
CD99: A Key Regulator in Immune Response and Tumor Microenvironment



Abstract
1. Introduction
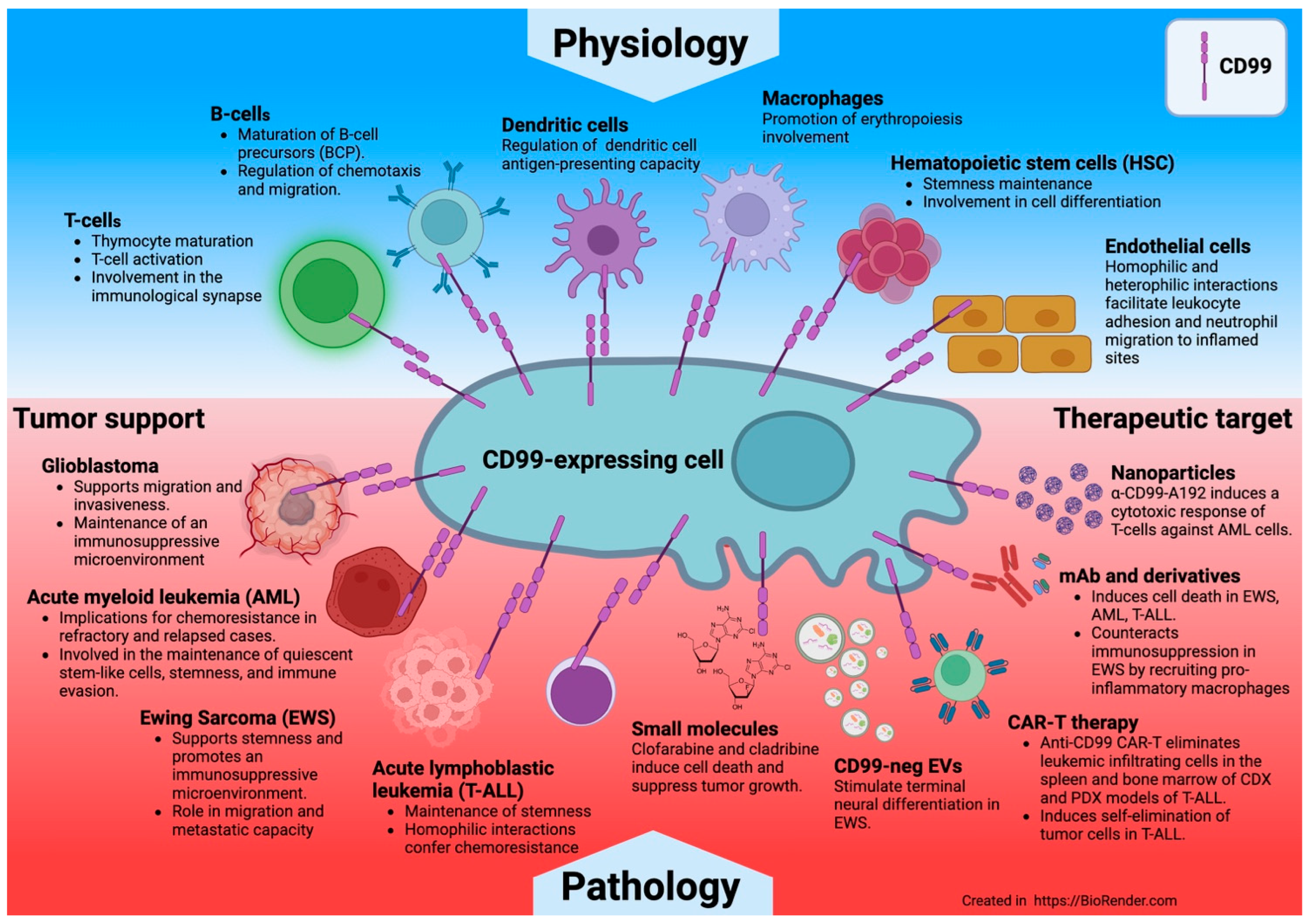
- CD99 is expressed in normal cells, particularly immune cells, and in some tumors.
- CD99 regulates various biological processes such as adhesion, transendothelial migration, differentiation, and cell death, thereby affecting immune function, inflammation, and cancer metastasis.
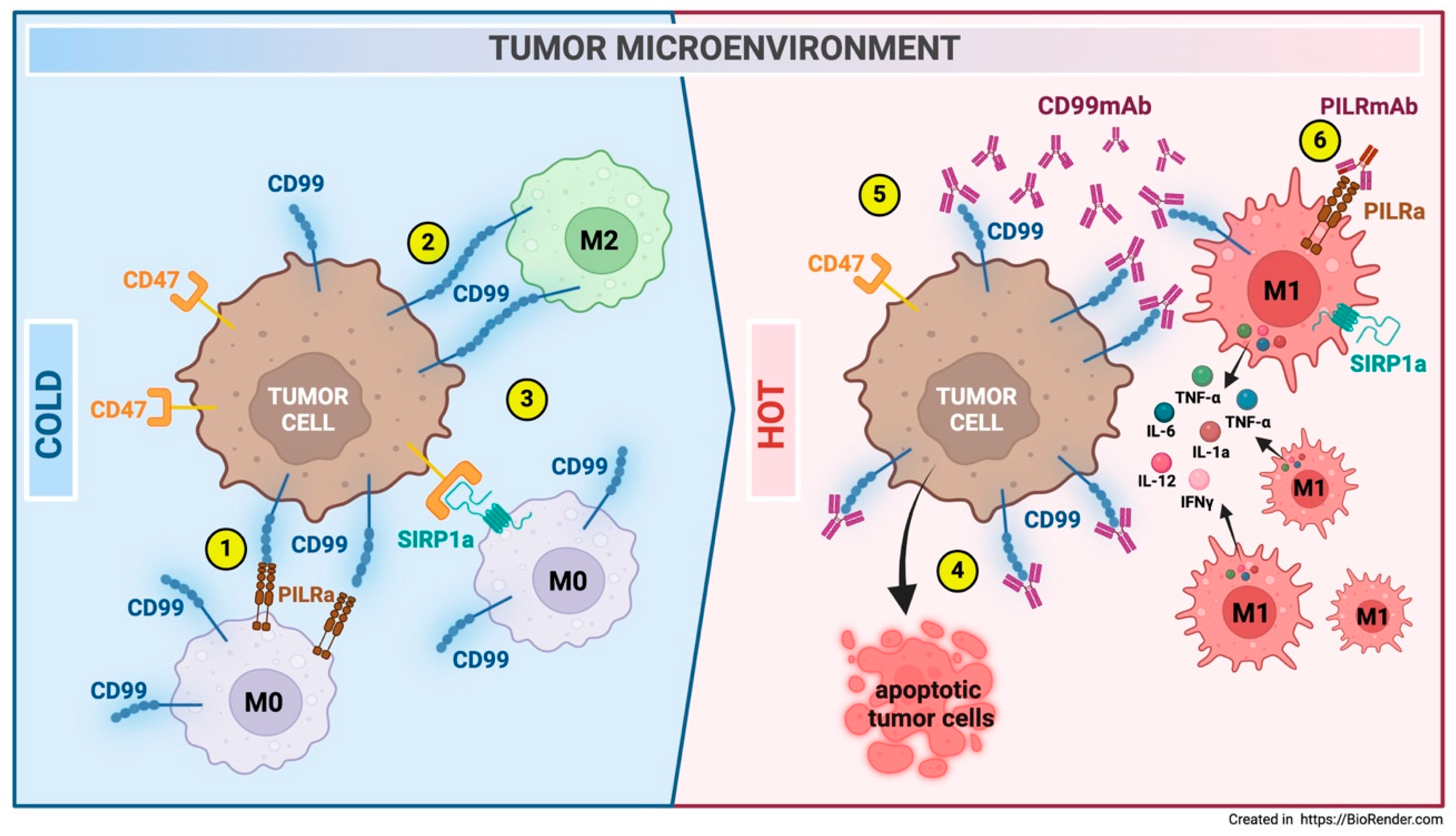
- The CD99 molecule is found at the interface between immune cells and tumors and may play a dual role as a modulator against CD99-expressing tumor cells and immune cells.
2. CD99 Antigen
3. CD99 Ligands
4. CD99 Functions in Immune Cells
4.1. The Role of CD99 in Immune T Cells
4.2. The Role of CD99 in Immune B Cells
4.3. The Role of CD99 in Other Immune Cells
4.4. The Role of CD99 in Cell Adhesion and Diapedesis
5. The Role of CD99 in Autoimmune Diseases
6. CD99 Functions in Tumors
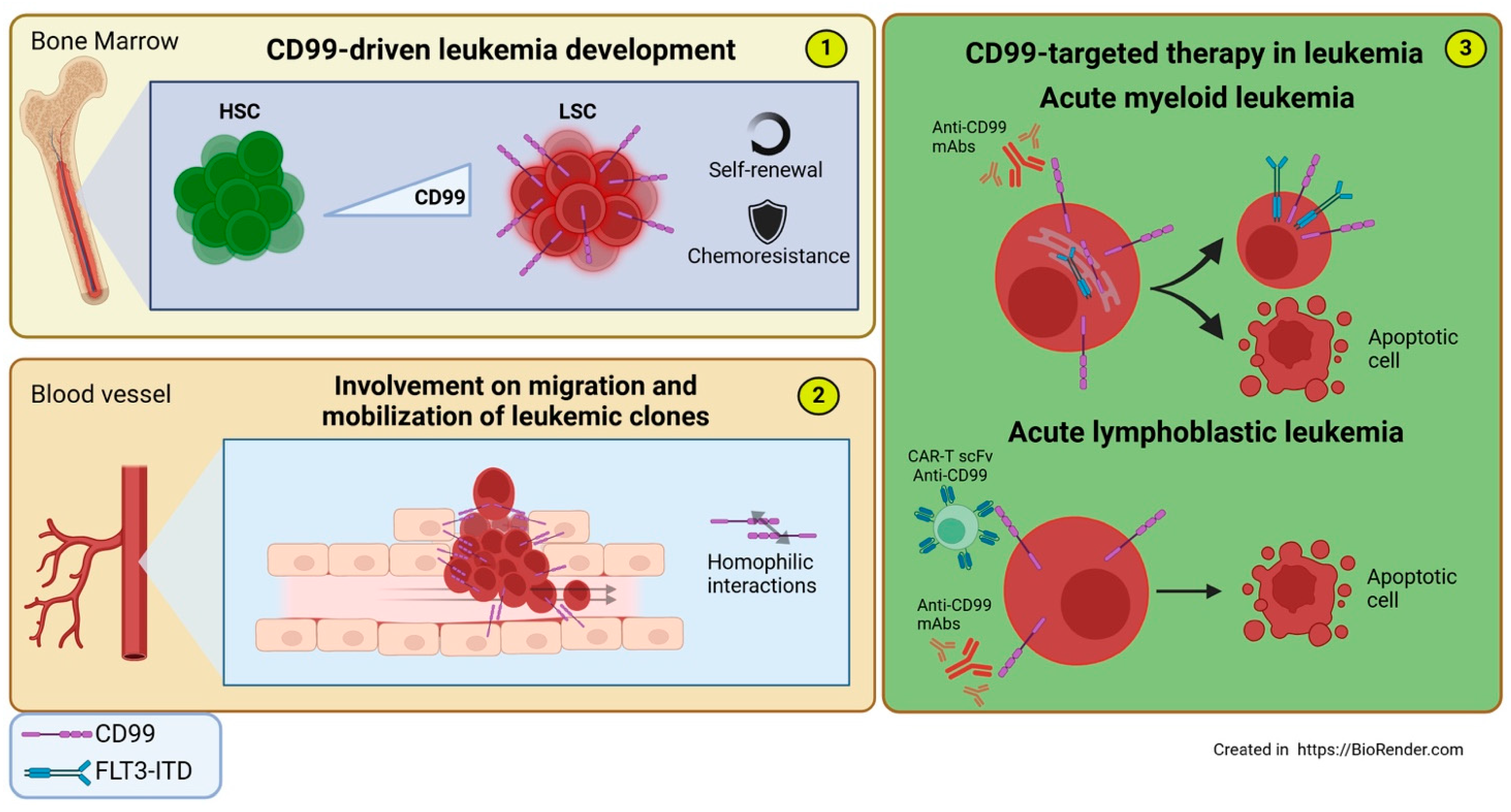
6.1. Leukemia
6.1.1. Acute Myeloid Leukemia
6.1.2. Acute Lymphoblastic Leukemia
6.2. Malignant Glioma/Glioblastoma
6.3. Ewing Sarcoma
7. Conclusions
- Information on the expression of the two isoforms in immune and tumor cells is still lacking, as is a shortage of specific antibodies capable of distinguishing between them. A clearer definition of these isoforms’ expression across cells could enhance our understanding of their function.
- The interplay between tumor cells and the immune microenvironment mediated by CD99 is a subject of great interest. A comprehensive understanding of how the presence or absence of CD99 may impact tumor infiltration and the communication between tumor cells and normal host cells is essential for accurately defining effective innovative therapeutic strategies.
- The potential to enhance the tumor microenvironment by triggering CD99 with antibodies or compounds that mimic CD99 in immune-cold tumors could provide new therapeutic perspectives for immune-evasive tumors. However, further studies are necessary to fully comprehend the benefits and minimize any collateral effects that this modulation may cause.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cellular cytotoxicity |
| AKT | AKT serine-threonine protein kinase |
| ALL | Acute linphoblastic leukemia |
| AML | Acute myeloid leukemia |
| AML | Acute myeloid leukemia |
| AS | Ankylosing spondylitis |
| AP-1 | Activating protein-1 |
| ARG1 | Arginase 1 |
| ATF-2 | Activating Transcription Factor 2 |
| AYAs | Adolescent-young adults |
| Baso-E | basophilic erythroblast |
| BBB | Blood–brain barrier |
| BFU-E | Burst-forming unit-erythroid |
| BMP | Bone morphogenetic protein |
| CAF | Cancer-associated fibroblast |
| CALR | Calreticulin |
| CAR-T | Chimeric Antigen Receptor T-Cell |
| CD | Crohn’s Disease |
| CD99 | Cluster of Differentiation 99 |
| CD99L2 | CD99 antigen-like protein 2 |
| CD99sh | CD99 short form |
| CD99wt | CD99 wild type |
| CDXs | Cell-derived xenografts |
| CFU-E | Colony Forming Unit-Erythroid |
| CLL | Chronic lymphocytic leukemia |
| CNS | Central nervous system |
| CREB-1 | cAMP response element-binding protein |
| CSK | C-terminal Src kinase |
| CTCs | Circulating tumor cells |
| CTF | C-terminal fragment |
| CTLA-4 | Cytotoxic T-Lymphocyte Antigen 4 |
| CuAS | Cuproptosis Activation Scoring |
| CXCL12 | CXC motif chemokine 12 |
| DMGs | Diffuse midline gliomas |
| EAE | Experimental autoimmune encephalomyelitis |
| EPCs | Endothelial progenitor cells |
| ERK | Extracellular signal-regulated kinase |
| EV | Extracellular vesicle |
| EWS | Ewing sarcoma |
| FDA | Food and Drug Administration |
| FLT3-ITD | Fms-like tyrosine kinase 3 internal tandem duplication |
| GBM | Glioblastoma |
| GDF6 | Growth and Differentiation Factor 6 |
| HAMA | Human anti-mouse antibody |
| HLA -I | Human leukocyte antigen |
| HMGA1 | High-mobility group protein A1 |
| HSC | Hematopoietic stem cells |
| IBD | Inflammatory bowel disease |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| ICAM-2 | Intercellular Adhesion Molecule 2 |
| ICD | Intracellular domain |
| iDC | In vitro endritic cells |
| IFNγ | Interferon gamma |
| IGF1-R | Insulin-like growth factor 1 receptor |
| IL6 | Interleukin-6 |
| JAK | Janus kinase |
| JAM-A | Junctional adhesion molecule A |
| JNK | c-Jun N-terminal kinases |
| kD | Kilo dalton |
| LFA-1 | Lymphocyte function-associated antigen 1 |
| LSC | Leukemic stem cells |
| LTB4 | Leukotriene B4 |
| mAbs | Monoclonal antibodies |
| MAPK | Mitogen-activated protein kinase |
| MARCO | Macrophage receptor with collagenous structure |
| MDM2 | Mouse double minute 2 homolog |
| MDS | Myelodysplastic syndrome |
| MDSCs | Myeloid-derived suppressor cells |
| MHC-1 | Major Histocompatibility complex type I |
| MHC-II | Major Histocompatibility complex type II |
| MIF | Macrophage migration inhibitory factor |
| MMP-9 | Matrix metalloproteinase-9 |
| MRD | Minimal residual disease |
| MSK1/2 | Mitogen- and stress-activated protein kinase |
| MTOR | Mammalian target of rapamycin |
| MS | Multiple sclerosis |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer |
| NTF | N-terminal fragment |
| OS | Overall survival |
| PCs | Plasma cells |
| PD-1 | Programmed cell death protein 1 |
| PDXs | Patient-derived xenografts |
| PECAM-1 | Platelet endothelial cell adhesion molecule |
| PI3K | Phosphoinositide 3-kinases |
| PILR | Paired Ig-like type 2 receptor |
| PKA | Protein kinase A |
| PKARIIα | cAMP-dependent Protein Kinase |
| PMT | Proneural–Mesenchymal transition |
| Poly-E | Polychromatophilic erythroblast |
| PPARG | Peroxisome proliferator-activated receptor gamma |
| Pro-E | Proerythroblast |
| PSCs | Pluripotent Stem Cells |
| scFv dAbd C7 | Single-chain fragment variable diabody |
| Sp1 | specificity protein 1 |
| SPP1 | Secreted Phosphoprotein 1 |
| SSC | Side scatter |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tumor-associated macrophages |
| TCM | T-cell-mediated cytotoxicity |
| TCR | T-cell receptors |
| TEM | tTansendothelial migration |
| TGFB1 | Transforming growth factor beta 1 |
| TGN | Trans-Golgi network |
| Th1 | Type 1 T helper |
| TME | Tumor microenvironment |
| TNFα | Tumor necrosis factor |
| UC | Ulcerative colitis |
| VCAM-1 | Vascular cell adhesion protein 1 |
| VEGF | Vascular endothelial growth factor |
References
- Pasello, M.; Manara, M.C.; Scotlandi, K. CD99 at the crossroads of physiology and pathology. J. Cell Commun. Signal 2018, 12, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Lee, G.K.; Kook, M.C.; Jung, K.C.; Kim, J.R.; Song, H.G.; Park, S.H.; Chi, J.G. Reduced expression of CD99 and functional disturbance in anencephalic cortical thymocytes. Virchows Arch. 1999, 434, 443–449. [Google Scholar] [CrossRef]
- Bernard, G.; Breittmayer, J.P.; de Matteis, M.; Trampont, P.; Hofman, P.; Senik, A.; Bernard, A. Apoptosis of immature thymocytes mediated by E2/CD99. J. Immunol. 1997, 158, 2543–2550. [Google Scholar] [CrossRef]
- Bernard, G.; Zoccola, D.; Deckert, M.; Breittmayer, J.P.; Aussel, C.; Bernard, A. The E2 molecule (CD99) specifically triggers homotypic aggregation of CD4+ CD8+ thymocytes. J. Immunol. 1995, 154, 26–32. [Google Scholar] [CrossRef]
- Takheaw, N.; Earwong, P.; Laopajon, W.; Pata, S.; Kasinrerk, W. Interaction of CD99 and its ligand upregulates IL-6 and TNF-alpha upon T cell activation. PLoS ONE 2019, 14, e0217393. [Google Scholar] [CrossRef] [PubMed]
- Wingett, D.; Forcier, K.; Nielson, C.P. A role for CD99 in T cell activation. Cell Immunol. 1999, 193, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Waclavicek, M.; Majdic, O.; Stulnig, T.; Berger, M.; Sunder-Plassmann, R.; Zlabinger, G.J.; Baumruker, T.; Stockl, J.; Ebner, C.; Knapp, W.; et al. CD99 engagement on human peripheral blood T cells results in TCR/CD3-dependent cellular activation and allows for Th1-restricted cytokine production. J. Immunol. 1998, 161, 4671–4678. [Google Scholar] [CrossRef]
- Manara, M.C.; Manferdini, C.; Cristalli, C.; Carrabotta, M.; Santi, S.; De Feo, A.; Caldoni, G.; Pasello, M.; Landuzzi, L.; Lollini, P.L.; et al. Engagement of CD99 Activates Distinct Programs in Ewing Sarcoma and Macrophages. Cancer Immunol. Res. 2024, 12, 247–260. [Google Scholar] [CrossRef]
- O’Neill, A.F.; Nguyen, E.M.; Maldonado, E.D.; Chang, M.R.; Sun, J.; Zhu, Q.; Marasco, W.A. Anti-CD99 Antibody Therapy Triggers Macrophage-Dependent Ewing Cell Death In Vitro and Myeloid Cell Recruitment In Vivo. Antibodies 2024, 13, 24. [Google Scholar] [CrossRef]
- Llombart-Bosch, A.; Machado, I.; Navarro, S.; Bertoni, F.; Bacchini, P.; Alberghini, M.; Karzeladze, A.; Savelov, N.; Petrov, S.; Alvarado-Cabrero, I.; et al. Histological heterogeneity of Ewing’s sarcoma/PNET: An immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009, 455, 397–411. [Google Scholar] [CrossRef]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9, eaaj2025. [Google Scholar] [CrossRef] [PubMed]
- Vaikari, V.; Jang, M.; Akhtari, M.; Alachkar, H. CD99 Is Highly Expressed in Acute Myeloid Leukemia (AML) and Presents a Viable Therapeutic Target. Blood 2016, 128, 1540. [Google Scholar] [CrossRef]
- Vaikari, V.P.; Du, Y.; Wu, S.; Zhang, T.; Metzeler, K.; Batcha, A.M.N.; Herold, T.; Hiddemann, W.; Akhtari, M.; Alachkar, H. Clinical and preclinical characterization of CD99 isoforms in acute myeloid leukemia. Haematologica 2020, 105, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Dworzak, M.N.; Froschl, G.; Printz, D.; Zen, L.D.; Gaipa, G.; Ratei, R.; Basso, G.; Biondi, A.; Ludwig, W.D.; Gadner, H. CD99 expression in T-lineage ALL: Implications for flow cytometric detection of minimal residual disease. Leukemia 2004, 18, 703–708. [Google Scholar] [CrossRef]
- Seol, H.J.; Chang, J.H.; Yamamoto, J.; Romagnuolo, R.; Suh, Y.; Weeks, A.; Agnihotri, S.; Smith, C.A.; Rutka, J.T. Overexpression of CD99 Increases the Migration and Invasiveness of Human Malignant Glioma Cells. Genes Cancer 2012, 3, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Urias, U.; Marie, S.K.; Uno, M.; da Silva, R.; Evagelinellis, M.M.; Caballero, O.L.; Stevenson, B.J.; Silva, W.A., Jr.; Simpson, A.J.; Oba-Shinjo, S.M. CD99 is upregulated in placenta and astrocytomas with a differential subcellular distribution according to the malignancy stage. J. Neurooncol 2014, 119, 59–70. [Google Scholar] [CrossRef]
- Aubrit, F.; Gelin, C.; Pham, D.; Raynal, B.; Bernard, A. The biochemical characterization of E2, a T cell surface molecule involved in rosettes. Eur. J. Immunol. 1989, 19, 1431–1436. [Google Scholar] [CrossRef]
- Banting, G.S.; Pym, B.; Darling, S.M.; Goodfellow, P.N. The MIC2 gene product: Epitope mapping and structural prediction analysis define an integral membrane protein. Mol. Immunol. 1989, 26, 181–188. [Google Scholar] [CrossRef]
- Ellis, N.A.; Tippett, P.; Petty, A.; Reid, M.; Weller, P.A.; Ye, T.Z.; German, J.; Goodfellow, P.N.; Thomas, S.; Banting, G. PBDX is the XG blood group gene. Nat. Genet. 1994, 8, 285–290. [Google Scholar] [CrossRef]
- Ellis, N.A.; Ye, T.Z.; Patton, S.; German, J.; Goodfellow, P.N.; Weller, P. Cloning of PBDX, an MIC2-related gene that spans the pseudoautosomal boundary on chromosome Xp. Nat. Genet. 1994, 6, 394–400. [Google Scholar] [CrossRef]
- Fouchet, C.; Gane, P.; Huet, M.; Fellous, M.; Rouger, P.; Banting, G.; Cartron, J.P.; Lopez, C. A study of the coregulation and tissue specificity of XG and MIC2 gene expression in eukaryotic cells. Blood 2000, 95, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, P.J.; Darling, S.M.; Thomas, N.S.; Goodfellow, P.N. A pseudoautosomal gene in man. Science 1986, 234, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.H.; Kim, M.K.; Choi, E.Y.; Kim, S.H.; Sohn, H.W.; Ham, D.I.; Chung, D.H.; Kim, T.J.; Lee, W.J.; Park, C.K.; et al. CD99 (MIC2) regulates the LFA-1/ICAM-1-mediated adhesion of lymphocytes, and its gene encodes both positive and negative regulators of cellular adhesion. J. Immunol. 1997, 159, 2250–2258. [Google Scholar] [CrossRef]
- Lee, M.K.; Kim, H.S.; Kim, S.S.; Cho, M.H.; Lee, I.S. Analysis of the dimerization of human CD99 using bimolecular fluorescence complementation technique. J. Microbiol. Biotechnol. 2008, 18, 472–476. [Google Scholar]
- Smith, M.J.; Goodfellow, P.J.; Goodfellow, P.N. The genomic organisation of the human pseudoautosomal gene MIC2 and the detection of a related locus. Hum. Mol. Genet. 1993, 2, 417–422. [Google Scholar] [CrossRef]
- Suh, Y.H.; Shin, Y.K.; Kook, M.C.; Oh, K.I.; Park, W.S.; Kim, S.H.; Lee, I.S.; Park, H.J.; Huh, T.L.; Park, S.H. Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs. Gene 2003, 307, 63–76. [Google Scholar] [CrossRef]
- Nam, G.; Lee, Y.K.; Lee, H.Y.; Ma, M.J.; Araki, M.; Araki, K.; Lee, S.; Lee, I.S.; Choi, E.Y. Interaction of CD99 with its paralog CD99L2 positively regulates CD99L2 trafficking to cell surfaces. J. Immunol. 2013, 191, 5730–5742. [Google Scholar] [CrossRef] [PubMed]
- Laopajon, W.; Pata, S.; Takheaw, N.; Surinkaew, S.; Khummuang, S.; Kasinrerk, W. Triggering of CD99 on monocytes by a specific monoclonal antibody regulates T cell activation. Cell Immunol. 2019, 335, 51–58. [Google Scholar] [CrossRef]
- Takheaw, N.; Pata, S.; Laopajon, W.; Roytrakul, S.; Kasinrerk, W. The presence of membrane bound CD99 ligands on leukocyte surface. BMC Res. Notes 2020, 13, 496. [Google Scholar] [CrossRef]
- Wilkerson, A.E.; Glasgow, M.A.; Hiatt, K.M. Immunoreactivity of CD99 in invasive malignant melanoma. J. Cutan. Pathol. 2006, 33, 663–666. [Google Scholar] [CrossRef]
- Diaz, M.J.; Tran, J.T.; Samia, A.M.; Forouzandeh, M.; Grant-Kels, J.M.; Montanez-Wiscovich, M.E. Integrated Analysis of Single-Cell and Bulk RNA Data Reveals Complexity and Significance of the Melanoma Interactome. Cancers 2025, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liang, H.; Tang, X.; Ling, X.; Yang, Y. Single-cell RNA sequencing reveals distinct transcriptomic profiles and evolutionary patterns in lung cancer brain metastasis. Heliyon 2024, 10, e27071. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Jin, T.; Chen, B.; Dong, Q.; Liu, M.; Guo, J.; Song, X.; Li, Y.; Chen, T.; Han, H.; et al. Multi-omics integration analysis unveils heterogeneity in breast cancer at the individual level. Cell Cycle 2023, 22, 2229–2244. [Google Scholar] [CrossRef]
- Shiratori, I.; Ogasawara, K.; Saito, T.; Lanier, L.L.; Arase, H. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J. Exp. Med. 2004, 199, 525–533. [Google Scholar] [CrossRef]
- Goswami, D.; Marz, S.; Li, Y.T.; Artz, A.; Schafer, K.; Seelige, R.; Pacheco-Blanco, M.; Jing, D.; Bixel, M.G.; Araki, M.; et al. Endothelial CD99 supports arrest of mouse neutrophils in venules and binds to neutrophil PILRs. Blood 2017, 129, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Elzi, D.J.; Jayabal, P.; Ma, X.; Chiu, Y.C.; Chen, Y.; Blackman, B.; Weintraub, S.T.; Houghton, P.J.; Shiio, Y. GDF6-CD99 Signaling Regulates Src and Ewing Sarcoma Growth. Cell Rep. 2020, 33, 108332. [Google Scholar] [CrossRef]
- Alberti, I.; Bernard, G.; Rouquette-Jazdanian, A.K.; Pelassy, C.; Pourtein, M.; Aussel, C.; Bernard, A. CD99 isoforms expression dictates T cell functional outcomes. FASEB J. 2002, 16, 1946–1948. [Google Scholar] [CrossRef]
- Byun, H.J.; Hong, I.K.; Kim, E.; Jin, Y.J.; Jeoung, D.I.; Hahn, J.H.; Kim, Y.M.; Park, S.H.; Lee, H. A splice variant of CD99 increases motility and MMP-9 expression of human breast cancer cells through the AKT-, ERK-, and JNK-dependent AP-1 activation signaling pathways. J. Biol. Chem. 2006, 281, 34833–34847. [Google Scholar] [CrossRef]
- Pettersen, R.D.; Bernard, G.; Olafsen, M.K.; Pourtein, M.; Lie, S.O. CD99 signals caspase-independent T cell death. J. Immunol. 2001, 166, 4931–4942. [Google Scholar] [CrossRef]
- Aussel, C.; Bernard, G.; Breittmayer, J.P.; Pelassy, C.; Zoccola, D.; Bernard, A. Monoclonal antibodies directed against the E2 protein (MIC2 gene product) induce exposure of phosphatidylserine at the thymocyte cell surface. Biochemistry 1993, 32, 10096–10101. [Google Scholar] [CrossRef]
- Choi, E.Y.; Park, W.S.; Jung, K.C.; Kim, S.H.; Kim, Y.Y.; Lee, W.J.; Park, S.H. Engagement of CD99 induces up-regulation of TCR and MHC class I and II molecules on the surface of human thymocytes. J. Immunol. 1998, 161, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Genestier, L.; Prigent, A.F.; Paillot, R.; Quemeneur, L.; Durand, I.; Banchereau, J.; Revillard, J.P.; Bonnefoy-Berard, N. Caspase-dependent ceramide production in Fas- and HLA class I-mediated peripheral T cell apoptosis. J. Biol. Chem. 1998, 273, 5060–5066. [Google Scholar] [CrossRef]
- Pettersen, R.D.; Gaudernack, G.; Olafsen, M.K.; Lie, S.O.; Hestdal, K. The TCR-binding region of the HLA class I alpha2 domain signals rapid Fas-independent cell death: A direct pathway for T cell-mediated killing of target cells? J. Immunol. 1998, 160, 4343–4352. [Google Scholar] [CrossRef]
- Sohn, H.W.; Shin, Y.K.; Lee, I.S.; Bae, Y.M.; Suh, Y.H.; Kim, M.K.; Kim, T.J.; Jung, K.C.; Park, W.S.; Park, C.S.; et al. CD99 regulates the transport of MHC class I molecules from the Golgi complex to the cell surface. J. Immunol. 2001, 166, 787–794. [Google Scholar] [CrossRef]
- Bremond, A.; Meynet, O.; Mahiddine, K.; Coito, S.; Tichet, M.; Scotlandi, K.; Breittmayer, J.P.; Gounon, P.; Gleeson, P.A.; Bernard, A.; et al. Regulation of HLA class I surface expression requires CD99 and p230/golgin-245 interaction. Blood 2009, 113, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Choi, Y.L.; Kim, M.K.; Kim, S.H.; Choi, E.Y.; Park, W.S.; Bae, Y.M.; Woo, S.K.; Park, S.H. MHC class II engagement inhibits CD99-induced apoptosis and up-regulation of T cell receptor and MHC molecules in human thymocytes and T cell line. FEBS Lett. 2003, 546, 379–384. [Google Scholar] [CrossRef]
- Yoon, S.S.; Kim, H.J.; Chung, D.H.; Kim, T.J. CD99 costimulation up-regulates T cell receptor-mediated activation of JNK and AP-1. Mol. Cells 2004, 18, 186–191. [Google Scholar] [CrossRef]
- Yoon, S.S.; Jung, K.I.; Choi, Y.L.; Choi, E.Y.; Lee, I.S.; Park, S.H.; Kim, T.J. Engagement of CD99 triggers the exocytic transport of ganglioside GM1 and the reorganization of actin cytoskeleton. FEBS Lett. 2003, 540, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Dworzak, M.N.; Fritsch, G.; Buchinger, P.; Fleischer, C.; Printz, D.; Zellner, A.; Schollhammer, A.; Steiner, G.; Ambros, P.F.; Gadner, H. Flow cytometric assessment of human MIC2 expression in bone marrow, thymus, and peripheral blood. Blood 1994, 83, 415–425. [Google Scholar] [CrossRef]
- Park, C.K.; Shin, Y.K.; Kim, T.J.; Park, S.H.; Ahn, G.H. High CD99 expression in memory T and B cells in reactive lymph nodes. J. Korean Med. Sci. 1999, 14, 600–606. [Google Scholar] [CrossRef]
- Hahn, M.J.; Yoon, S.S.; Sohn, H.W.; Song, H.G.; Park, S.H.; Kim, T.J. Differential activation of MAP kinase family members triggered by CD99 engagement. FEBS Lett. 2000, 470, 350–354. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, E.; Jee, B.; Hahn, J.H.; Han, K.; Jung, K.C.; Park, S.H.; Lee, H. Functional involvement of src and focal adhesion kinase in a CD99 splice variant-induced motility of human breast cancer cells. Exp. Mol. Med. 2002, 34, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.I.; Kim, B.K.; Ban, Y.L.; Choi, E.Y.; Jung, K.C.; Lee, I.S.; Park, S.H. CD99 activates T cells via a costimulatory function that promotes raft association of TCR complex and tyrosine phosphorylation of TCR zeta. Exp. Mol. Med. 2007, 39, 176–184. [Google Scholar] [CrossRef]
- Pata, S.; Otahal, P.; Brdicka, T.; Laopajon, W.; Mahasongkram, K.; Kasinrerk, W. Association of CD99 short and long forms with MHC class I, MHC class II and tetraspanin CD81 and recruitment into immunological synapses. BMC Res. Notes 2011, 4, 293. [Google Scholar] [CrossRef]
- Husak, Z.; Printz, D.; Schumich, A.; Potschger, U.; Dworzak, M.N. Death induction by CD99 ligation in TEL/AML1-positive acute lymphoblastic leukemia and normal B cell precursors. J. Leukoc. Biol. 2010, 88, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Pak, H.K.; Lee, A.N.; Park, S.J.; Lee, Y.; Roh, J.; Lee, H.; Chung, Y.S.; Park, C.S. CD99 regulates CXCL12-induced chemotaxis of human plasma cells. Immunol. Lett. 2015, 168, 329–336. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Zhao, S.; Yan, H.; Xin, Z.; Cui, T.; Zang, R.; Zhao, L.; Wang, H.; Zhou, J.; et al. Decoding human in vitro terminal erythropoiesis originating from umbilical cord blood mononuclear cells and pluripotent stem cells. Cell Prolif. 2024, 57, e13614. [Google Scholar] [CrossRef]
- Mahiddine, K.; Mallavialle, A.; Bziouech, H.; Larbret, F.; Bernard, A.; Bernard, G. CD99 isoforms regulate CD1a expression in human monocyte-derived DCs through ATF-2/CREB-1 phosphorylation. Eur. J. Immunol. 2016, 46, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.; Raimondi, V.; Alberti, I.; Pourtein, M.; Widjenes, J.; Ticchioni, M.; Bernard, A. CD99 (E2) up-regulates alpha4beta1-dependent T cell adhesion to inflamed vascular endothelium under flow conditions. Eur. J. Immunol. 2000, 30, 3061–3065. [Google Scholar] [CrossRef]
- Lou, O.; Alcaide, P.; Luscinskas, F.W.; Muller, W.A. CD99 is a key mediator of the transendothelial migration of neutrophils. J. Immunol. 2007, 178, 1136–1143. [Google Scholar] [CrossRef]
- Schenkel, A.R.; Mamdouh, Z.; Chen, X.; Liebman, R.M.; Muller, W.A. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 2002, 3, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.P.; Seidman, M.A.; Muller, W.A. Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am. J. Pathol. 2013, 182, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.L.; Buck, J.; Levin, L.R.; Winger, R.C.; Wang, J.; Arase, H.; Muller, W.A. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J. Exp. Med. 2015, 212, 1021–1041. [Google Scholar] [CrossRef] [PubMed]
- Bixel, M.G.; Li, H.; Petri, B.; Khandoga, A.G.; Khandoga, A.; Zarbock, A.; Wolburg-Buchholz, K.; Wolburg, H.; Sorokin, L.; Zeuschner, D.; et al. CD99 and CD99L2 act at the same site as, but independently of, PECAM-1 during leukocyte diapedesis. Blood 2010, 116, 1172–1184. [Google Scholar] [CrossRef]
- Bixel, M.G.; Petri, B.; Khandoga, A.G.; Khandoga, A.; Wolburg-Buchholz, K.; Wolburg, H.; Marz, S.; Krombach, F.; Vestweber, D. A CD99-related antigen on endothelial cells mediates neutrophil but not lymphocyte extravasation in vivo. Blood 2007, 109, 5327–5336. [Google Scholar] [CrossRef]
- Li, W.; Luckstadt, W.; Wohner, B.; Bub, S.; Schulz, A.; Socher, E.; Arnold, P. Structural and functional properties of meprin beta metalloproteinase with regard to cell signaling. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119136. [Google Scholar] [CrossRef]
- Bedau, T.; Peters, F.; Prox, J.; Arnold, P.; Schmidt, F.; Finkernagel, M.; Kollmann, S.; Wichert, R.; Otte, A.; Ohler, A.; et al. Ectodomain shedding of CD99 within highly conserved regions is mediated by the metalloprotease meprin beta and promotes transendothelial cell migration. FASEB J. 2017, 31, 1226–1237. [Google Scholar] [CrossRef]
- Aguilera-Montilla, N.; Bailon, E.; Uceda-Castro, R.; Ugarte-Berzal, E.; Santos, A.; Gutierrez-Gonzalez, A.; Perez-Sanchez, C.; Van den Steen, P.E.; Opdenakker, G.; Garcia-Marco, J.A.; et al. MMP-9 affects gene expression in chronic lymphocytic leukemia revealing CD99 as an MMP-9 target and a novel partner in malignant cell migration/arrest. Oncogene 2019, 38, 4605–4619. [Google Scholar] [CrossRef]
- Torzicky, M.; Viznerova, P.; Richter, S.; Strobl, H.; Scheinecker, C.; Foedinger, D.; Riedl, E. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) and CD99 are critical in lymphatic transmigration of human dendritic cells. J. Investig. Dermatol. 2012, 132, 1149–1157. [Google Scholar] [CrossRef]
- Feng, X.; Wang, C.; Ji, B.; Qiao, J.; Xu, Y.; Zhu, S.; Ji, Z.; Zhou, B.; Tong, W.; Xu, W. CD_99 G1 neutrophils modulate osteogenic differentiation of mesenchymal stem cells in the pathological process of ankylosing spondylitis. Ann. Rheum. Dis. 2024, 83, 324–334. [Google Scholar] [CrossRef]
- Zhou, G.; Yang, W.; Yu, L.; Yu, T.; Liu, Z. CD99 refers to the activity of inflammatory bowel disease. Scand. J. Gastroenterol. 2017, 52, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Winger, R.C.; Harp, C.T.; Chiang, M.Y.; Sullivan, D.P.; Watson, R.L.; Weber, E.W.; Podojil, J.R.; Miller, S.D.; Muller, W.A. Cutting Edge: CD99 Is a Novel Therapeutic Target for Control of T Cell-Mediated Central Nervous System Autoimmune Disease. J. Immunol. 2016, 196, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Samus, M.; Seelige, R.; Schafer, K.; Sorokin, L.; Vestweber, D. CD99L2 deficiency inhibits leukocyte entry into the central nervous system and ameliorates neuroinflammation. J. Leukoc. Biol. 2018, 104, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Soelberg Sorensen, P.; Cook, S.; Rammohan, K.; Rieckmann, P.; Comi, G.; Dangond, F.; Adeniji, A.K.; Vermersch, P. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study. Mult. Scler. 2018, 24, 1594–1604. [Google Scholar] [CrossRef]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Rorie, C.J.; Thomas, V.D.; Chen, P.; Pierce, H.H.; O’Bryan, J.P.; Weissman, B.E. The Ews/Fli-1 fusion gene switches the differentiation program of neuroblastomas to Ewing sarcoma/peripheral primitive neuroectodermal tumors. Cancer Res. 2004, 64, 1266–1277. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Zhang, J.; Wu, L.; Shimada, H.; Schofield, D.E.; Triche, T.J. EWS-FLI1 fusion protein up-regulates critical genes in neural crest development and is responsible for the observed phenotype of Ewing’s family of tumors. Cancer Res. 2005, 65, 4633–4644. [Google Scholar] [CrossRef]
- Franzetti, G.A.; Laud-Duval, K.; Bellanger, D.; Stern, M.H.; Sastre-Garau, X.; Delattre, O. MiR-30a-5p connects EWS-FLI1 and CD99, two major therapeutic targets in Ewing tumor. Oncogene 2013, 32, 3915–3921. [Google Scholar] [CrossRef]
- Kreppel, M.; Aryee, D.N.; Schaefer, K.L.; Amann, G.; Kofler, R.; Poremba, C.; Kovar, H. Suppression of KCMF1 by constitutive high CD99 expression is involved in the migratory ability of Ewing’s sarcoma cells. Oncogene 2006, 25, 2795–2800. [Google Scholar] [CrossRef]
- Rocchi, A.; Manara, M.C.; Sciandra, M.; Zambelli, D.; Nardi, F.; Nicoletti, G.; Garofalo, C.; Meschini, S.; Astolfi, A.; Colombo, M.P.; et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J. Clin. Investig. 2010, 120, 668–680. [Google Scholar] [CrossRef]
- Ventura, S.; Aryee, D.N.; Felicetti, F.; De Feo, A.; Mancarella, C.; Manara, M.C.; Picci, P.; Colombo, M.P.; Kovar, H.; Care, A.; et al. CD99 regulates neural differentiation of Ewing sarcoma cells through miR-34a-Notch-mediated control of NF-kappaB signaling. Oncogene 2016, 35, 3944–3954. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.E.; Ahn, H.H.; Kye, Y.C.; Kim, S.N. Precursor B-cell lymphoblastic lymphoma presenting as solitary infiltrative plaque in a child. Acta Derm. Venereol. 2008, 88, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, S.; Ottone, T.; Angelini, D.F.; Fiori, V.; Dominici, S.; Noguera, N.I.; Sniegocka, M.; Antonelli, S.; Irno Consalvo, M.A.; De Bardi, M.; et al. CD99 as a novel therapeutic target on leukemic progenitor cells in FLT3-ITD(mut) AML. Leukemia 2022, 36, 1685–1688. [Google Scholar] [CrossRef]
- Jung, K.C.; Park, W.S.; Bae, Y.M.; Hahn, J.H.; Hahn, K.; Lee, H.; Lee, H.W.; Koo, H.J.; Shin, H.J.; Shin, H.S.; et al. Immunoreactivity of CD99 in stomach cancer. J. Korean Med. Sci. 2002, 17, 483–489. [Google Scholar] [CrossRef]
- Scotlandi, K.; Zuntini, M.; Manara, M.C.; Sciandra, M.; Rocchi, A.; Benini, S.; Nicoletti, G.; Bernard, G.; Nanni, P.; Lollini, P.L.; et al. CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene 2007, 26, 6604–6618. [Google Scholar] [CrossRef]
- Cantiani, L.; Manara, M.C.; Zucchini, C.; De Sanctis, P.; Zuntini, M.; Valvassori, L.; Serra, M.; Olivero, M.; Di Renzo, M.F.; Colombo, M.P.; et al. Caveolin-1 reduces osteosarcoma metastases by inhibiting c-Src activity and met signaling. Cancer Res. 2007, 67, 7675–7685. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, Y.; Ye, L.; Jiang, W.G. The FERM family proteins in cancer invasion and metastasis. Front. Biosci. (Landmark Ed.) 2011, 16, 1536–1550. [Google Scholar] [CrossRef]
- Zucchini, C.; Manara, M.C.; Pinca, R.S.; De Sanctis, P.; Guerzoni, C.; Sciandra, M.; Lollini, P.L.; Cenacchi, G.; Picci, P.; Valvassori, L.; et al. CD99 suppresses osteosarcoma cell migration through inhibition of ROCK2 activity. Oncogene 2014, 33, 1912–1921. [Google Scholar] [CrossRef]
- Sciandra, M.; Marino, M.T.; Manara, M.C.; Guerzoni, C.; Grano, M.; Oranger, A.; Lucarelli, E.; Lollini, P.L.; Dozza, B.; Pratelli, L.; et al. CD99 drives terminal differentiation of osteosarcoma cells by acting as a spatial regulator of ERK 1/2. J. Bone Miner. Res. 2014, 29, 1295–1309. [Google Scholar] [CrossRef]
- Manara, M.C.; Pasello, M.; Scotlandi, K. CD99: A Cell Surface Protein with an Oncojanus Role in Tumors. Genes 2018, 9, 159. [Google Scholar] [CrossRef]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A Leukemia-Associated CD34/CD123/CD25/CD99+ Immunophenotype Identifies FLT3-Mutated Clones in Acute Myeloid Leukemia. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Liang, Z.; Zhang, L.; Song, J.; Huang, M.; Xie, K.; Gale, R.; Liang, Y. 824P FLT3-ITD induces immune escape in AML via up-regulating CD47 expression and decreased phagocytic ability of macrophages. Ann. Oncol. 2024, 35, S605. [Google Scholar] [CrossRef]
- Li, K.; Du, Y.; Cai, Y.; Liu, W.; Lv, Y.; Huang, B.; Zhang, L.; Wang, Z.; Liu, P.; Sun, Q.; et al. Single-cell analysis reveals the chemotherapy-induced cellular reprogramming and novel therapeutic targets in relapsed/refractory acute myeloid leukemia. Leukemia 2023, 37, 308–325. [Google Scholar] [CrossRef]
- Huang, Y.; Kapti, E.G.; Ji, Y.; Thomas, T.; Dempsey, J.; Mims, K.; Berezniuk, I.; Guo, L.; Kroger, B.; Hu, W.; et al. CD99 Promotes Self-renewal in Hematopoietic Stem Cells and Leukemia Stem Cells by Regulating Protein Synthesis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 2017, 129, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Mallet, F.; Gaugler, B.; Sainty, D.; Arnoulet, C.; Gastaut, J.A.; Olive, D. Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000, 60, 4403–4411. [Google Scholar]
- Guerzoni, C.; Fiori, V.; Terracciano, M.; Manara, M.C.; Moricoli, D.; Pasello, M.; Sciandra, M.; Nicoletti, G.; Gellini, M.; Dominici, S.; et al. CD99 triggering in Ewing sarcoma delivers a lethal signal through p53 pathway reactivation and cooperates with doxorubicin. Clin. Cancer Res. 2015, 21, 146–156. [Google Scholar] [CrossRef]
- Yamawaki, K.; Shiina, I.; Murata, T.; Tateyama, S.; Maekawa, Y.; Niwa, M.; Shimonaka, M.; Okamoto, K.; Suzuki, T.; Nishida, T.; et al. FLT3-ITD transduces autonomous growth signals during its biosynthetic trafficking in acute myelogenous leukemia cells. Sci. Rep. 2021, 11, 22678. [Google Scholar] [CrossRef]
- Reiter, K.; Polzer, H.; Krupka, C.; Maiser, A.; Vick, B.; Rothenberg-Thurley, M.; Metzeler, K.H.; Dorfel, D.; Salih, H.R.; Jung, G.; et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia 2018, 32, 313–322. [Google Scholar] [CrossRef]
- Ali, A.; Phan, A.; Vaikari, V.; Park, M.; Pospiech, M.; Chu, R.; Meng, Y.; MacKay, J.A.; Alachkar, H. FLT3/CD99 Bispecific Antibody-Based Nanoparticles for Acute Myeloid Leukemia. Cancer Res. Commun. 2024, 4, 1946–1962. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.; Ali, A.; Pospiech, M.; Onyemaechi, S.; Meng, Y.; Dhuri, K.; MacKay, J.A.; Alachkar, H. Enhanced T cell activation and cytotoxicity against AML via targeted anti-CD99 nanoparticle treatment. Biomed. Pharmacother. 2024, 179, 117265. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.V.; Diamanti, P.; Moppett, J.P.; Blair, A. Investigating CD99 Expression in Leukemia Propagating Cells in Childhood T Cell Acute Lymphoblastic Leukemia. PLoS ONE 2016, 11, e0165210. [Google Scholar] [CrossRef]
- Enein, A.A.; Rahman, H.A.; Sharkawy, N.E.; Elhamid, S.A.; Abbas, S.M.; Abdelfaatah, R.; Khalil, M.; Fathalla, L.A. Significance of CD99 expression in T-lineage acute lymphoblastic leukemia. Cancer Biomark. 2016, 17, 117–123. [Google Scholar] [CrossRef]
- Kotemul, K.; Kasinrerk, W.; Takheaw, N. CD99 tumor associated antigen is a potential target for antibody therapy of T-cell acute lymphoblastic leukemia. Explor. Target. Antitumor Ther. 2024, 5, 96–107. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Z.; Cen, H.; Wu, H.; Zhang, S.; Liu, J.; Leng, Y.; Ren, A.; Liu, X.; Zhang, Z.; et al. CAR T cells targeting CD99 as an approach to eradicate T-cell acute lymphoblastic leukemia without normal blood cells toxicity. J. Hematol. Oncol. 2021, 14, 162. [Google Scholar] [CrossRef]
- Ren, A.; Zhao, Y.; Zhu, H. T-ALL Cells as Tool Cells for CAR T Therapy. Vaccines 2023, 11, 854. [Google Scholar] [CrossRef]
- Ebadi, M.; Jonart, L.M.; Ostergaard, J.; Gordon, P.M. CD99 antibody disrupts T-cell acute lymphoblastic leukemia adhesion to meningeal cells and attenuates chemoresistance. Sci. Rep. 2021, 11, 24374. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.C.; Soares, R.D.S.; Laurentino, T.S.; Lerario, A.M.; Marie, S.K.N.; Oba-Shinjo, S.M. CD99 Expression in Glioblastoma Molecular Subtypes and Role in Migration and Invasion. Int. J. Mol. Sci. 2019, 20, 1137. [Google Scholar] [CrossRef]
- Ishizawa, K.; Komori, T.; Shimada, S.; Hirose, T. Olig2 and CD99 are useful negative markers for the diagnosis of brain tumors. Clin. Neuropathol. 2008, 27, 118–128. [Google Scholar] [CrossRef]
- Jung, T.Y.; Choi, Y.D.; Kim, Y.H.; Lee, J.J.; Kim, H.S.; Kim, J.S.; Kim, S.K.; Jung, S.; Cho, D. Immunological characterization of glioblastoma cells for immunotherapy. Anticancer. Res. 2013, 33, 2525–2533. [Google Scholar] [PubMed]
- Shang, E.; Sun, S.; Zhang, R.; Cao, Z.; Chen, Q.; Shi, L.; Wu, J.; Wu, S.; Liu, Y.; Zheng, Y. Overexpression of CD99 is associated with tumor adaptiveness and indicates the tumor recurrence and therapeutic responses in gliomas. Transl. Oncol. 2023, 37, 101759. [Google Scholar] [CrossRef]
- Rocca, A.; Giudici, F.; Donofrio, C.A.; Bottin, C.; Pinamonti, M.; Ferrari, B.; Schettini, F.; Pineda, E.; Panni, S.; Cominetti, M.; et al. CD99 Expression and Prognostic Impact in Glioblastoma: A Single-Center Cohort Study. Cells 2024, 13, 597. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xiao, D.; Jiang, X.; Nie, C. EREG is the core onco-immunological biomarker of cuproptosis and mediates the cross-talk between VEGF and CD99 signaling in glioblastoma. J. Transl. Med. 2023, 21, 28. [Google Scholar] [CrossRef]
- Balakrishnan, I.; Madhavan, K.; Pierce, A.M.; Michlin, J.; Brunt, B.; Chetty, S.L.; Wang, D.; DeSisto, J.; Nuss, Z.; Davidson, N.; et al. Targeting Diffuse Midline Glioma with a novel anti-CD99 Antibody. bioRxiv 2024. bioRxiv:2024.03.19.585814. [Google Scholar] [CrossRef]
- Ambros, I.M.; Ambros, P.F.; Strehl, S.; Kovar, H.; Gadner, H.; Salzer-Kuntschik, M. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer 1991, 67, 1886–1893. [Google Scholar] [CrossRef]
- Fellinger, E.J.; Garin-Chesa, P.; Triche, T.J.; Huvos, A.G.; Rettig, W.J. Immunohistochemical analysis of Ewing’s sarcoma cell surface antigen p30/32MIC2. Am. J. Pathol. 1991, 139, 317–325. [Google Scholar]
- Martinelli, M.; Parra, A.; Scapoli, L.; De Sanctis, P.; Chiadini, V.; Hattinger, C.; Picci, P.; Zucchini, C.; Scotlandi, K. CD99 polymorphisms significantly influence the probability to develop Ewing sarcoma in earlier age and patient disease progression. Oncotarget 2016, 7, 77958–77967. [Google Scholar] [CrossRef]
- Miyagawa, Y.; Okita, H.; Nakaijima, H.; Horiuchi, Y.; Sato, B.; Taguchi, T.; Toyoda, M.; Katagiri, Y.U.; Fujimoto, J.; Hata, J.; et al. Inducible expression of chimeric EWS/ETS proteins confers Ewing’s family tumor-like phenotypes to human mesenchymal progenitor cells. Mol. Cell Biol. 2008, 28, 2125–2137. [Google Scholar] [CrossRef]
- Riggi, N.; Suva, M.L.; Suva, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.M.; Stehle, J.C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.T.; Manara, M.C.; Berghuis, D.; Ordonez, J.L.; Biscuola, M.; Lopez-Garcia, M.A.; Osuna, D.; Lucarelli, E.; Alviano, F.; Lankester, A.; et al. Characterization of human mesenchymal stem cells from ewing sarcoma patients. Pathogenetic implications. PLoS ONE 2014, 9, e85814. [Google Scholar] [CrossRef]
- Pasello, M.; Laginestra, M.A.; Manara, M.C.; Landuzzi, L.; Ruzzi, F.; Maioli, M.; Pellegrini, E.; De Feo, A.; Lollini, P.L.; Scotlandi, K. CD99 contributes to the EWS::FLI1 transcriptome by specifically affecting FOXM1-targets involved in the G2/M cell cycle phase, thus influencing the Ewing sarcoma genetic landscape. J. Cell Commun. Signal 2024, 18, e12047. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Alberts, I.; Li, X. The role of ERK1/2 in the regulation of proliferation and differentiation of astrocytes in developing brain. Int. J. Dev. Neurosci. 2013, 31, 783–789. [Google Scholar] [CrossRef]
- De Feo, A.; Pazzaglia, L.; Ciuffarin, L.; Mangiagli, F.; Pasello, M.; Simonetti, E.; Pellegrini, E.; Ferrari, C.; Bianchi, G.; Spazzoli, B.; et al. miR-214-3p Is Commonly Downregulated by EWS-FLI1 and by CD99 and Its Restoration Limits Ewing Sarcoma Aggressiveness. Cancers 2022, 14, 1762. [Google Scholar] [CrossRef]
- De Feo, A.; Sciandra, M.; Ferracin, M.; Felicetti, F.; Astolfi, A.; Pignochino, Y.; Picci, P.; Care, A.; Scotlandi, K. Exosomes from CD99-deprived Ewing sarcoma cells reverse tumor malignancy by inhibiting cell migration and promoting neural differentiation. Cell Death Dis. 2019, 10, 471. [Google Scholar] [CrossRef]
- De Feo, A.; Manfredi, M.; Mancarella, C.; Maqueda, J.J.; De Giorgis, V.; Pignochino, Y.; Sciandra, M.; Cristalli, C.; Donadelli, M.; Scotlandi, K. CD99 Modulates the Proteomic Landscape of Ewing Sarcoma Cells and Related Extracellular Vesicles. Int. J. Mol. Sci. 2024, 25, 1588. [Google Scholar] [CrossRef]
- Samuel, G.; Crow, J.; Klein, J.B.; Merchant, M.L.; Nissen, E.; Koestler, D.C.; Laurence, K.; Liang, X.; Neville, K.; Staggs, V.; et al. Ewing sarcoma family of tumors-derived small extracellular vesicle proteomics identify potential clinical biomarkers. Oncotarget 2020, 11, 2995–3012. [Google Scholar] [CrossRef]
- Benini, S.; Gamberi, G.; Cocchi, S.; Garbetta, J.; Alberti, L.; Righi, A.; Gambarotti, M.; Picci, P.; Ferrari, S. Detection of circulating tumor cells in liquid biopsy from Ewing sarcoma patients. Cancer Manag. Res. 2018, 10, 49–60. [Google Scholar] [CrossRef]
- Cerisano, V.; Aalto, Y.; Perdichizzi, S.; Bernard, G.; Manara, M.C.; Benini, S.; Cenacchi, G.; Preda, P.; Lattanzi, G.; Nagy, B.; et al. Molecular mechanisms of CD99-induced caspase-independent cell death and cell-cell adhesion in Ewing’s sarcoma cells: Actin and zyxin as key intracellular mediators. Oncogene 2004, 23, 5664–5674. [Google Scholar] [CrossRef]
- Balestra, T.; Manara, M.C.; Laginestra, M.A.; Pasello, M.; De Feo, A.; Bassi, C.; Guerzoni, C.; Landuzzi, L.; Lollini, P.L.; Donati, D.M.; et al. Targeting CD99 Compromises the Oncogenic Effects of the Chimera EWS-FLI1 by Inducing Reexpression of Zyxin and Inhibition of GLI1 Activity. Mol. Cancer Ther. 2022, 21, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Manara, M.C.; Terracciano, M.; Mancarella, C.; Sciandra, M.; Guerzoni, C.; Pasello, M.; Grilli, A.; Zini, N.; Picci, P.; Colombo, M.P.; et al. CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1R/RAS/Rac1 signaling. Oncotarget 2016, 7, 79925–79942. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Perdichizzi, S.; Bernard, G.; Nicoletti, G.; Nanni, P.; Lollini, P.L.; Curti, A.; Manara, M.C.; Benini, S.; Bernard, A.; et al. Targeting CD99 in association with doxorubicin: An effective combined treatment for Ewing’s sarcoma. Eur. J. Cancer 2006, 42, 91–96. [Google Scholar] [CrossRef]
- Moricoli, D.; Carbonella, D.C.; Dominici, S.; Fiori, V.; Balducci, M.C.; Guerzoni, C.; Manara, M.C.; Pasello, M.; Laguardia, M.E.; Cianfriglia, M.; et al. Process development of a human recombinant diabody expressed in E. coli: Engagement of CD99-induced apoptosis for target therapy in Ewing’s sarcoma. Appl. Microbiol. Biotechnol. 2016, 100, 3949–3963. [Google Scholar] [CrossRef]
- Kim, H.J.; Chong, K.H.; Kang, S.W.; Lee, J.R.; Kim, J.Y.; Hahn, M.J.; Kim, T.J. Identification of cyclophilin A as a CD99-binding protein by yeast two-hybrid screening. Immunol. Lett. 2004, 95, 155–159. [Google Scholar] [CrossRef]
- Celik, H.; Sciandra, M.; Flashner, B.; Gelmez, E.; Kayraklioglu, N.; Allegakoen, D.V.; Petro, J.R.; Conn, E.J.; Hour, S.; Han, J.; et al. Clofarabine inhibits Ewing sarcoma growth through a novel molecular mechanism involving direct binding to CD99. Oncogene 2018, 37, 2181–2196. [Google Scholar] [CrossRef]
- Sevim, H.; Celik, H.; Dusunceli, L.; Ceyhan, C.S.; Molotkova, A.; Nakazawa, K.; Graham, G.T.; Petro, J.R.; Toretsky, J.A.; Uren, A. Clofarabine induces ERK/MSK/CREB activation through inhibiting CD99 on Ewing sarcoma cells. PLoS ONE 2021, 16, e0253170. [Google Scholar] [CrossRef]
- Balaraman, K.; Deniz, E.; Nelson, E.; Pilicer, S.L.; Atasoy, S.; Molotkova, A.; Sevim, H.; Tiwari, P.B.; Uren, A.; Wolf, C. Design, synthesis and biological evaluation of Nucleosidic CD99 inhibitors that selectively reduce Ewing sarcoma viability. Eur. J. Med. Chem. 2023, 251, 115244. [Google Scholar] [CrossRef]
- Evdokimova, V.; Gassmann, H.; Radvanyi, L.; Burdach, S.E.G. Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma. Cancers 2022, 15, 272. [Google Scholar] [CrossRef]
- Luo, W.; Hoang, H.; Miller, K.E.; Zhu, H.; Xu, S.; Mo, X.; Garfinkle, E.A.R.; Costello, H.; Wijeratne, S.; Chemnitz, W.; et al. Combinatorial macrophage induced innate immunotherapy against Ewing sarcoma: Turning “Two Keys” simultaneously. J. Exp. Clin. Cancer Res. 2024, 43, 193. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manara, M.C.; Fiori, V.; Sparti, A.; Scotlandi, K. CD99: A Key Regulator in Immune Response and Tumor Microenvironment. Biomolecules 2025, 15, 632. https://doi.org/10.3390/biom15050632
Manara MC, Fiori V, Sparti A, Scotlandi K. CD99: A Key Regulator in Immune Response and Tumor Microenvironment. Biomolecules. 2025; 15(5):632. https://doi.org/10.3390/biom15050632
Chicago/Turabian StyleManara, Maria Cristina, Valentina Fiori, Angelo Sparti, and Katia Scotlandi. 2025. "CD99: A Key Regulator in Immune Response and Tumor Microenvironment" Biomolecules 15, no. 5: 632. https://doi.org/10.3390/biom15050632
APA StyleManara, M. C., Fiori, V., Sparti, A., & Scotlandi, K. (2025). CD99: A Key Regulator in Immune Response and Tumor Microenvironment. Biomolecules, 15(5), 632. https://doi.org/10.3390/biom15050632