

Microbial Composition, Disease Trajectory and Genetic Background in a Slow Onset Model of Frontotemporal Lobar Degeneration


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Transgenic Mice
2.2. Immunohistochemistry
2.3. Quantitative Pathology
2.4. RNA Extraction and Nanostring Analysis
2.5. DNA Extraction
2.6. Library Construction and Sequencing
2.7. Bioinformatics
2.8. Colonization of Mice by Fecal Microbiotal Transplant (FMT)
2.9. Statistical Analyses
3. Results
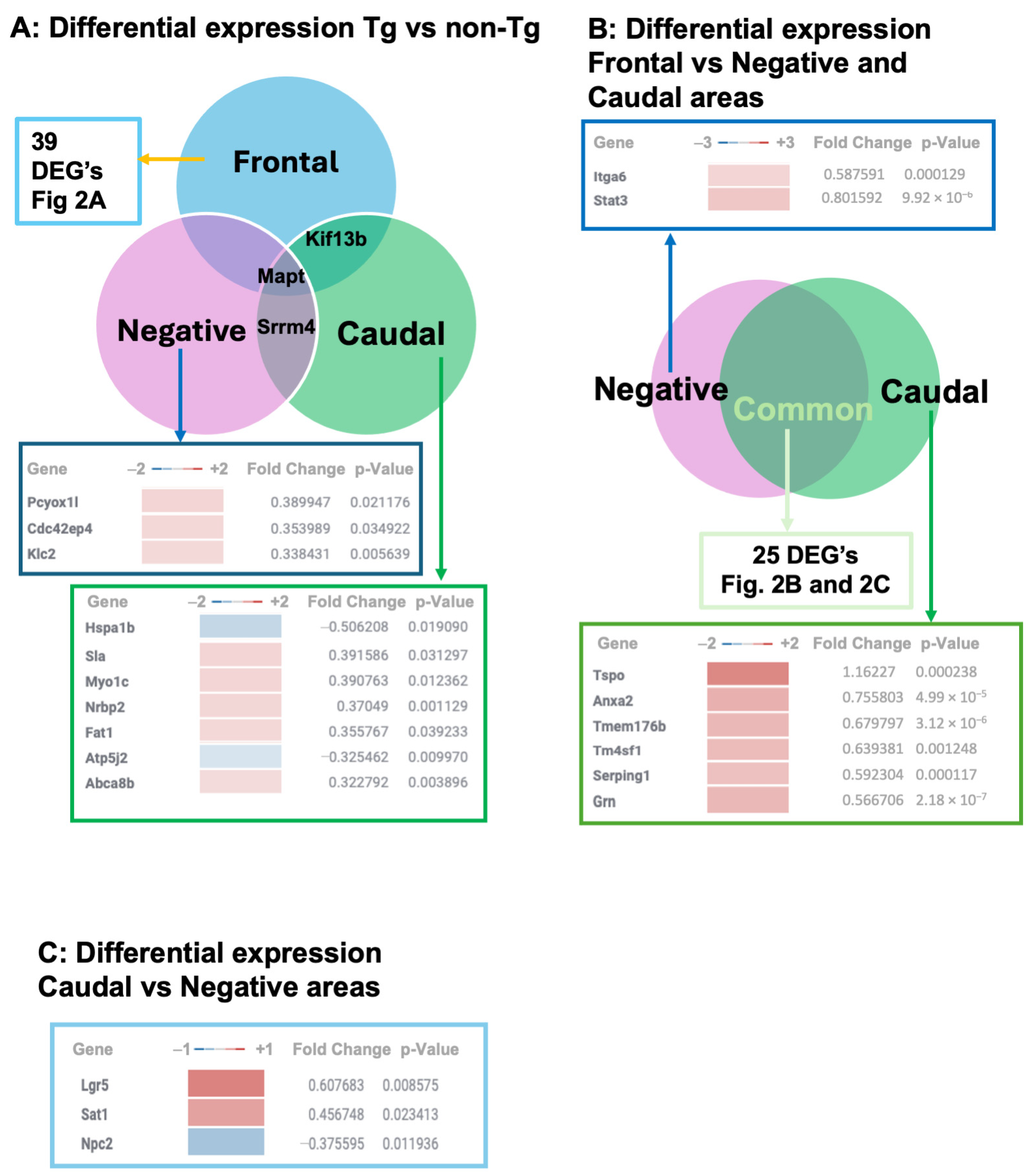
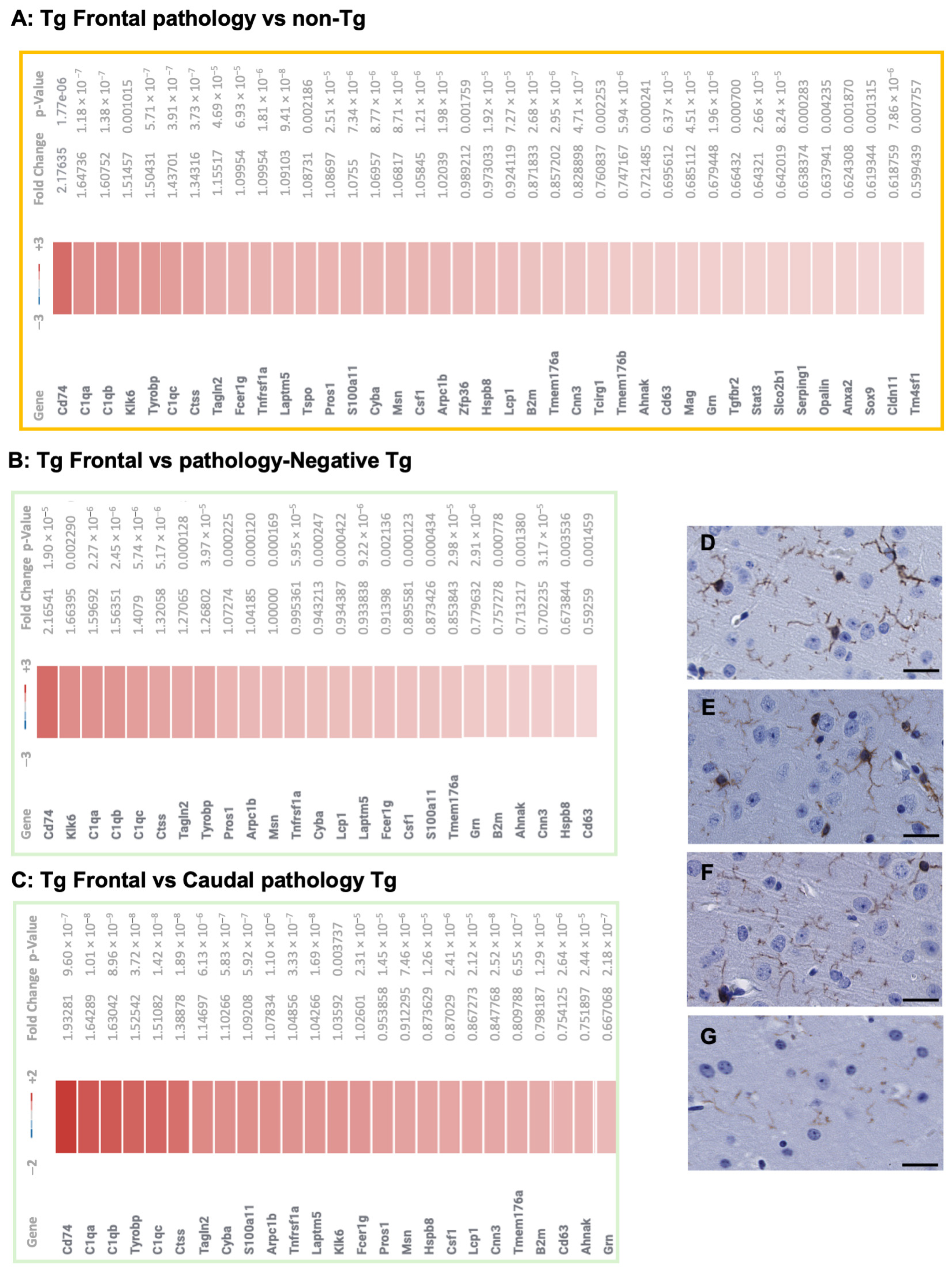
3.1. Transcriptional Profiles in TgTauP301L Mice with Frontal or Caudal Tau Deposition
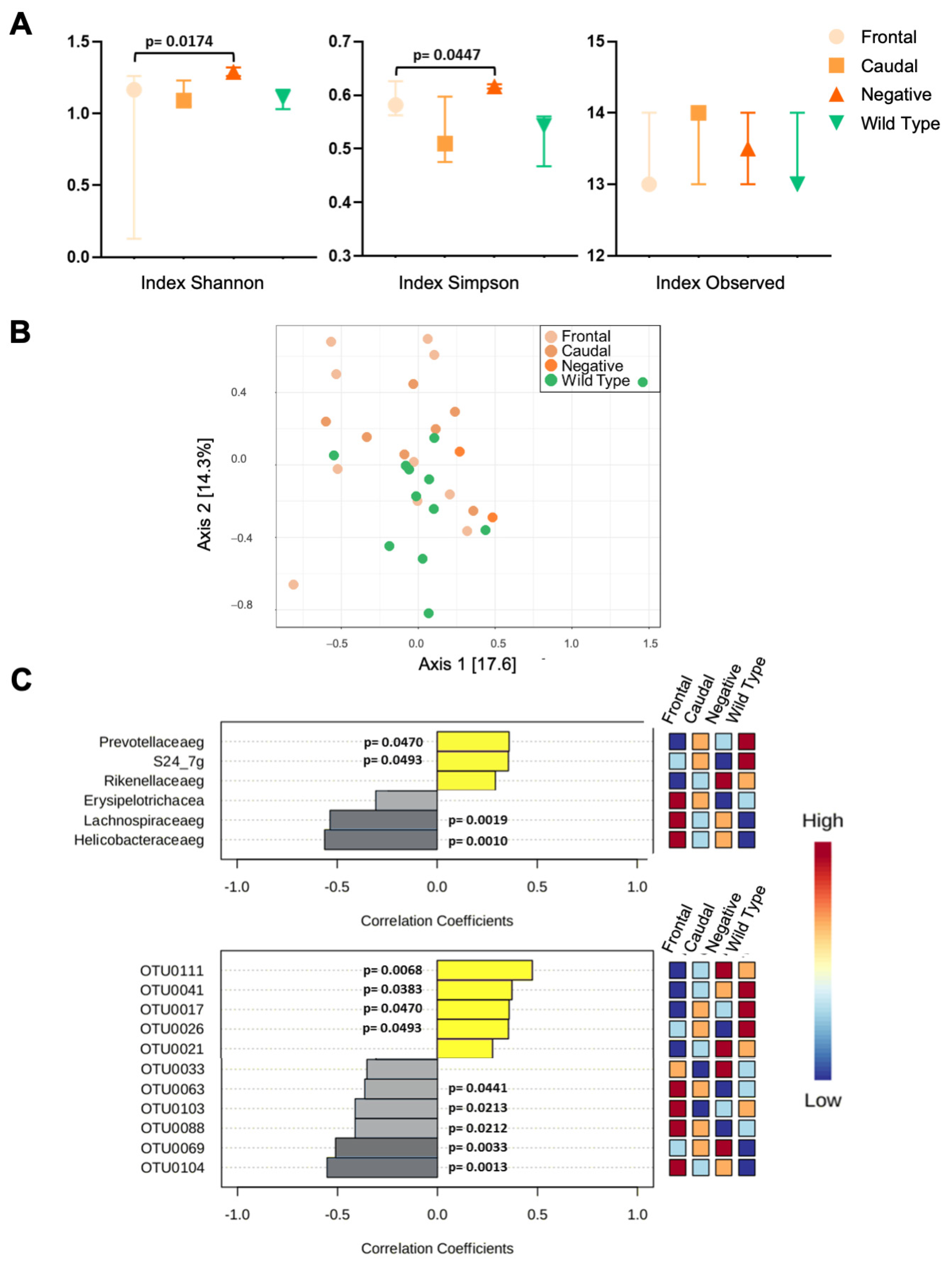
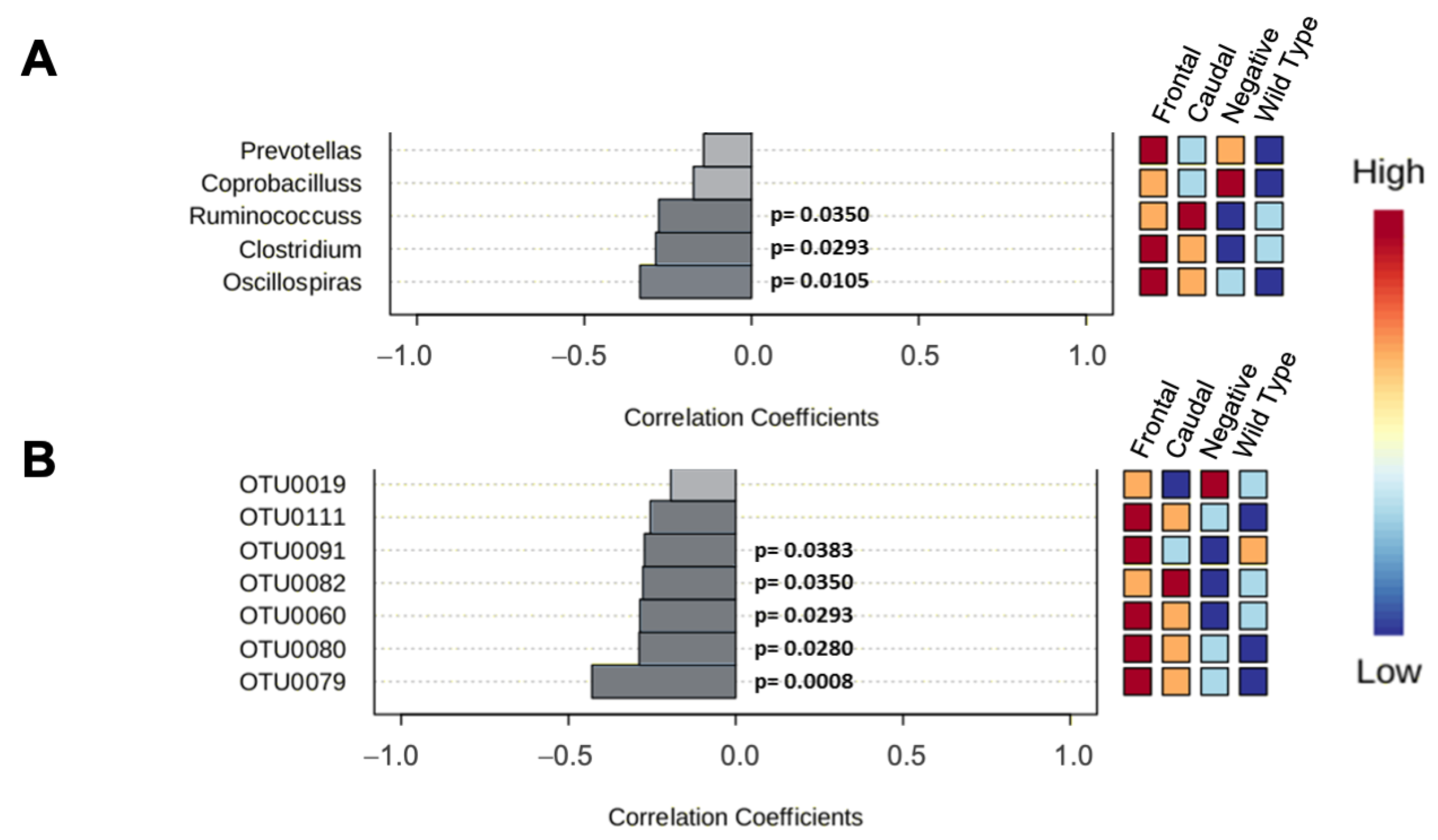
3.2. 16S rRNA Analysis of Gut Microbiota
3.2.1. 16rRNA from C57BL6/Tac Samples
3.2.2. 16rRNA from 129SvEv/Tac Samples
3.3. Pathology Outcomes After Microbial Transplantation
4. Discussion
4.1. Phenotypic Variation Within Models of FTLD-MAPT
4.2. Microbial Profiles and Phenotypic Variation in a Genetic Tauopathy
4.3. Mouse Inbred Strains and Modeling of Protein Folding Diseases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [CrossRef]
- Avan, A.; Hachinski, V. Global, regional, and national trends of dementia incidence and risk factors, 1990-2019: A Global Burden of Disease study. Alzheimers Dement. 2023, 19, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Mollah, S.A.; Nayak, A.; Barhai, S.; Maity, U. A comprehensive review on frontotemporal dementia: Its impact on language, speech and behavior. Dement. Neuropsychol. 2024, 18, e20230072. [Google Scholar] [CrossRef] [PubMed]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef] [PubMed]
- Munoz, D.G.; Dickson, D.W.; Bergeron, C.; Mackenzie, I.R.; Delacourte, A.; Zhukareva, V. The neuropathology and biochemistry of frontotemporal dementia. Ann. Neurol. 2003, 54 (Suppl. 5), S24–S28. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Mirra, S.S.; Murrell, J.R.; Gearing, M.; Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Levey, A.I.; Jones, R.; Green, J.; Shoffner, J.M.; et al. Tau pathology in a family with dementia and a P301L mutation in tau. J. Neuropathol. Exp. Neurol. 1999, 58, 335–345. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Phinney, A.L.; Horne, P.; Yang, J.; Janus, C.; Bergeron, C.; Westaway, D. Mouse models of Alzheimer’s disease: The long and filamentous road. Neurol. Res. 2003, 25, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Eskandari-Sedighi, G.; Westaway, D. Murine models of tauopathies: A platform to study neurodegenerative diseases associated with aging. In The Neuroscience of Aging: Assessments Treatments and Modeling in Aging and Neurological Diseases; Martin, C., Preedy, V.R., Rajendram, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 506–515. [Google Scholar]
- Eskandari-Sedighi, G.; Daude, N.; Gapeshina, H.; Sanders, D.W.; Kamali-Jamil, R.; Yang, J.; Shi, B.; Wille, H.; Ghetti, B.; Diamond, M.I.; et al. The CNS in inbred transgenic models of 4-repeat Tauopathy develops consistent tau seeding capacity yet focal and diverse patterns of protein deposition. Mol. Neurodegener. 2017, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Paitel, E.; Kawarabayashi, T.; Ikeda, M.; Chishti, M.A.; Janus, C.; Matsubara, E.; Sasaki, A.; Kawarai, T.; Phinney, A.L.; et al. Cortical neuronal and glial pathology in TgTauP301L transgenic mice: Neuronal degeneration, memory disturbance, and phenotypic variation. Am. J. Pathol. 2006, 169, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Daude, N.; Kim, C.; Kang, S.G.; Eskandari-Sedighi, G.; Haldiman, T.; Yang, J.; Fleck, S.C.; Gomez-Cardona, E.; Han, Z.Z.; Borrego-Ecija, S.; et al. Diverse, evolving conformer populations drive distinct phenotypes in frontotemporal lobar degeneration caused by the same MAPT-P301L mutation. Acta Neuropathol. 2020, 139, 1045–1070. [Google Scholar] [CrossRef]
- Chung, D.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 2021, 16, 57. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef]
- Geng, Z.H.; Zhu, Y.; Li, Q.L.; Zhao, C.; Zhou, P.H. Enteric Nervous System: The Bridge Between the Gut Microbiota and Neurological Disorders. Front. Aging Neurosci. 2022, 14, 810483. [Google Scholar] [CrossRef]
- Ghezzi, L.; Cantoni, C.; Rotondo, E.; Galimberti, D. The Gut Microbiome-Brain Crosstalk in Neurodegenerative Diseases. Biomedicines 2022, 10, 1486. [Google Scholar] [CrossRef]
- Mittal, A.; Sharma, R.; Sardana, S.; Goyal, P.K.; Piplani, M.; Pandey, A. A Systematic Review of updated mechanistic insights towards Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2022, 22, 1232–1242. [Google Scholar] [CrossRef]
- Wang, Y. The Role of the Gut Microbiota and Microbial Metabolites in the Pathogenesis of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2022, 22, 577–598. [Google Scholar] [CrossRef]
- Zhong, S.R.; Kuang, Q.; Zhang, F.; Chen, B.; Zhong, Z.G. Functional roles of the microbiota-gut-brain axis in Alzheimer’s disease: Implications of gut microbiota-targeted therapy. Transl. Neurosci. 2021, 12, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.R.; Dawes, J.M.; McMahon, S.B.; Bennett, D.L.H.; Orengo, C.; Kohl, M. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT-qPCR quantification cycle (Cq) data. BMC Genom. 2012, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Hennig, C. An empirical comparison and characterisation of nine popular clustering methods. Adv. Data Anal. Classif. 2022, 16, 201–229. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-116. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Personett, A.R.; Turner, G.; Dorfmeyer, R.A.; Franklin, C.L. Variable Colonization after Reciprocal Fecal Microbiota Transfer between Mice with Low and High Richness Microbiota. Front. Microbiol. 2017, 8, 196. [Google Scholar] [CrossRef]
- Jimeno, R.; Brailey, P.M.; Barral, P. Quantitative Polymerase Chain Reaction-based Analyses of Murine Intestinal Microbiota After Oral Antibiotic Treatment. J. Vis. Exp. 2018, 141, e58481. [Google Scholar] [CrossRef]
- Saha, U.S.; Misra, R.; Tiwari, D.; Prasad, K.N. A cost-effective anaerobic culture method & its comparison with a standard method. Indian J. Med. Res. 2016, 144, 611–613. [Google Scholar] [CrossRef]
- Benson, A.; Pifer, R.; Behrendt, C.L.; Hooper, L.V.; Yarovinsky, F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 2009, 6, 187–196. [Google Scholar] [CrossRef]
- Aredo, B.; Zhang, K.; Chen, X.; Wang, C.X.; Li, T.; Ufret-Vincenty, R.L. Differences in the distribution, phenotype and gene expression of subretinal microglia/macrophages in C57BL/6N (Crb1 rd8/rd8) versus C57BL6/J (Crb1 wt/wt) mice. J. Neuroinflamm. 2015, 12, 6. [Google Scholar] [CrossRef]
- Ma, L.; Lu, H.; Chen, R.; Wu, M.; Jin, Y.; Zhang, J.; Wang, S. Identification of Key Genes and Potential New Biomarkers for Ovarian Aging: A Study Based on RNA-Sequencing Data. Front. Genet. 2020, 11, 590660. [Google Scholar] [CrossRef]
- Mohammadi, F.; Beauparlant, C.J.; Bianco, S.; Droit, A.; Bertrand, N.; Rudkowska, I. Ruminant Trans Fatty Acid Intake Modulates Inflammation Pathways in the Adipose Tissue Transcriptome of C57BL/6 Mice. Mol. Nutr. Food Res. 2024, 68, e2400290. [Google Scholar] [CrossRef] [PubMed]
- Reichwald, J.; Danner, S.; Wiederhold, K.H.; Staufenbiel, M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflamm. 2009, 6, 35. [Google Scholar] [CrossRef]
- Virgone-Carlotta, A.; Uhlrich, J.; Akram, M.N.; Ressnikoff, D.; Chrétien, F.; Domenget, C.; Gherardi, R.; Despars, G.; Jurdic, P.; Honnorat, J.; et al. Mapping and kinetics of microglia/neuron cell-to-cell contacts in the 6-OHDA murine model of Parkinson’s disease. Glia 2013, 61, 1645–1658. [Google Scholar] [CrossRef]
- Parker, C.; Zhao, J.; Pearce, D.A.; Kovács, A.D. Comparative analysis of the gut microbiota composition in the Cln1(R151X) and Cln2(R207X) mouse models of Batten disease and in three wild-type mouse strains. Arch. Microbiol. 2020, 203, 85–96. [Google Scholar] [CrossRef]
- Allen, B.; Ingram, E.; Takao, M.; Smith, M.J.; Jakes, R.; Virdee, K.; Yoshida, H.; Holzer, M.; Craxton, M.; Emson, P.C.; et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 2002, 22, 9340–9351. [Google Scholar] [CrossRef]
- Johnson, N.R.; Condello, C.; Guan, S.; Oehler, A.; Becker, J.; Gavidia, M.; Carlson, G.A.; Giles, K.; Prusiner, S.B. Evidence for sortilin modulating regional accumulation of human tau prions in transgenic mice. Proc. Natl. Acad. Sci. USA 2017, 114, E11029–E11036. [Google Scholar] [CrossRef]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.; Trojanowski, J.Q. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015, 129, 221–237. [Google Scholar] [CrossRef]
- Shen, L.; Liu, L.; Ji, H.F. Alzheimer’s Disease Histological and Behavioral Manifestations in Transgenic Mice Correlate with Specific Gut Microbiome State. J. Alzheimer’s Dis. 2017, 56, 385–390. [Google Scholar] [CrossRef]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 4907. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Lutz, H.L.; Weigle, I.Q.; Patel, P.; Michalkiewicz, J.; Roman-Santiago, C.J.; Zhang, C.M.; Liang, Y.; Srinath, A.; Zhang, X.; et al. Gut microbiota-driven brain Aβ amyloidosis in mice requires microglia. J. Exp. Med. 2022, 219, e20200895. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Seo, D.O.; O’Donnell, D.; Jain, N.; Ulrich, J.D.; Herz, J.; Li, Y.; Lemieux, M.; Cheng, J.; Hu, H.; Serrano, J.R.; et al. ApoE isoform- and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science 2023, 379, eadd1236. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.K.; Borchelt, D.R.; Olson, K.; Johannsdottir, R.; Kitt, C.; Yunis, W.; Xu, S.; Eckman, C.; Younkin, S.; Price, D.; et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 1995, 15, 1203–1218. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Staufenbiel, M.; Paganetti, P.A. Electrophoretic separation and immunoblotting of aβ(1-40) and aβ (1-42). Methods Mol. Med. 2000, 32, 91–99. [Google Scholar] [CrossRef]
- Balogh, S.A.; McDowell, C.S.; Stavnezer, A.J.; Denenberg, V.H. A behavioral and neuroanatomical assessment of an inbred substrain of 129 mice with behavioral comparisons to C57BL/6J mice. Brain Res. 1999, 836, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Carlson, G.A.; Borchelt, D.R.; Dake, A.; Turner, S.; Danielson, V.; Coffin, J.D.; Eckman, C.; Meiners, J.; Nilsen, S.P.; Younkin, S.G.; et al. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum. Mol. Genet. 1997, 6, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Zhang, F.; Niwa, K.; Eckman, C.; Turner, S.K.; Fischer, E.; Younkin, S.; Borchelt, D.R.; Hsiao, K.K.; Carlson, G.A. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 1999, 2, 157–161. [Google Scholar] [CrossRef]
- Weber, E.M.; Algers, B.; Hultgren, J.; Olsson, I.A. Pup mortality in laboratory mice—Infanticide or not? Acta Vet. Scand. 2013, 55, 83. [Google Scholar] [CrossRef] [PubMed]
- Brajon, S.; Morello, G.M.; Capas-Peneda, S.; Hultgren, J.; Gilbert, C.; Olsson, A. All the Pups We Cannot See: Cannibalism Masks Perinatal Death in Laboratory Mouse Breeding but Infanticide Is Rare. Animals 2021, 11, 2327. [Google Scholar] [CrossRef]
- Morello, G.M.; Hultgren, J.; Capas-Peneda, S.; Wiltshire, M.; Thomas, A.; Wardle-Jones, H.; Brajon, S.; Gilbert, C.; Olsson, I.A.S. High laboratory mouse pre-weaning mortality associated with litter overlap, advanced dam age, small and large litters. PLoS ONE 2020, 15, e0236290. [Google Scholar] [CrossRef]
- Moore, S.J.; Murphy, G.G.; Cazares, V.A. Turning strains into strengths for understanding psychiatric disorders. Mol. Psychiatry 2020, 25, 3164–3177. [Google Scholar] [CrossRef]
- Yoshiki, A.; Moriwaki, K. Mouse phenome research: Implications of genetic background. Ilar J. 2006, 47, 94–102. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daude, N.; Machado, I.; Arce, L.; Yang, J.; Westaway, D. Microbial Composition, Disease Trajectory and Genetic Background in a Slow Onset Model of Frontotemporal Lobar Degeneration. Biomolecules 2025, 15, 636. https://doi.org/10.3390/biom15050636
Daude N, Machado I, Arce L, Yang J, Westaway D. Microbial Composition, Disease Trajectory and Genetic Background in a Slow Onset Model of Frontotemporal Lobar Degeneration. Biomolecules. 2025; 15(5):636. https://doi.org/10.3390/biom15050636
Chicago/Turabian StyleDaude, Nathalie, Ivana Machado, Luis Arce, Jing Yang, and David Westaway. 2025. "Microbial Composition, Disease Trajectory and Genetic Background in a Slow Onset Model of Frontotemporal Lobar Degeneration" Biomolecules 15, no. 5: 636. https://doi.org/10.3390/biom15050636
APA StyleDaude, N., Machado, I., Arce, L., Yang, J., & Westaway, D. (2025). Microbial Composition, Disease Trajectory and Genetic Background in a Slow Onset Model of Frontotemporal Lobar Degeneration. Biomolecules, 15(5), 636. https://doi.org/10.3390/biom15050636