
Time-Dependent Kinetic Complexities in Enzyme Assays: A Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Theoretical Background
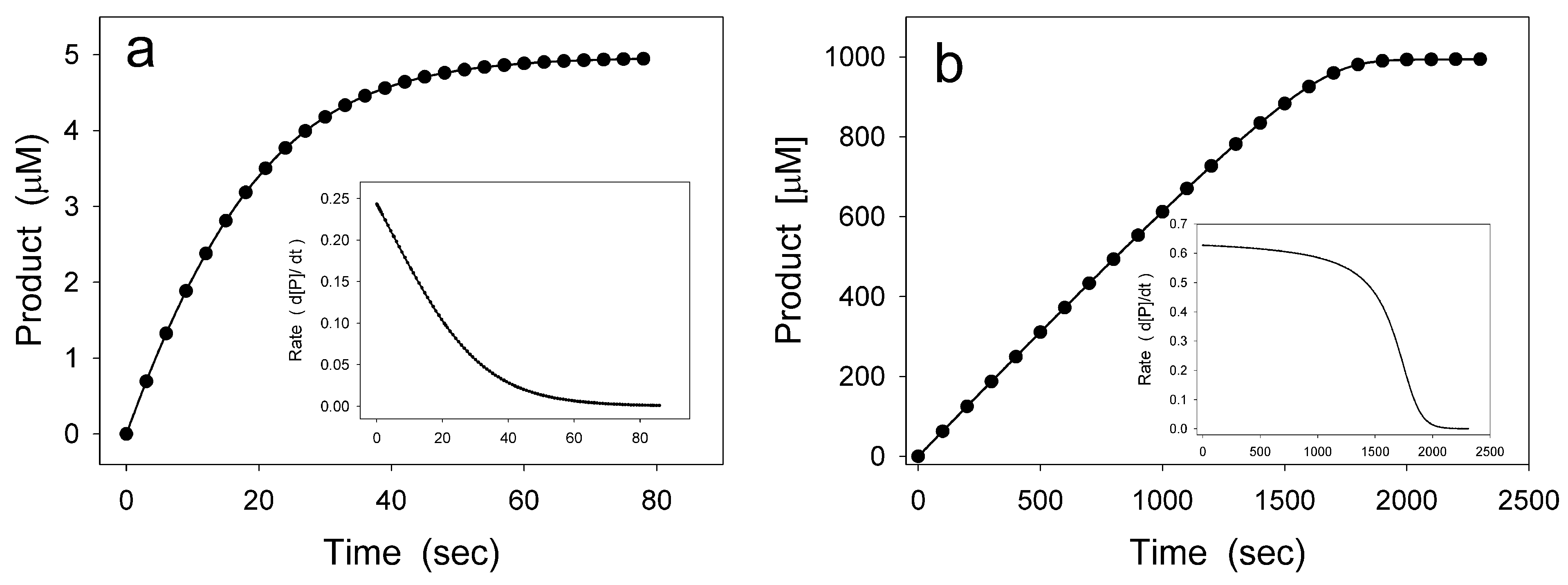
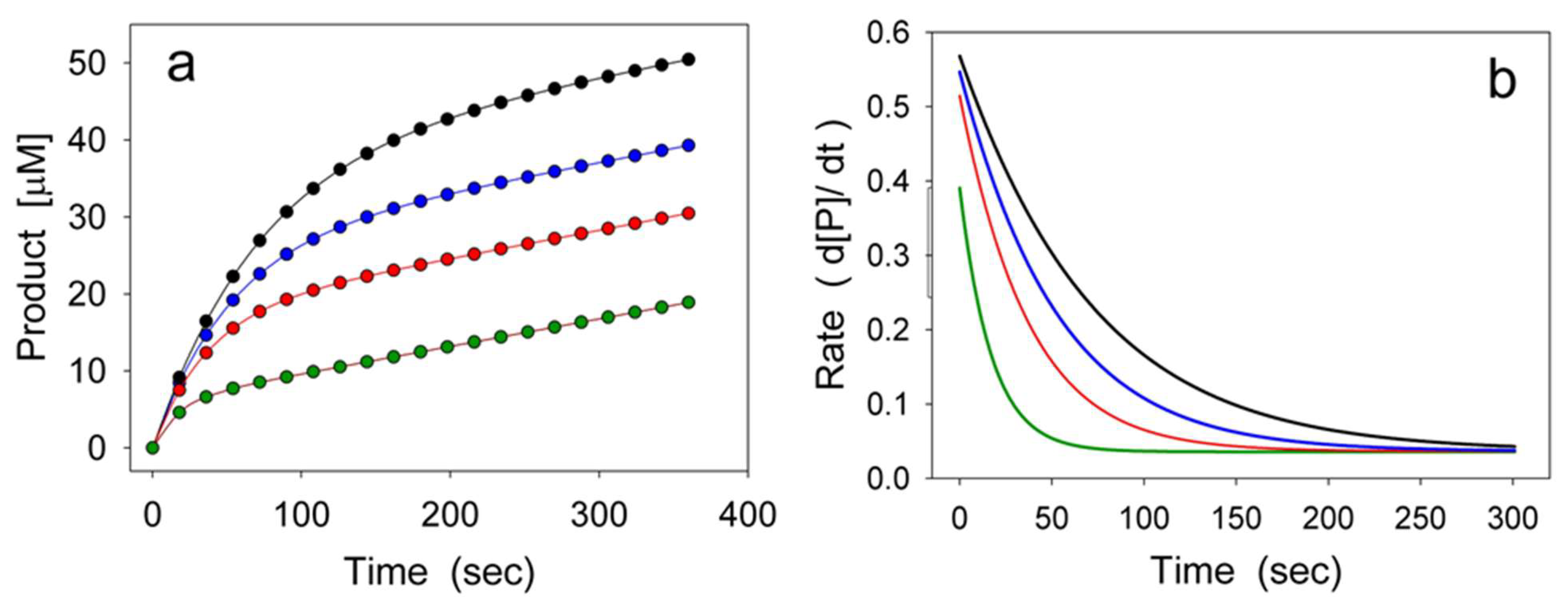
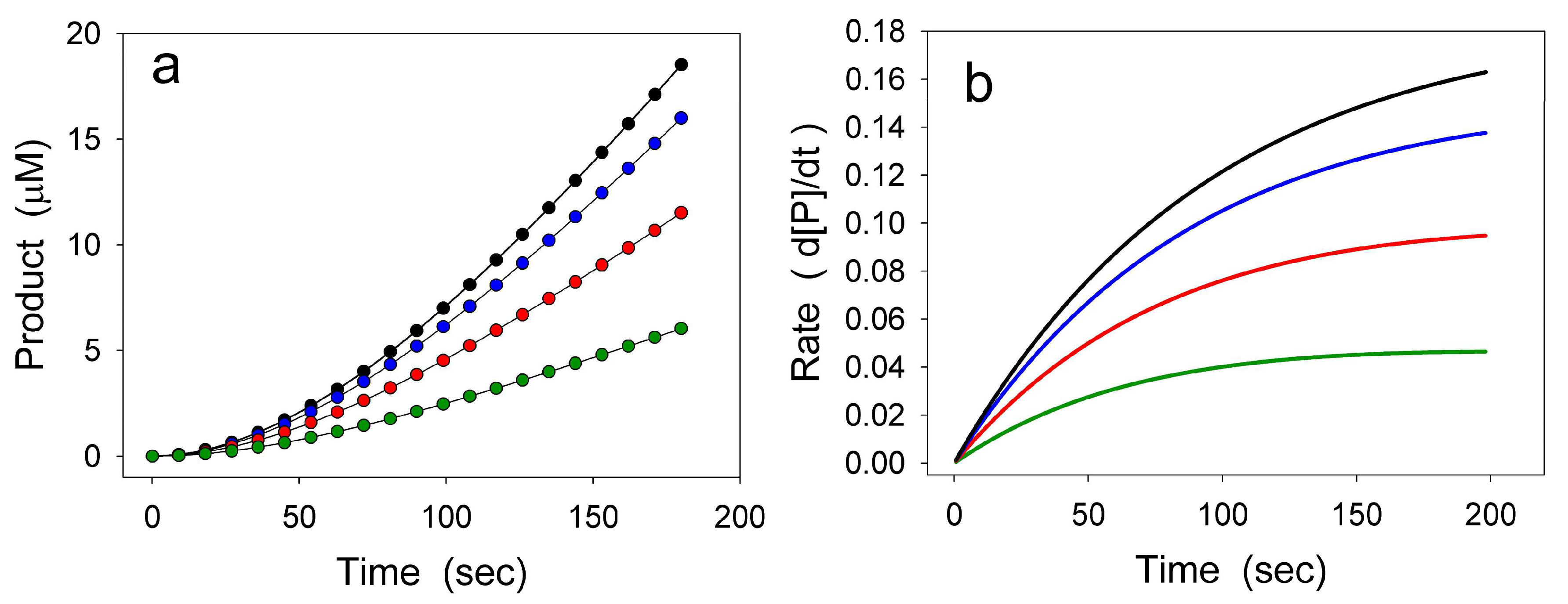
2. Hysteretic Enzymes
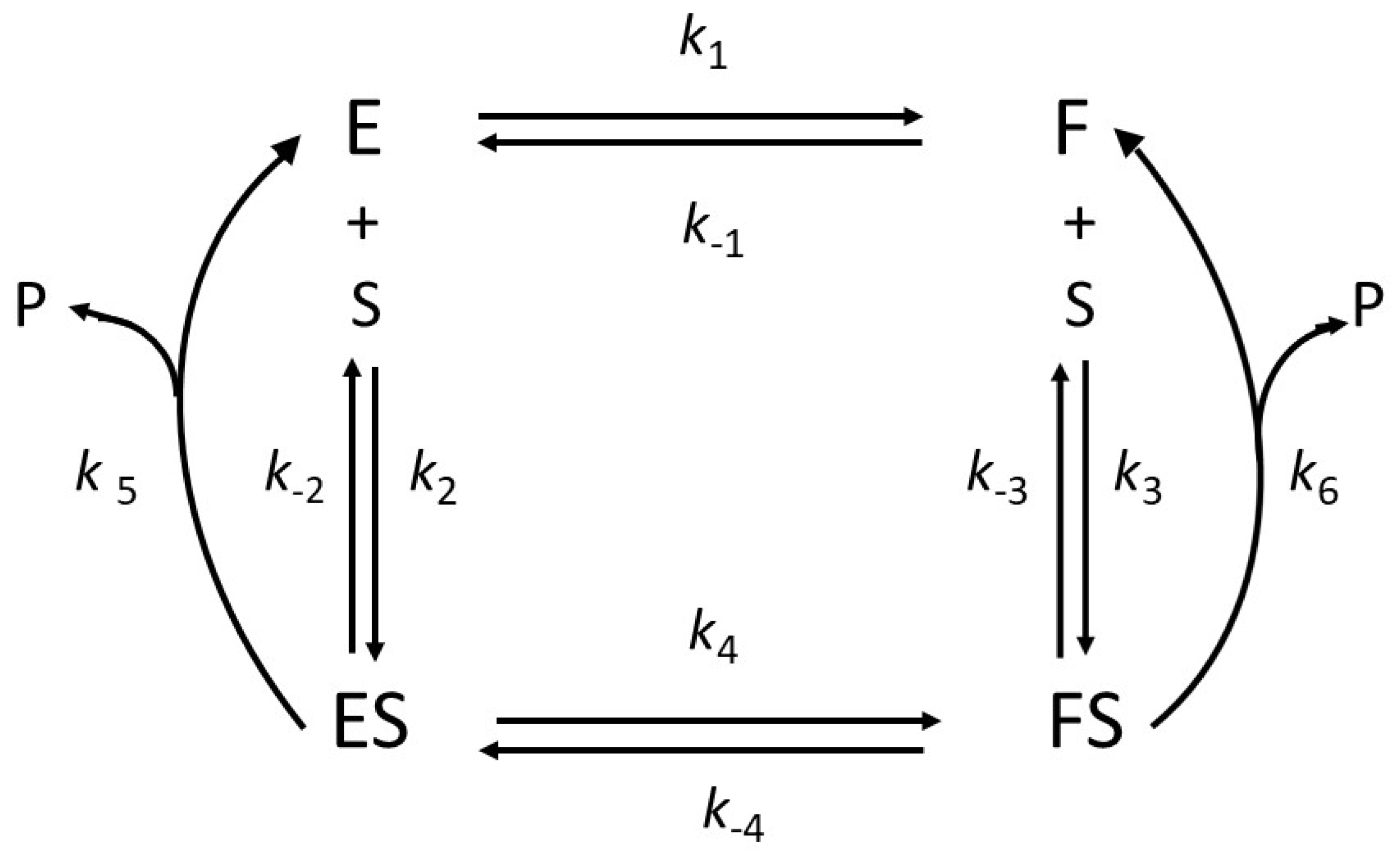
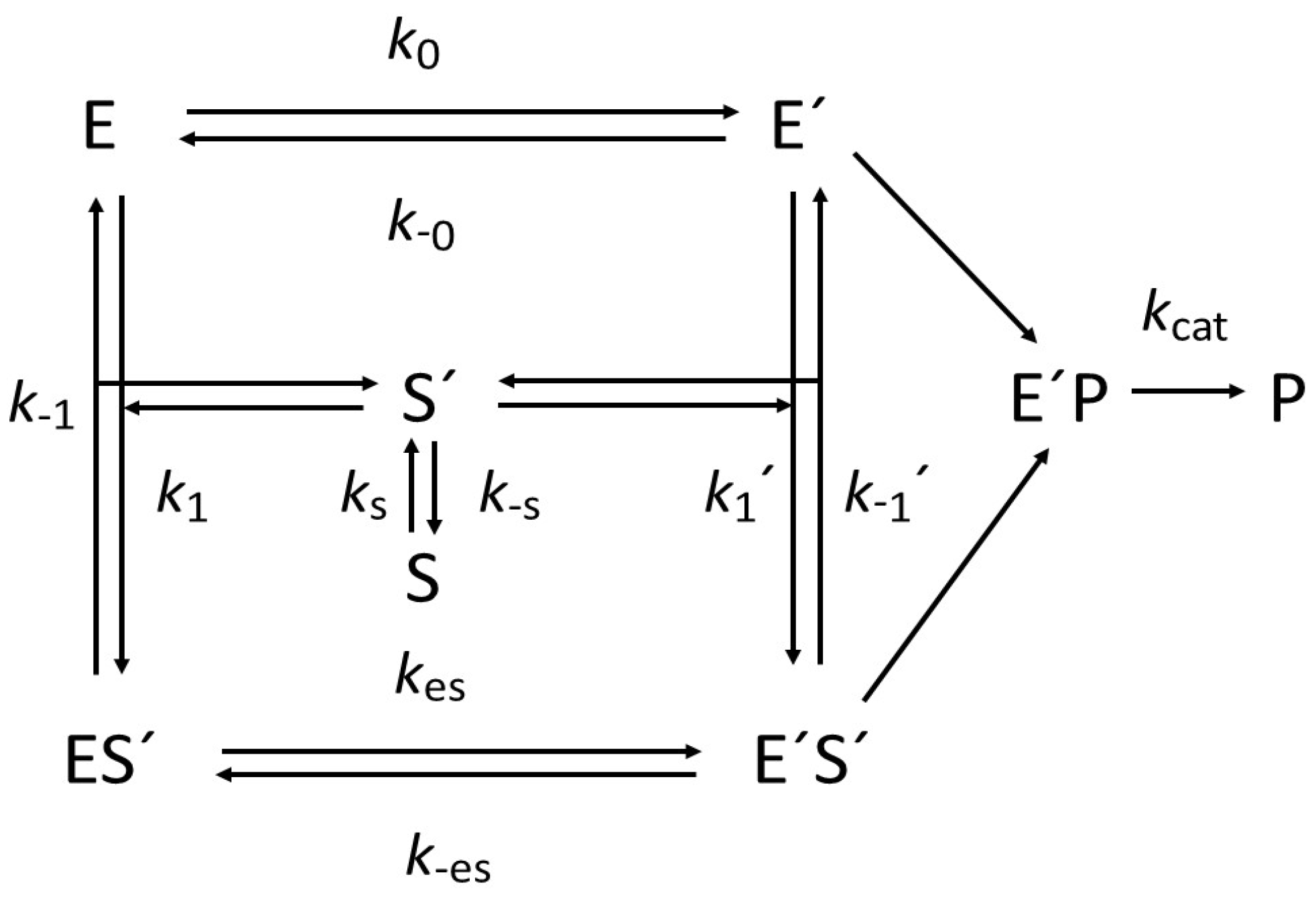
2.1. General Model of Hysteretic Behavior
2.2. Molecular Origin of Hysteretic Behavior
- (i)
- A slow conformational change: The addition of the ligand involved in the hysteretic phenomenon will displace the conformational equilibrium toward either the “ES” or “FS” complexes. Ligand binding occurs under rapid equilibrium conditions, so the observed transition in the activity assay will be the result of the slow conformational transition. Such structural transitions can be directly detected by following changes in the intrinsic fluorescence of the protein [44]. Examples of hysteretic enzymes showing a slow conformational change include pyruvate carboxylase [42], fructose 1,6-bisphosphatase [29], ribulose 1,5-bisphosphate carboxylase [45], methane monooxygenase [46], and 5-aminolevulinate synthase [47].
- (ii)
- A change in the aggregation state of the enzyme: In this case, binding of the ligand involved in hysteretic behavior will result in either dissociation or aggregation of the enzyme. For protein aggregation, the velocity of the transition is strongly influenced by protein concentration in the enzyme assay. Like the conformational changes, ligand binding occurs under rapid equilibrium conditions. When the change in the aggregation state of the enzyme is very slow, on the scale of several minutes, the transition can be analyzed using a combination of covalent cross-linkers and denaturing electrophoresis [48]. Examples of enzymes showing changes in their aggregation states, concomitant with hysteretic kinetics, include phosphofructokinase [49], CTP synthetase [50], glutamine phosphoribosylpyrophosphate amidotransferase [30], malic enzyme [51], and hexokinase [52].
- (iii)
- Displacement of a tightly bound ligand: In this model of hysteretic behavior, a compound with a very small dissociation constant, in the range of 10−7 M or less, is displaced from the enzyme in the presence of an alternative ligand, typically the enzyme substrate. As a result, the progress curve of reaction shows a gradual increase in velocity. Glutamate dehydrogenase from bovine liver displays this kind of behavior with GTP as an inhibitor [19].
- (iv)
- Hysteretic transitions can also arise from sudden changes in either pH [31,53,54] or temperature [32,55]. Such transitions may potentially be observed when the enzyme assay conditions differ from those of its storage [56]. The underlying molecular cause can be attributed to either conformational or aggregation changes. In the case of sudden changes in pH, differences in the protonation state of ionizable groups are the origin of the structural changes.
- (v)
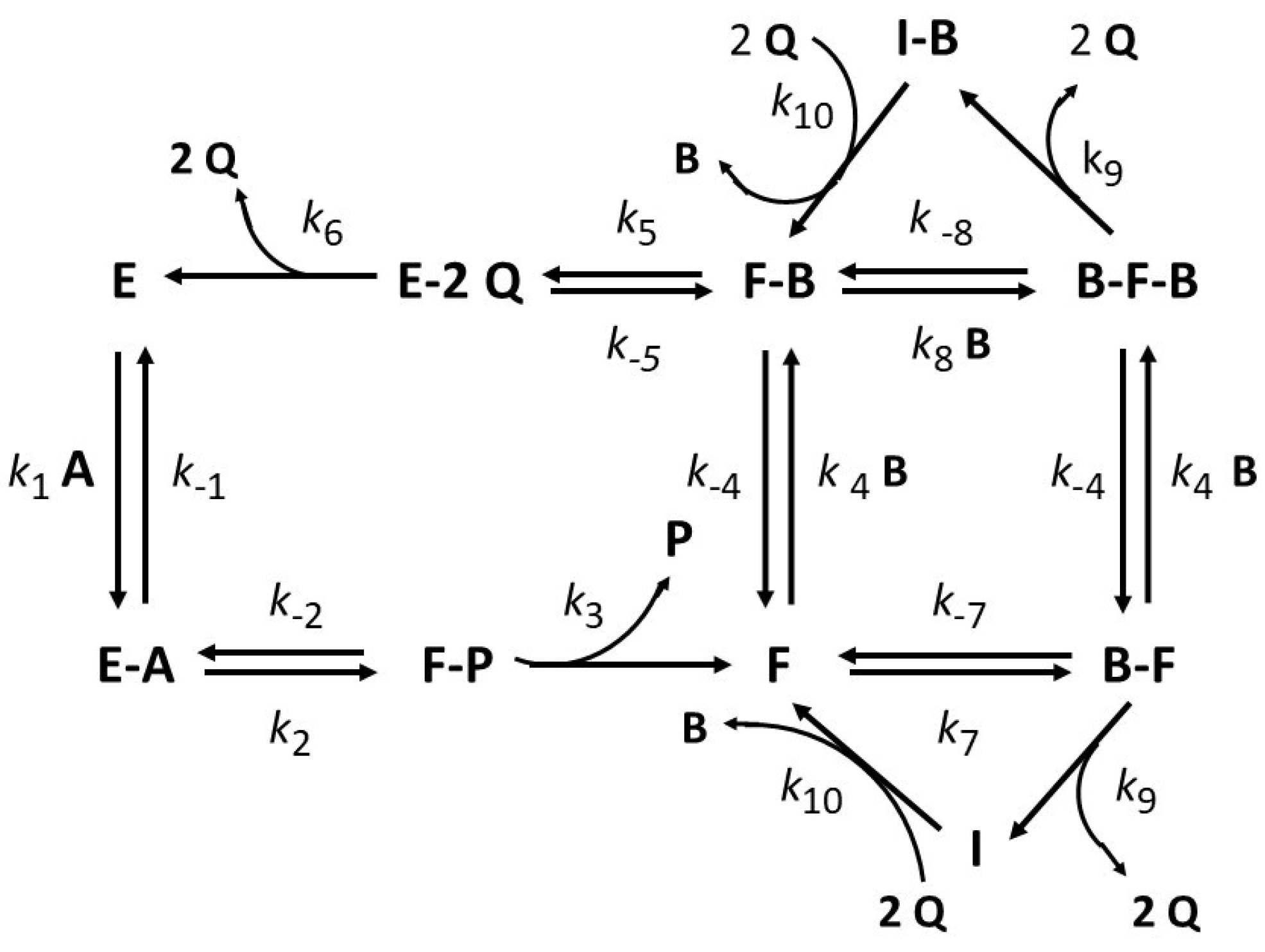
- Hysteresis by substrate channeling: In some Gram-negative bacteria, such as Escherichia coli and Helicobacter pylori, proline catabolism is dependent on a bifunctional flavoenzyme named as proline utilization A (PutA) [21,57]. The enzyme catalyzes the NAD-dependent oxidative conversion of proline into glutamate through the activity of two physically separated but functionally coupled active sites: proline dehydrogenase (PRODH) and glutamate semialdehyde dehydrogenase (GSAL) [21]. The corresponding reactions are as follows:
2.3. Hysteretic-like Progress Curves
2.4. Is It Possible to Avoid Hysteretic Behavior in an Enzyme Activity Assay?
3. Physiological Significance of Hysteretic Behavior
3.1. Hysteresis in Metabolic Pathways
3.2. Hysteresis as an Adaptation to Environmental Changes
3.3. Hysteresis in Medicine
4. Non-Hysteretic Time-Dependent Kinetic Complexities
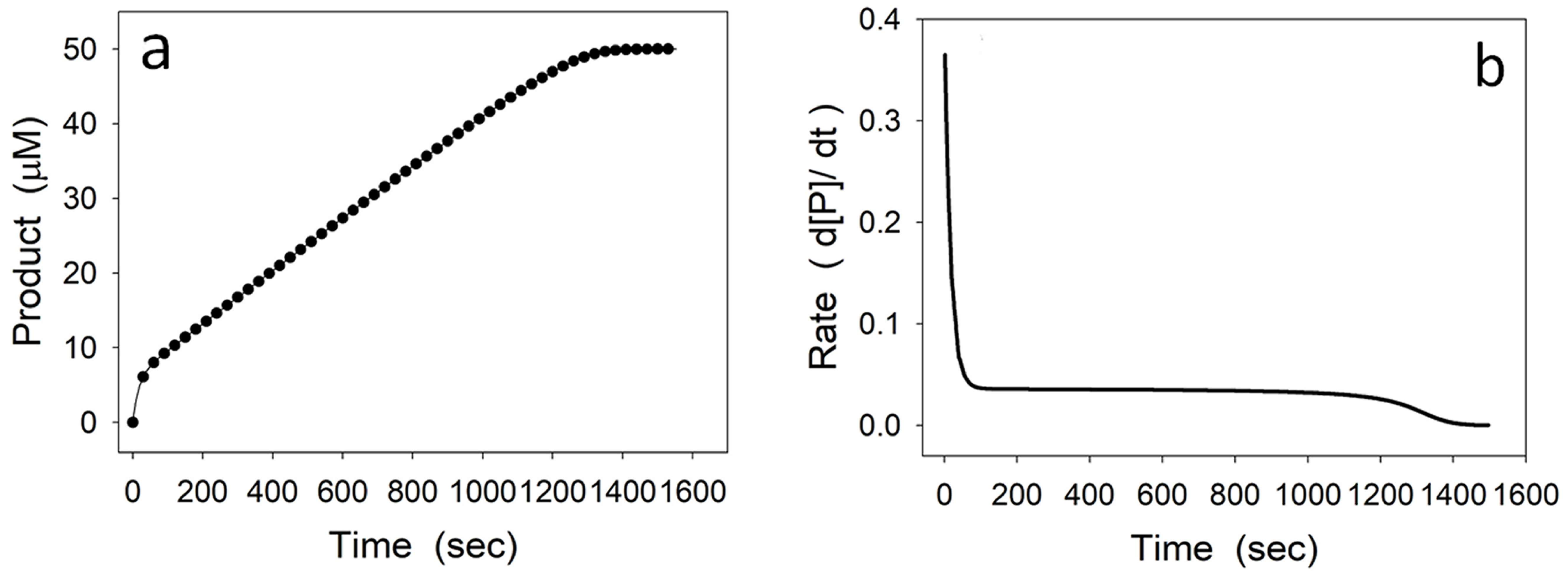
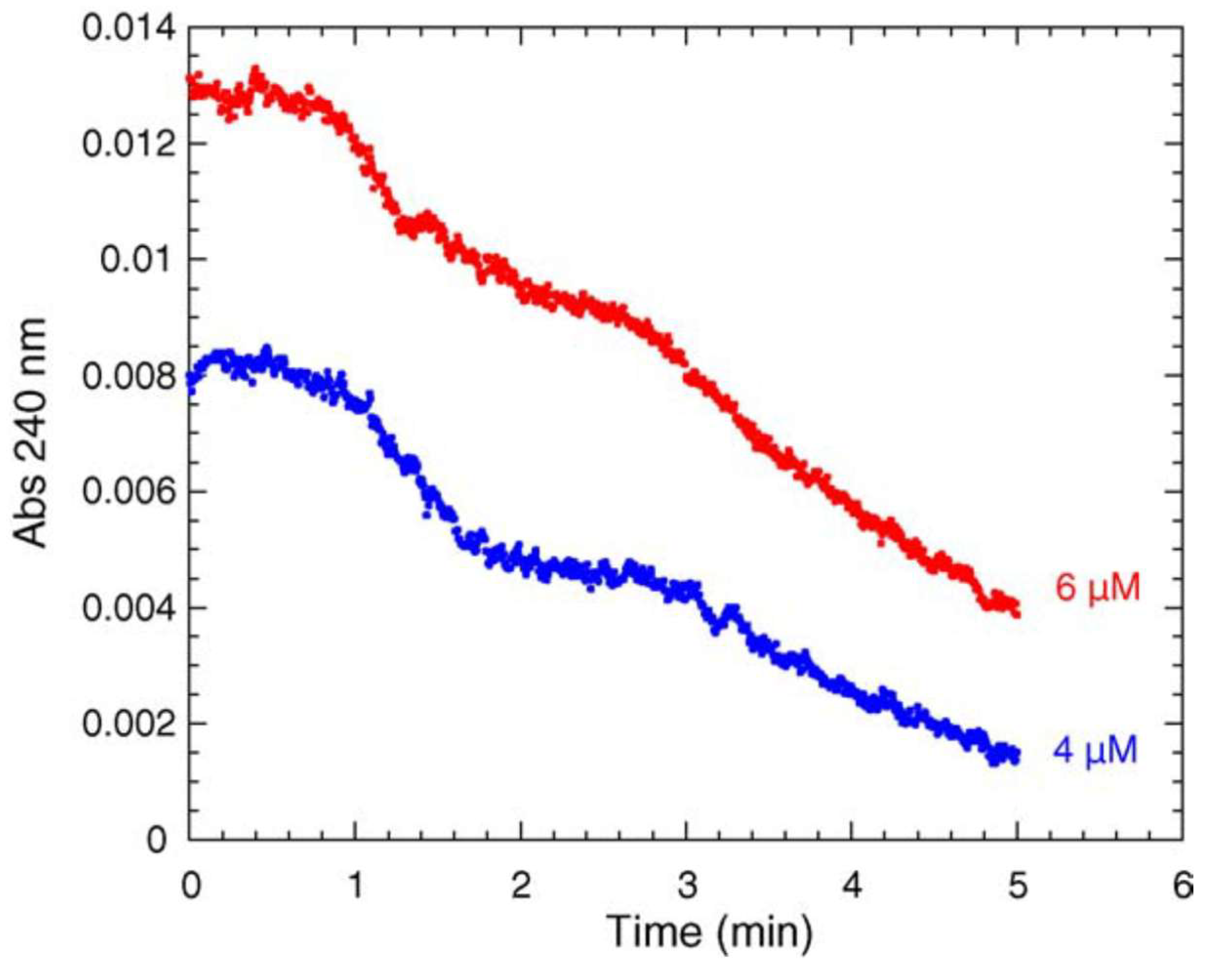
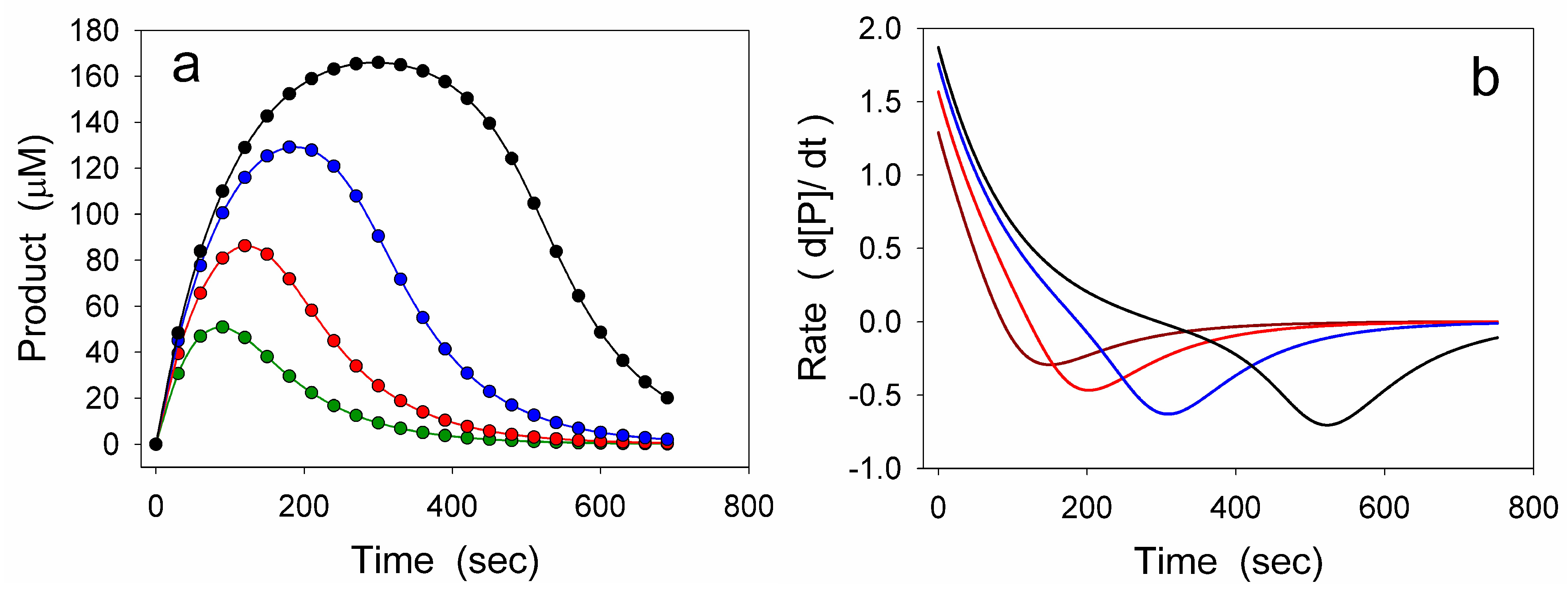
4.1. Damping Oscillatory Hysteresis
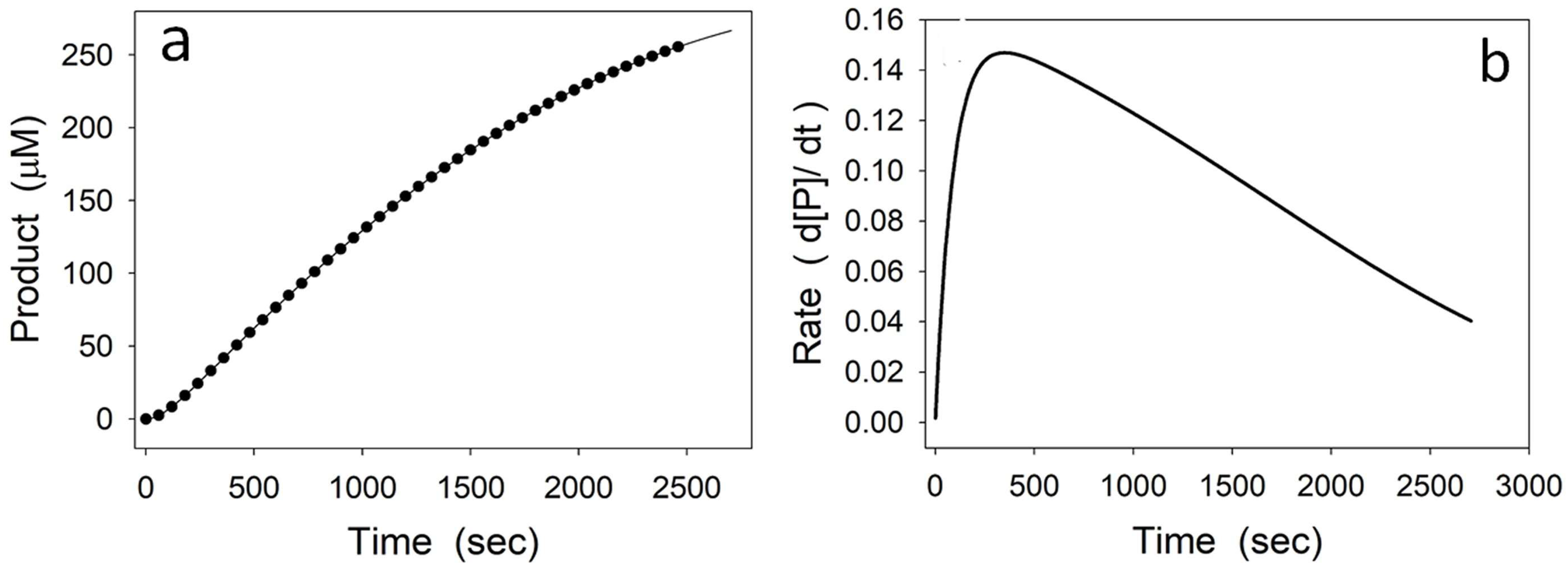
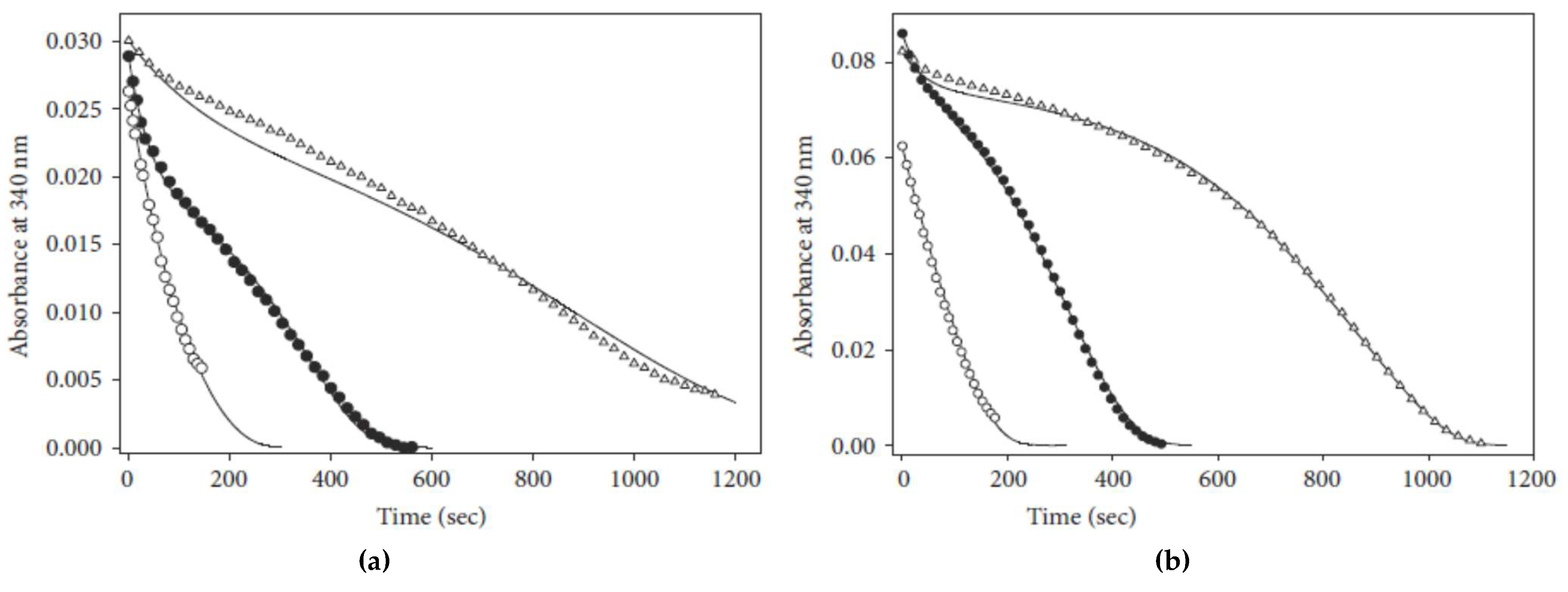
4.2. Instability of a Reaction Intermediary
4.3. Substrate Inhibition Followed by Product Reactivation (Kinetic Competence)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
References
- Segel, I.H. Enzyme Kinetics. Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; Wiley Interscience: New York, NY, USA, 1975. [Google Scholar]
- Cook, P.F.; Cleland, W.W. Enzyme assays. In Enzyme Kinetics and Mechanism; Garland Science: London, UK; New York, NY, USA, 2007; pp. 19–33. [Google Scholar]
- Purich, D.L. Initial-rate kinetics of one-substrate enzyme-catalyzed reactions. In Enzyme Kinetics: Catalysis & Control; Academic Press: London, UK, 2010; pp. 287–310. [Google Scholar]
- Zimmerle, C.T.; Frieden, C. Analysis of progress curves by simulations generated by numerical integration. Biochem. J. 1989, 258, 381–387. [Google Scholar] [CrossRef]
- Duggleby, R.G. Estimation of the initial velocity of enzyme-catalyzed reactions by non-linear regression analysis of progress curves. Biochem. J. 1985, 228, 55–60. [Google Scholar] [CrossRef]
- Hong, H.; Choi, B.; Kim, J.K. Beyond the Michaelis-Menten: Bayesian inference for enzyme kinetic analysis. In Computational Methods for Estimating the Kinetic Parameters of Biological Systems; Vanhaelen, Q., Ed.; Humana Press: New York, NY, USA, 2022; pp. 47–64. [Google Scholar]
- Bevc, S.; Konc, J.; Stojan, J.; Hodoscek, M.; Penca, M.; Praprotnik, M.; Janezic, D. ENZO: A web tool for derivation and evaluation of kinetic models of enzyme catalyzed reactions. PLoS ONE 2011, 6, e22265. [Google Scholar] [CrossRef]
- Reytor-González, M.L.; Cornell-Kennon, S.; Schaefer, E.; Kuzmic, P. An algebraic model to determine substrate kinetic parameters by global non-linear fit of progress curves. Anal. Biochem. 2017, 518, 16–24. [Google Scholar] [CrossRef]
- Purich, D.L. Regulatory behavior of enzymes. In Enzyme Kinetics: Catalysis & Control; Academic Press: London, UK, 2010; pp. 685–726. [Google Scholar]
- Morrison, J.F.; Walsh, C.T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988, 59, 201–301. [Google Scholar]
- Cha, S. Tight-binding inhibitors. I: Kinetic behavior. Biochem. Pharmacol. 1975, 24, 2177–2185. [Google Scholar] [CrossRef]
- Williams, J.; Morrison, J. The kinetics of reversible tight-binding inhibitors. Meth. Enzymol. 1979, 63, 437–467. [Google Scholar]
- Taylor, K.B. Steady-state kinetics. In Enzyme Kinetics and Mechanisms; Kluwer Academic Publishers, London, UK, 2002; pp. 1–6.
- Zechel, D.L.; Konermann, L.; Withers, S.G.; Douglas, D.J. Pre-steady state kinetic analysis of an enzymatic reaction monitored by time-resolved electrospray ionization mass spectrometry. Biochemistry 1998, 37, 7664–7669. [Google Scholar] [CrossRef]
- Purich, D.L. Probing fast enzyme processes. In Enzyme Kinetics: Catalysis & Control; Academic Press: London, UK, 2010; pp. 637–683. [Google Scholar]
- Cook, P.F.; Cleland, W.W. Pre-steady-state and relaxation kinetics. In Enzyme Kinetics and Mechanism; Garland Science: London, UK; New York, NY, USA, 2007; pp. 205–223. [Google Scholar]
- Fierke, C.A.; Hammes, G.G. Transient kinetics approaches to enzyme mechanisms. Methods Enzymol. 1995, 249, 3–37. [Google Scholar]
- Kuzmic, P. Dynafit, a software package for enzymology. Meth. Enzymol. 2009, 467, 247–280. [Google Scholar]
- Frieden, C. Kinetic aspects of regulation of metabolic processes. The hysteretic enzyme concept. J. Biol. Chem. 1970, 245, 5788–5799. [Google Scholar] [CrossRef]
- Frieden, C. Slow transitions and hysteretic behavior in enzymes. Ann. Rev. Biochem. 1979, 48, 471–489. [Google Scholar] [CrossRef]
- Liu, L.K.; Becker, D.F.; Tanner, J.J. Structure, function, and mechanism of proline utilization A (PutA). Arch. Biochem. Biophys. 2017, 632, 142–157. [Google Scholar] [CrossRef]
- Badiou, A.; Froment, M.T.; Fournier, D.; Masson, P.; Belzunces, L.P. Hysteresis of insect acetylcholinesterase. Chem.-Biol. Interact. 2008, 175, 410–412. [Google Scholar] [CrossRef]
- Beattie, N.R.; Keul, N.D.; Hicks Sirmans, T.N.; McDonald, W.E.; Talmadge, T.M.; Taujale, R.; Kannan, N.; Wood, Z.A. Conservation of atypical allostery in C. elegans UDP-glucose dehydrogenase. ACS Omega 2019, 4, 16318–16329. [Google Scholar] [CrossRef]
- Nelson-Rossow, K.L.; Sukalski, K.A.; Nordlie, R.C. Hysteresis at near-physiologic substrate concentrations underlies apparent sigmoid kinetics of the glucose-6-phosphatase system. Biochim. Biophys. Acta 1993, 1163, 297–302. [Google Scholar] [CrossRef]
- Soulie, J.M.; Riviere, M.; Ricard, J. Enzymes as biosensors 2. Hysteretic response of chloroplast fructose-1,6-bis Phosphatase. Eur. J. Biochem. 1988, 176, 111–117. [Google Scholar] [CrossRef]
- Iglesias, A.A.; Andreo, C.S. Hysteretic properties of NADP-malic enzyme from sugar cane leaves. Photosynt. Res. 1992, 31, 89–97. [Google Scholar] [CrossRef]
- Ngam-ek, A.; Seery, T.A.P.; Amis, E.J.; Grover, S.D. Malate-induced hysteresis of phosphoenolpyruvate carboxylase from Crassula argentea. Plant. Physiol. 1989, 91, 954–960. [Google Scholar] [CrossRef]
- Davies, J.S.; Coombes, D.; Horne, C.R.; Pearce, F.G.; Frieman, R.; North, R.A.; Dobson, R.C.J. Functional and solution structure studies of amino sugar deacetylase and deaminase enzyme from Staphylococcus aureus. FEBS Lett. 2019, 593, 52–66. [Google Scholar] [CrossRef]
- Soulie, J.M.; Riviere, M.; Baldet, P.; Ricard, J. Kinetic of the conformational transition of the spinach chloroplast fructose-1,6-bisphosphatase induced by fructose 2,6-bisphosphate. Eur. J. Biochem. 1991, 195, 671–678. [Google Scholar] [CrossRef]
- Singer, S.C.; Holmes, E.W. Human glutamine phosphoribosylpyrophosphate amidotransferase. J. Biol. Chem. 1977, 252, 7959–7963. [Google Scholar] [CrossRef]
- Baici, A.; Szedlacsek, S.E.; Früh, H.; Michel, B.A. pH-dependent hysteretic behavior of human myeloblastin (leucocyte proteinase 3). Biochem. J. 1996, 317, 901–905. [Google Scholar] [CrossRef]
- Herrera, J.E.; Thompson, J.A.; Rimmer, M.A.; Nadeau, O.W.; Carlson, G.M. Activation of phosphorylase kinase by physiological temperature. Biochemistry 2015, 54, 7524–7530. [Google Scholar] [CrossRef]
- King, M.M.; Carlson, G.M. Synergistic activation by Ca++ and Mg++ as the primary cause for hysteresis in the phosphorylase kinase reactions. J. Biol. Chem. 1981, 256, 11058–11064. [Google Scholar] [CrossRef]
- Karr, D.B. Bradyrhizobium japonicum isocitrate dehydrogenae exhibits Ca++ dependent hysteresis. Arch. Biochem. Biophys. 2000, 376, 101–108. [Google Scholar] [CrossRef]
- Lloyd, A.C.; Carpenter, C.A.; Saggerson, E.D. Intertissue differences in the hysteretic behavior of carnitine palmityl transferase in the presence of malonyl-CoA. Biochem. J. 1986, 237, 289–291. [Google Scholar] [CrossRef]
- Acosta, H.; Cáceres, A.; González-Marcano, E.; Quiñones, W.; Avilán, L.; Dubourdieu, M.; Concepción, J.L. Hysteresis and positive cooperativity as possible regulatory mechanisms of Trypanosoma cruzi hexokinase activity. Mol. Biochem. Parasitol. 2014, 198, 82–91. [Google Scholar] [CrossRef]
- Le Goff, J.M.; Martin, P.M.; Ojasoo, T.; Raynaud, J.P. Non-Michaelian behavior of 5 alpha-reductase in human prostate. J. Steroid Biochem. 1989, 33, 155–163. [Google Scholar] [CrossRef]
- Gates, E.W.J.; Prince-Hallée, A.; Heidari, Y.; Sedighi, A.; Keillor, J.W. High-affinity fluorogenic substrate for tissue transglutaminase reveals enzymatic hysteresis. Biochemistry 2023, 62, 3085–3095. [Google Scholar] [CrossRef]
- Cheron, G.; Noat, G.; Ricard, J. Hysteresis of plant cell-wall β-glucosidase. Biochem. J. 1990, 269, 389–392. [Google Scholar] [CrossRef]
- Chow, S.F.; Horowitz, P.M.; Westley, J.; Jarabak, R. Evidence for dynamically determined conformational states in rhodanase. J. Biol. Chem. 1985, 260, 2763–2770. [Google Scholar] [CrossRef]
- Valero, E.; García-Carmona, F. Hysteresis and cooperative behavior of plant polyphenol oxidase. Plant. Physiol. 1992, 98, 774–776. [Google Scholar] [CrossRef]
- Yip, B.P.; Rudolph, F.B. Hysteretic kinetic behavior of beef liver pyruvate carboxylase. Arch. Biochem. Biophys. 1978, 191, 657–665. [Google Scholar] [CrossRef]
- Neet, K.E.; Ainslie, G.R. Hysteretic enzymes. Meth. Enzymol. 1980, 64, 192–226. [Google Scholar]
- Brand, L.; Witholt, B. Fluorescence measurements. Meth. Enzymol. 1967, 11, 776–856. [Google Scholar]
- Yokota, A.; Wadano, A.; Murayama, H. Modeling of continuously and directly analyzed biphasic reaction courses of ribulose 1,5 bisphosphate carboxylase. J. Biochem. 1996, 119, 487–499. [Google Scholar] [CrossRef]
- Blazyk, J.L.; Gassner, G.T.; Lippard, S.J. Intermolecular electron-transfer reactions in soluble methane monooxygenase: A role for hysteresis in protein function. J. Am. Chem. Soc. 2005, 127, 17364–17376. [Google Scholar] [CrossRef]
- Stojanovski, B.M.; Hunter, G.A.; Jahn, M.; Jahn, D.; Ferreira, G.C. Unstable reaction intermediates and hysteresis during the catalytic cycle of 5-Aminolevulinate synthase: Implications from using pseudo and alternate substrates and a promiscuous enzyme variant. J. Biol. Chem. 2014, 289, 22915–22925. [Google Scholar] [CrossRef]
- Rojo-Domínguez, A.; Hernández-Arana, A.; Mendoza-Hernández, G.; Rendón, J.L. Thermal denaturation of glutathione reductase from cyanobacterium Spirulina máxima. Biochem. Mol. Biol. Int. 1997, 42, 631–639. [Google Scholar] [CrossRef]
- Lad, P.M.; Hill, D.E.; Hammes, G.G. Influence of allosteric ligands on the activity and aggregation of rabbit muscle phosphofructokinase. Biochemistry 1973, 12, 4303–4309. [Google Scholar] [CrossRef]
- Anderson, P.M. CTP synthetase from Escherichia coli: An improved purification procedure and characterization of hysteretic and enzyme concentration effects on kinetic properties. Biochemistry 1983, 22, 3285–3292. [Google Scholar] [CrossRef]
- Avilán, L.; García, P. Hysteresis of cytosolic NADP-malic enzyme II from Trypanosoma cruzi. Mol. Biochem. Parasitol. 1994, 65, 225–232. [Google Scholar] [CrossRef]
- Hoggett, J.G.; Kellet, G.L. Kinetics of the monomer-dimer reaction of yeast hexokinase. Biochem. J. 1992, 287, 567–572. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Hervagault, J.F.; Thomas, D. pH-induced bistable dynamic behavior in the reaction catalyzed by glucose 6-phosphate dehydrogenase and conformational hysteresis of the enzyme. Biochem. J. 1989, 262, 795–800. [Google Scholar] [CrossRef]
- Jiménez, M.; García-Carmona, F. Kinetics of the slow pH-mediated transition of polyphenol oxidase. Arch. Biochem. Biophys. 1996, 331, 15–22. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Morrow, B.R.; Pothukuchy, A.; Gollihar, J.; Novak, R.; Reilly, C.B.; Ellington, A.D.; Walt, D.R. Single molecule mechanistic study of enzyme hysteresis. ACS Cent. Sci. 2019, 5, 1691–1698. [Google Scholar] [CrossRef]
- Deits, T.; Shapiro, B.M. pH-induced hysteretic transitions of ovoperoxidase. J. Biol. Chem. 1985, 260, 7882–7888. [Google Scholar] [CrossRef]
- Tanner, J.J. Structural biology of proline catabolism. Amino Acids. 2008, 35, 719–730. [Google Scholar] [CrossRef]
- Luo, M.; Gamage, T.T.; Arentson, B.W.; Schlasner, K.N.; Becker, D.F.; Tanner, J.J. Structures of Proline Utilization A (PutA) Reveal the Fold and Functions of the Aldehyde Dehydrogenase Superfamily Domain of Unknown Function. J. Biol. Chem. 2016, 291, 24065–24075. [Google Scholar] [CrossRef]
- Moxley, M.A.; Sanyal, N.; Krishnan, N.; Tanner, J.J.; Becker, D.F. Evidence for hysteretic substrate channeling in the Proline dehydrogenase and ∆ 1 -Pyrroline-5-carboxylate dehydrogenase coupled reaction of proline utilization (PutA). J. Biol. Chem. 2014, 289, 3639–3651. [Google Scholar] [CrossRef]
- Nagata, K.; Nagata, Y.; Sato, T.; Fujino, M.A.; Nakajima, K.; Tamura, T. L-Serine, D- and L-proline and alanine as respiratory substrates of Helicobacter pylori: Correlation between in vitro and in vivo amino acid levels. Microbiology 2003, 149, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, G.R.; Shill, J.P.; Neet, K.E. Transients and cooperativity. J. Biol. Chem. 1972, 247, 7088–7096. [Google Scholar] [CrossRef] [PubMed]
- McClure, W.R. A kinetic analysis of coupled enzyme assays. Biochemistry 1969, 8, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, I.E.; Manson, G.A. A general expression for coupled enzyme assays of hysteretic enzymes. Biochem. J. 1984, 220, 173–178. [Google Scholar] [CrossRef]
- Brushia, R.J.; Walsh, D.A. Phosphorylase kinase: The complexity of its regulation is reflected in the complexity of its structure. Front. Biosci. 1999, 4, 618–641. [Google Scholar] [CrossRef]
- Wang, J.H.; Stull, J.T.; Huang, T.S.; Krebs, E.G. A study on the autoactivation of rabbit muscle phosphorylase kinase. J. Biol. Chem. 1976, 251, 4521–4527. [Google Scholar] [CrossRef]
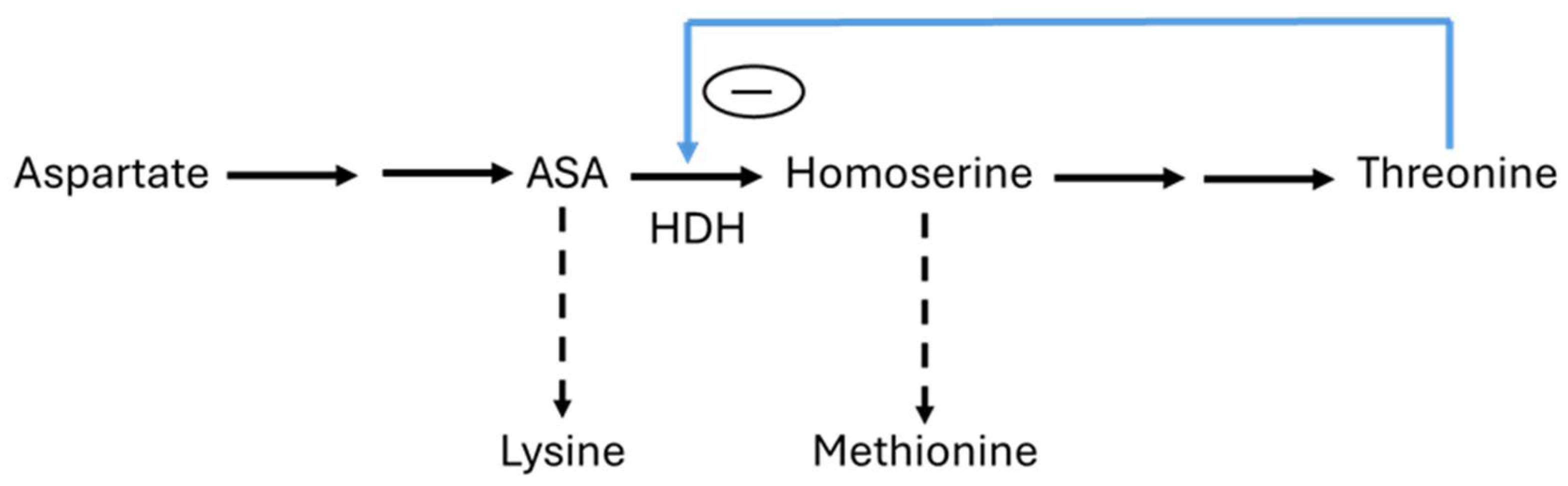
- Chassagnole, C.; Rais, B.; Quentin, E.; Fell, D.A.; Mazat, J.P. An integrated study of threonine-pathway enzyme kinetics in Escherichia coli. Biochem. J. 2001, 356, 415–423. [Google Scholar] [CrossRef]
- Barber, E.D.; Bright, H.J. The rate of an allosteric process: Inhibition of homoserine dehydrogenase I from E. coli by threonine. Proc. Natl. Acad. Sci. USA 1968, 60, 1363–1370. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Li, J.; Ilangovan, U.; Daubner, S.C.; Hinck, A.P.; Fitzpatrick, P.F. Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase. Arch. Biochem. Biophys. 2011, 505, 250–255. [Google Scholar] [CrossRef]
- Jaffe, E.K.; Stith, L.; Lawrence, S.H.; Andrake, M.; Dunbrack, R.L., Jr. A new model for allosteric regulation of phenylalanine hydroxylase: Implications for disease and therapeutics. Arch. Biochem. Biophys. 2013, 530, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Erlandsen, H.; Fusetti, F.; Martínez, A.; Hough, E.; Flatmark, T.; Stevens, R.C. Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nat. Struct. Biol. 1997, 4, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Shiman, R.; Gray, D.W. Substrate activation of phenylalanine hydroxylase. A kinetic characterization. J. Biol. Chem. 1980, 255, 4793–4800. [Google Scholar] [CrossRef]
- Roberts, K.M.; Khan, C.A.; Hinck, C.S.; Fitzpatrick, P.F. Activation of phenylalanine hydroxylase by phenylalanine does not require binding in the active site. Biochemistry 2014, 53, 7846–7853. [Google Scholar] [CrossRef]
- Patil, Y.N.; Marden, B.; Brand, M.D.; Hand, S.C. Metabolic downregulation and inhibition of carbohydrate metabolism during diapause in embryos of Artemia franciscana. Physiol. Biochem. Zool. 2012, 86, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Hand, S.C.; Denlinger, D.L.; Podrabsky, J.E.; Roy, R. Mechanisms of animal diapause: Recent developments from nematodes, crustaceans, insects and fish. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1193–R1211. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Hand, S.C. Arrestment of carbohydrate-metabolism during anaerobic dormancy and aerobic acidosis in Artemia embryos: Determination of pH-sensitive control points. J. Comp. Physiol. B 1986, 156, 451–459. [Google Scholar] [CrossRef]
- Hand, S.C.; Carpenter, J.F. pH-induced metabolic transitions in Artemia embryos mediated by a novel hysteretic trehalase. Science 1986, 232, 1535–1537. [Google Scholar] [CrossRef]
- Chee, Y.L. Coagulation. J. R. Coll. Physicians Edinb. 2014, 44, 42–45. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, S.-I.; Morita, T.; Iwanaga, S.; Igarashi, H. Enzymatic properties of staphylothrombin, an active molecular complex formed between staphylocoagulase and human prothrombin. J. Biochem. 1985, 98, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Bjerketorp, J.; Nilsson, M.; Ljungh, A.; Flock, J.I.; Jacobsson, K.; Frykberg, L. A novel von Willebrand factor binding protein expressed by Staphylococcus aureus. Microbiology 2002, 148, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Panizzi, P.; Friedrich, R.; Fuentes-Prior, P.; Richter, K.; Bock, P.E.; Bode, W. Fibrinogen substrate recognition by staphylocoagulase-(Pro)thrombin complexes. J. Biol. Chem. 2006, 281, 1179–1187. [Google Scholar] [CrossRef]
- Kroh, H.K.; Panizzi, P.; Bock, P.E. Von Willebrand factor-binding protein is a hysteretic conformational activator of prothrombin. Proc. Natl. Acad. Sci. USA 2009, 106, 7786–7791. [Google Scholar] [CrossRef]
- Korzeniowski, O.M.; Kaye, D. Heart Disease: A Textbook of Cardiovascular Medicine, 4th ed.; Braunwald, E., Ed.; W. B. Saunders: Philadelphia, PA, USA, 1992; pp. 1078–1105. [Google Scholar]
- Gabay, J.E.; Scott, R.W.; Campanelli, D.; Griffith, J.; Wilde, C.; Marra, M.N.; Seeger, M.; Nathan, C.F. Antibiotic proteins of human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 5610–5614. [Google Scholar] [CrossRef]
- Kao, R.C.; Wehner, N.G.; Skubitz, K.M.; Gray, B.H.; Hoidal, J.R. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J. Clin. Investig. 1988, 82, 1963–1973. [Google Scholar] [CrossRef]
- Cech, P.; Lehrer, R.I. Phagolysosomal pH of human neutrophils. Blood 1984, 63, 88–95. [Google Scholar] [CrossRef]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef]
- Lockridge, O. Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinyl-choline. Pharmacol. Ther. 1990, 47, 35–60. [Google Scholar] [CrossRef]
- Masson, P.; Nachon, F.; Bartels, C.F.; Froment, M.-T.; Ribes, F.; Matthews, C.; Lockridge, O. High activity of human butyrylcholinesterase at low pH in the presence of excess butyryl-thiocholine. Eur. J. Biochem. 2003, 270, 315–324. [Google Scholar] [CrossRef]
- Johnson, J.L.; Cusack, B.; Davies, M.P.; Fauq, A.; Rosenberry, T.L. Unmasking tandem site interaction in human acetylcholinesterase. Substrate activation with a cationic acetanilide substrate. Biochemistry 2003, 42, 5438–5452. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Froment, M.T.; Fort, S.; Ribes, F.; Bec, N.; Balny, C.; Schopfer, L.M. Butyrylcholinesterase-catalyzed hydrolysis of N-methylindoxyl acetate: Analysis of volume changes upon reaction and hysteretic behavior. Biochim. Biophys. Acta 2002, 1597, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Froment, M.T.; Guillon, E.; Nachon, F.; Darvesh, S.; Schopfer, L.M. Kinetic analysis of butyrylcholineterase -catalyzed hydrolysis of acetanilides. Biochim. Biophys. Acta 2007, 1774, 1139–1147. [Google Scholar] [CrossRef]
- Masson, P.; Goldstein, B.N.; Debouzy, J.-C.; Froment, M.-T.; Lockridge, O.; Schopfer, L.M. Damped oscillatory hysteretic behavior of butyrylcholinesterase with benzoylcholine as substrate. Eur. J. Biochem. 2004, 271, 220–234. [Google Scholar] [CrossRef]
- Masson, P.; Schopfer, L.M.; Froment, M.T.; Debouzy, J.C.; Nachon, F.; Gillon, E.; Lockridge, O.; Hrabovska, A.; Goldstein, B.N. Hysteresis of butyrylcholinesterase in the approach to steady-state kinetics. Chem.-Biol. Interact. 2005, 157–158, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Frere, J.M.; Dormans, C.; Lenzini, V.M.; Duyckaerts, C. Interaction of clavulanate with the β-lactamases of Streptomyces albus G and Actinomadura R39. Biochem. J. 1982, 207, 429–436. [Google Scholar] [CrossRef]
- Jiménez, M.; García-Carmona, F.; García-Cánovas, F.; Iborra, J.L.; Lozano, J.A.; Martínez, F. Chemical intermediates in dopamine oxidation by tyrosinase, and kinetic studies of the process. Arch. Biochem. Biophys. 1984, 235, 438–448. [Google Scholar] [CrossRef]
- Garrido-del Solo, C.; Moruno, A.; Havsteen, B.H.; Varón Castellanos, R. The kinetic effect of product instability in a Michaelis-Menten mechanism with competitive inhibition. BioSystems 2000, 56, 75–82. [Google Scholar] [CrossRef]
- Garrido-del Solo, C.; García-Cánovas, F.; Havsteen, B.H.; Valero, E.; Varón, R. Kinetics of an enzyme reaction in which both the enzyme-substrate complex and the product are unstable or only the product is unstable. Biochem. J. 1994, 303, 435–440. [Google Scholar] [CrossRef]
- Garrido-del Solo, C.; García-Cánovas, F.; Havsteen, B.H.; Varón Castellanos, R. Kinetic analysis of a Michaelis-Menten mechanism in which the enzyme is unstable. Biochem. J. 1993, 294, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Corbalán-García, S.; Teruel, J.A.; Varón, R.; García-Canovas, F.; Gómez-Fernández, J.C. A kinetic study of an unstable enzyme measured through coupling reactions. Application to the self-inactivation of detergent-solubilized Ca2+ ATPase from sarcoplasmic reticulum. Biochim. Biophys. Acta 1993, 1203, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ferrer, A.; Rodriguez-López, J.N.; García-Cánovas, F.; García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Garrido-del Solo, C.; García-Cánovas, F.; Havsteen, B.H.; Varón, R. Analysis of progress curves of enzymatic reactions with unstable species. Int. J. Biochem. Cell Biol. 1996, 28, 1371–1379. [Google Scholar] [CrossRef]
- Sun, Q.A.; Kirnarsky, L.; Sherman, S.; Gladyshev, V.N. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. USA 2001, 98, 3673–3678. [Google Scholar] [CrossRef] [PubMed]
- Salinas, G.; Selkirk, M.E.; Chalar, C.; Maizels, R.M.; Fernández, C. Linked thioredoxin-glutathione systems in platyhelminths. Trends Parasitol. 2004, 20, 340–346. [Google Scholar] [CrossRef]
- Angelucci, F.; Miele, A.E.; Boumis, G.; Dimastrogiovanni, D.; Brunori, M.; Bellelli, A. Glutathione reductase and thioredoxin reductase at the crossroad: The structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins 2008, 72, 936–945. [Google Scholar] [CrossRef]
- Sun, Q.A.; Su, D.; Novoselov, S.V.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Reaction mechanism and regulation of mammalian thioredoxin/glutathione reductase. Biochemistry 2005, 44, 14528–14537. [Google Scholar] [CrossRef]
- Bonilla, M.; Denicola, A.; Novoselov, S.V.; Turanov, A.A.; Protasio, A.; Izmendi, S.; Gladyshev, V.; Salinas, G. Platyhelminth mitochondrial and cytosolic redox homeostasis is controlled by a single thioredoxin glutathione reductase and dependent on selenium and glutathione. J. Biol. Chem. 2008, 283, 17898–17907. [Google Scholar] [CrossRef]
- Huang, H.H.; Day, L.; Cass, C.L.; Ballou, D.P.; Williams, C.H.; Williams, D.L. Investigations on the catalytic mechanism of thioredoxin glutathione reductase (TGR) from Schistosoma mansoni. Biochemistry 2011, 50, 5870–5882. [Google Scholar] [CrossRef]
- Dou, Q.; Turanov, A.A.; Mariotti, M.; Hwang, J.Y.; Wang, H.; Lee, S.G.; Paulo, J.A.; Yim, S.H.; Gygi, S.P.; Chung, J.J.; et al. Selenoprotein TXNRD3 supports male fertility via the redox regulation of spermatogenesis. J. Biol. Chem. 2022, 298, 102183. [Google Scholar] [CrossRef]
- Alger, H.M.; Williams, D.L. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol. Biochem. Parasitol. 2002, 121, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Rendón, J.L.; del Arenal, I.P.; Guevara-Flores, A.; Uribe, A.; Plancarte, A.; Mendoza-Hernández, G. Purification, characterization, and kinetic properties of the multifunctional thioredoxin-glutathione reductase from Taenia crassiceps metacestode (cysticerci). Mol. Biochem. Parasitol. 2004, 133, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Flores, A.; Pardo, J.P.; Rendón, J.L. Hysteresis in thioredoxin-glutathione reductase (TGR) from the adult stage of the liver fluke Fasciola hepatica. Parasitol. Int. 2011, 60, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Plancarte, A.; Nava, G. Purification and kinetic analysis of cytosolic and mitochondrial thioredoxin glutathione reductase extracted from Taenia solium cysticerci. Experim. Parasitol. 2015, 149, 65–73. [Google Scholar] [CrossRef]
- Rendón, J.L.; Miranda-leyva, M.; Guevara-Flores, A.; Martínez-González, J.J.; del Arenal, I.P.; Flores-Herrera, O.; Pardo, J.P. Insight into the mechanistic basis of the hysteretic-like kinetic behavior of thioredoxin-glutathione reductase. Enzym. Res. 2018, 3215462. [Google Scholar] [CrossRef]
- Angelucci, F.; Dimastrogiovanni, D.; Boumis, G.; Brunori, M.; Miele, A.E.; Saccoccia, F.; Bellelli, A. Mapping the catalytic cycle of Schistosoma mansoni thioredoxin glutathione reductase by X-ray crystallography. J. Biol. Chem. 2010, 285, 32557–32567. [Google Scholar] [CrossRef]
- Pruett, W.A.; Hester, R.L.; Coleman, T.G. The apparent hysteresis in hormone-agonist relationships. J. Theor. Biol. 2012, 296, 1–5. [Google Scholar] [CrossRef]
- Louizos, C.; Yañez, J.A.; Forrest, L.; Davies, N.M. Understanding the hysteresis loop conundrum in pharmacokinetic/pharmacodynamic relationships. J. Pharm. Pharm. Sci. 2014, 17, 34–91. [Google Scholar] [CrossRef]
- Danhof, M. Systems pharmacology. Toward the modeling of network interactions. Eur. J. Pharmac. Sci. 2016, 94, 4–14. [Google Scholar] [CrossRef]
- Roemhild, R.; Gokhale, C.S.; Dirksen, P.; Blake, C.; Rosenstiel, P.; Traulsen, A.; Andersson, D.I.; Schulenburg, H. Cellular hysteresis as a principle to maximize the efficacy of antibiotic therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 9767–9772. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rendón, J.L.; Pardo, J.P. Time-Dependent Kinetic Complexities in Enzyme Assays: A Review. Biomolecules 2025, 15, 641. https://doi.org/10.3390/biom15050641
Rendón JL, Pardo JP. Time-Dependent Kinetic Complexities in Enzyme Assays: A Review. Biomolecules. 2025; 15(5):641. https://doi.org/10.3390/biom15050641
Chicago/Turabian StyleRendón, Juan Luis, and Juan Pablo Pardo. 2025. "Time-Dependent Kinetic Complexities in Enzyme Assays: A Review" Biomolecules 15, no. 5: 641. https://doi.org/10.3390/biom15050641
APA StyleRendón, J. L., & Pardo, J. P. (2025). Time-Dependent Kinetic Complexities in Enzyme Assays: A Review. Biomolecules, 15(5), 641. https://doi.org/10.3390/biom15050641