Google Scholar] [CrossRef]
- Gluud, M.; Pallesen, E.M.H.; Buus, T.B.; Gjerdrum, L.M.R.; Lindahl, L.M.; Kamstrup, M.R.; Bzorek, M.; Danielsen, M.; Bech, R.; Monteiro, M.N.; et al. Malignant T cells induce skin barrier defects through cytokine-mediated JAK/STAT signaling in cutaneous T-cell lymphoma. Blood 2023, 141, 180–193. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, F.; Zhu, X.; Leone, V.A.; Dalal, S.; Tao, Y.; Messer, J.S.; Chang, E.B. Distinct roles of intracellular heat shock protein 70 in maintaining gastrointestinal homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G164–G178. [Google Scholar] [CrossRef]
- Wachstein, J.; Tischer, S.; Figueiredo, C.; Limbourg, A.; Falk, C.; Immenschuh, S.; Blasczyk, R.; Eiz-Vesper, B. HSP70 enhances immunosuppressive function of CD4+CD25+FoxP3+ T regulatory cells and cytotoxicity in CD4+CD25− T cells. PLoS ONE 2012, 7, e51747. [Google Scholar] [CrossRef]
- Steinke, J.W.; Borish, L. Th2 cytokines and asthma. Interleukin-4: Its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir. Res. 2001, 2, 66–70. [Google Scholar] [CrossRef]
- Patel, N.; Kreider, T.; Urban, J.F., Jr.; Gause, W.C. Characterisation of effector mechanisms at the host: Parasite interface during the immune response to tissue-dwelling intestinal nematode parasites. Int. J. Parasitol. 2009, 39, 13–21. [Google Scholar] [CrossRef]
- Webster, H.C.; Gamino, V.; Andrusaite, A.T.; Ridgewell, O.J.; McCowan, J.; Shergold, A.L.; Heieis, G.A.; Milling, S.W.; Maizels, R.M.; Perona-Wright, G. Tissue-based IL-10 signalling in helminth infection limits IFNgamma expression and promotes the intestinal Th2 response. Mucosal Immunol. 2022, 15, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Murphy, K.M.; Sher, A. Functional diversity of helper T lymphocytes. Nature 1996, 383, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin. Cancer Res. 2013, 19, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Kemp, K.; Poe, C. Stressed: The Unfolded Protein Response in T Cell Development, Activation, and Function. Int. J. Mol. Sci. 2019, 20, 1792. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Q.; Wang, H.; Xia, S. Endoplasmic reticulum stress in T cell-mediated diseases. Scand. J. Immunol. 2023, 98, e13307. [Google Scholar] [CrossRef]
- Thaxton, J.E.; Wallace, C.; Riesenberg, B.; Zhang, Y.; Paulos, C.M.; Beeson, C.C.; Liu, B.; Li, Z. Modulation of Endoplasmic Reticulum Stress Controls CD4+ T-Cell Activation and Antitumor Function. Cancer Immunol. Res. 2017, 5, 666–675. [Google Scholar] [CrossRef]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef]
- Schleicher, S.M.; Moretti, L.; Varki, V.; Lu, B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: Implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 79–86. [Google Scholar] [CrossRef]
- Stone, K.R.; Smith, R.E.; Joklik, W.K. Changes in membrane polypeptides that occur when chick embryo fibroblasts and NRK cells are transformed with avian sarcoma viruses. Virology 1974, 58, 86–100. [Google Scholar] [CrossRef]
- Dong, D.; Stapleton, C.; Luo, B.; Xiong, S.; Ye, W.; Zhang, Y.; Jhaveri, N.; Zhu, G.; Ye, R.; Liu, Z.; et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011, 71, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Liu, Y.; Wang, X.; Ren, M. Multidimensional pan-cancer analysis of HSPA5 and its validation in the prognostic value of bladder cancer. Heliyon 2024, 10, E27184. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Parsian, A.J.; Sheren, J.E.; Tao, T.Y.; Goswami, P.C.; Malyapa, R.; Van Rheeden, R.; Watson, M.S.; Hunt, C.R. The human Hsp70B gene at the HSPA7 locus of chromosome 1 is transcribed but non-functional. Biochim. Biophys. Acta 2000, 1494, 201–205. [Google Scholar] [CrossRef]
- Leung, T.K.; Rajendran, M.Y.; Monfries, C.; Hall, C.; Lim, L. The human heat-shock protein family. Expression of a novel heat-inducible HSP70 (HSP70B’) and isolation of its cDNA and genomic DNA. Biochem. J. 1990, 267, 125–132. [Google Scholar] [CrossRef]
- Hammad, S.M.; Twal, W.O.; Barth, J.L.; Smith, K.J.; Saad, A.F.; Virella, G.; Argraves, W.S.; Lopes-Virella, M.F. Oxidized LDL immune complexes and oxidized LDL differentially affect the expression of genes involved with inflammation and survival in human U937 monocytic cells. Atherosclerosis 2009, 202, 394–404. [Google Scholar] [CrossRef]
- Smith, K.J.; Twal, W.O.; Soodavar, F.; Virella, G.; Lopes-Virella, M.F.; Hammad, S.M. Heat shock protein 70B’ (HSP70B’) expression and release in response to human oxidized low density lipoprotein immune complexes in macrophages. J. Biol. Chem. 2010, 285, 15985–15993. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef]
- McConkey, D.J.; Zhu, K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. Updates 2008, 11, 164–179. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark. Res. 2013, 1, 13. [Google Scholar] [CrossRef]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Song, N.J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2019, 10, 3154. [Google Scholar] [CrossRef] [PubMed]
- Staron, M.; Yang, Y.; Liu, B.; Li, J.; Shen, Y.; Zúñiga-Pflücker, J.C.; Aguila, H.L.; Goldschneider, I.; Li, Z. gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood 2010, 115, 2380–2390. [Google Scholar] [CrossRef]
- Lee, W.H.; Park, Y.M.; Kim, J.; Jang, J.J.; Seo, J.S. Expression of heat shock protein 70 blocks thymic differentiation of T cells in transgenic mice. Immunology 1998, 95, 559–565. [Google Scholar] [CrossRef]
- Mycko, M.P.; Cwiklinska, H.; Walczak, A.; Libert, C.; Raine, C.S.; Selmaj, K.W. A heat shock protein gene (Hsp70.1) is critically involved in the generation of the immune response to myelin antigen. Eur. J. Immunol. 2008, 38, 1999–2013. [Google Scholar] [CrossRef]
- Mansilla, M.J.; Costa, C.; Eixarch, H.; Tepavcevic, V.; Castillo, M.; Martin, R.; Lubetzki, C.; Aigrot, M.-S.; Montalban, X.; Espejo, C. Hsp70 regulates immune response in experimental autoimmune encephalomyelitis. PLoS ONE 2014, 9, e105737. [Google Scholar] [CrossRef]
- Pramanik, J.; Chen, X.; Kar, G.; Henriksson, J.; Gomes, T.; Park, J.-E.; Natarajan, K.; Meyer, K.B.; Miao, Z.; McKenzie, A.N.J.; et al. Genome-wide analyses reveal the IRE1a-XBP1 pathway promotes T helper cell differentiation by resolving secretory stress and accelerating proliferation. Genome Med. 2018, 10, 76. [Google Scholar] [CrossRef]
- Weiner, D.M.; Durgin, J.S.; Wysocka, M.; Rook, A.H. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: Current and future approaches. J. Am. Acad. Dermatol. 2021, 84, 597–604. [Google Scholar] [CrossRef]
- Tendler, C.L.; Burton, J.D.; Jaffe, J.; Danielpour, D.; Charley, M.; McCoy, J.P.; Pittelkow, M.R.; Waldmann, T.A. Abnormal cytokine expression in Sezary and adult T-cell leukemia cells correlates with the functional diversity between these T-cell malignancies. Cancer Res. 1994, 54, 4430–4435. [Google Scholar]
- Anthony, R.M.; Rutitzky, L.I.; Urban, J.F.; Jr Stadecker, M.J.; Gause, W.C. Protective immune mechanisms in helminth infection. Nat. Rev. Immunol. 2007, 7, 975–987. [Google Scholar] [CrossRef]
- Chen, F.; Liu, Z.; Wu, W.; Rozo, C.; Bowdridge, S.; Millman, A.; Van Rooijen, N.; Urban, J.F., Jr.; Wynn, T.A.; Gause, W.C. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med. 2012, 18, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; Rosenstein, R.K.; Medzhitov, R. Allergic host defences. Nature 2012, 484, 465–472. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.; Kent, A.; Sharp, E.; Schmidt, F.; Turini, M. Mycosis fungoides-type cutaneous T-cell lymphoma (MF-CTCL) epidemiology and treatment pathway in Spain: New insights for an accurate description. Drugs Context 2020, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goldgeier, M.H.; Cohen, S.R.; Braverman, I.M.; Stenn, K.S. An unusual and fatal case of disseminated cutaneous herpes simplex. Infection in a patient with cutaneous T cell lymphoma (mycosis fungoides). J. Am. Acad. Dermatol. 1981, 4, 176–180. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | ||
|---|---|---|
| Protein Target | Vendor | Catalog # |
| β-actin | Sigma | A5441 |
| ATF3 | Cell Signaling | 33593S |
| ATF4 | Cell Signaling | 11815S |
| BiP | Cell Signaling | 3183S |
| CHOP | Cell Signaling | 2895S |
| eIF2α | Cell Signaling | 5324T |
| p-eIF2α | Cell Signaling | 3398S |
| HERPUD1 | Cell Signaling | 26730 |
| HSC70/HSPA8 | Santa Cruz | 71270 |
| HSPA6 | Santa Cruz | 374589 |
| HSP70 | Cell Signaling | 4872S |
| Tubulin | Cell Signaling | 3873S |
| Ubiquitin | Cell Signaling | 3936S |
| XBP1S | Cell Signaling | 12782S |
| Secondary Antibodies | Vendor | Catalog # |
| Goat anti-Mouse IgG-H + L | Invitrogen | 31430 |
| Goat anti-Rabbit IgG-H + L | Invitrogen | 31480 |
| Goat anti-Rat IgG-H + L | Invitrogen | 9520 |
| Target | Fwd (5′-3′) | Rvs (5′-3′) |
|---|---|---|
| ATF3 | GGAGTGCCTGCAGAAAGAGT | CCATTCTGAGCCCGG ACAAT |
| ATF4 | GACGGAGCGCTTTCCTCTT | TCCACAAAATGGACGCTCAC |
| DDIT3 | GGAAACAGAGTGGTCATTCCC | CTGCTTGAGCCGTTCATTCTC |
| DNAJB1 | CCAGTCACCCACGACCTTC | CCCTTCTTCACTTCGATGGTCA |
| HERPUD1 | CCGGTTACACACCCTATGGG | TGAGGAGCAGCATTCTGATTG |
| HSPA1A | GGCCTTGAGGACTTTGGGTTA | TGGGAATGCAAAGCACACG |
| HSPA1B | GGGAGGACTTCGACAACAGG | GACAAGGTTCTCTTGGCCCG |
| HSPA1L | AAAGCAGGTCAGGGAGAGCGA | GGAGGGATTCCAGTCAGGTCA |
| HSPA5 | GGGAGGTGTCATGACCAAAC | GCAGGAGGAATTCCAGTCAG |
| HSPA6 | GATGTGTCGGTTCTCTCCATTG | CTTCCATGAAGTGGTTCACGA |
| IL2 | AGAATCCCAAACTCACCAGGATGC | AGATGTTTCAGTTCTGTGGCCTTC |
| IL4 | ACAGCCTCACAGAGCAGAAGAC | TCTCATGGTGGCTGTAGAACTGC |
| IL5 | GGCACTGCTTTCTACTCATCGA | AGTTGGTGATTTTTATGTACAGGAACA |
| IL6 | TCTCCACAAGCGCCTTCG | CTCAGGGCTGAGATGCCG |
| IL10 | GCTGGAGGACTTTAAGGGTTACCT | CTTGATGTCTGGGTCTTGGTTCT |
| IL12b | TCATCAAACCTGACCCACCCAAGA | TTTCTCTCTTGCTCTTGCCCTGGA |
| IL13 | GAAGGCTCCGCTCTGCAAT | TCTGGGTCTTCTCGATGGCA |
| IL22 | GCAGGCTTGACAAGTCCAACT | GCCTCCTTAGCCAGCATGAA |
| IL23a | ACTCAGCAGATTCCAAGCCTCAGT | TGGAGATCTGAGTGCCATCCTTGA |
| IFNg | TCCAAGTGATGGCTGAACTGTCG | ACCTCGAAACAGCATCTGACTCC |
| TGFb1 | CAAGCAGAGTACACACAGCAT | TGCTCCACTTTTAACTTGAGCC |
| TNFa | CCAGGCAGTCAGATCATCTTCTCG | ATCTCTCAGCTCCACGCCATTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
St. Thomas, N.; Christopher, B.N.; Reyes, L.; Robinson, R.M.; Golick, L.; Zhu, X.; Chapman, E.; Dolloff, N.G. Pharmacological Modulation of the Unfolded Protein Response as a Therapeutic Approach in Cutaneous T-Cell Lymphoma. Biomolecules 2025, 15, 76. https://doi.org/10.3390/biom15010076
St. Thomas N, Christopher BN, Reyes L, Robinson RM, Golick L, Zhu X, Chapman E, Dolloff NG. Pharmacological Modulation of the Unfolded Protein Response as a Therapeutic Approach in Cutaneous T-Cell Lymphoma. Biomolecules. 2025; 15(1):76. https://doi.org/10.3390/biom15010076
Chicago/Turabian StyleSt. Thomas, Nadia, Benjamin N. Christopher, Leticia Reyes, Reeder M. Robinson, Lena Golick, Xiaoyi Zhu, Eli Chapman, and Nathan G. Dolloff. 2025. "Pharmacological Modulation of the Unfolded Protein Response as a Therapeutic Approach in Cutaneous T-Cell Lymphoma" Biomolecules 15, no. 1: 76. https://doi.org/10.3390/biom15010076
APA StyleSt. Thomas, N., Christopher, B. N., Reyes, L., Robinson, R. M., Golick, L., Zhu, X., Chapman, E., & Dolloff, N. G. (2025). Pharmacological Modulation of the Unfolded Protein Response as a Therapeutic Approach in Cutaneous T-Cell Lymphoma. Biomolecules, 15(1), 76. https://doi.org/10.3390/biom15010076