


Probenecid Inhibits NLRP3 Inflammasome Activity and Mitogen-Activated Protein Kinases (MAPKs)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. Cell Lysates and Western Blots
2.3. Canonical NLRP3 Inflammasome Activation
2.4. IL-1β, TNFα, and LDH Detection
2.5. Chemicals Used
2.6. Statistical Analysis
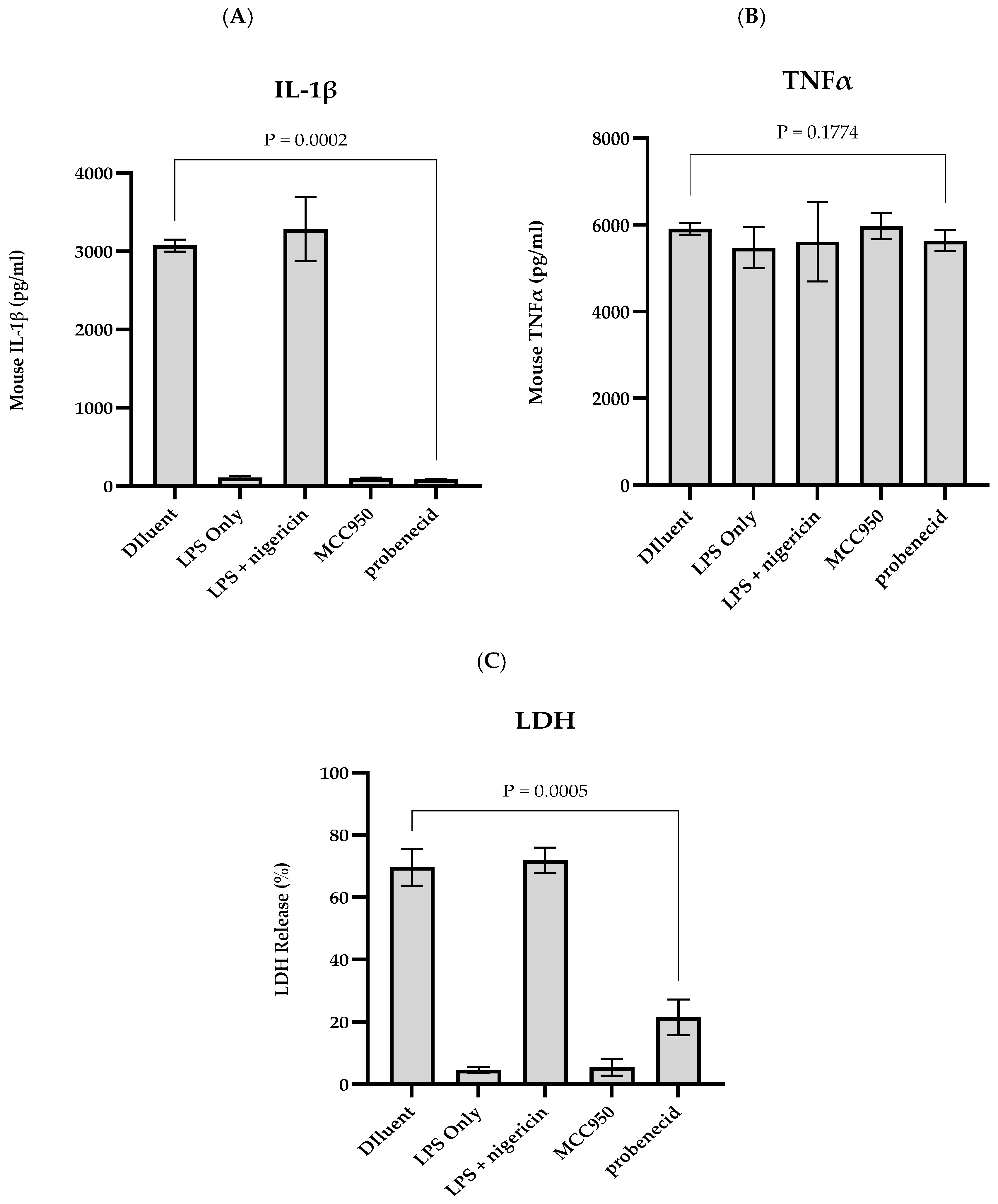
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, L.P.; Bergeron, H.C.; Martin, D.E.; Murray, J.; Sancilio, F.D.; Tripp, R.A. Probenecid Inhibits Extracellular Signal-Regulated Kinase and c-Jun N-Terminal Kinase Mitogen-Activated Protein Kinase Pathways in Regulating Respiratory Syncytial Virus Response. Int. J. Mol. Sci. 2024, 25, 12452. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Martin, D.E.; Sancilio, F.D.; Tripp, R.A. Antiviral Activity of Probenecid and Oseltamivir on Influenza Virus Replication. Viruses 2023, 15, 2366. [Google Scholar] [CrossRef]
- Tripp, R.A.; Martin, D.E. Screening Drugs for Broad-Spectrum, Host-Directed Antiviral Activity: Lessons from the Development of Probenecid for COVID-19. Viruses 2023, 15, 2254. [Google Scholar] [CrossRef]
- Tripp, R.A.; Martin, D.E. Repurposing Probenecid to Inhibit SARS-CoV-2, Influenza Virus, and Respiratory Syncytial Virus (RSV) Replication. Viruses 2022, 14, 612. [Google Scholar] [CrossRef]
- Martin, D.E.; Pandey, N.; Chavda, P.; Singh, G.; Sutariya, R.; Sancilio, F.; Tripp, R.A. Oral Probenecid for Nonhospitalized Adults with Symptomatic Mild-to-Moderate COVID-19. Viruses 2023, 15, 1508. [Google Scholar] [CrossRef]
- Murray, J.; Bergeron, H.C.; Jones, L.P.; Reener, Z.B.; Martin, D.E.; Sancilio, F.D.; Tripp, R.A. Probenecid Inhibits Respiratory Syncytial Virus (RSV) Replication. Viruses 2022, 14, 912. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, H.C.; Crabtree, J.; Nagy, T.; Martin, D.E.; Tripp, R.A. Probenecid Inhibits Human Metapneumovirus (HMPV) Replication In Vitro and in BALB/c Mice. Viruses 2024, 16, 1087. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Hogan, R.J.; Martin, D.E.; Blahunka, K.; Sancilio, F.D.; Balyan, R.; Lovern, M.; Still, R.; Tripp, R.A. Probenecid inhibits SARS-CoV-2 replication in vivo and in vitro. Sci. Rep. 2021, 11, 18085. [Google Scholar] [CrossRef]
- Perwitasari, O.; Yan, X.; Johnson, S.; White, C.; Brooks, P.; Tompkins, S.M.; Tripp, R.A. Targeting organic anion transporter 3 with probenecid as a novel anti-influenza a virus strategy. Antimicrob. Agents Chemother. 2013, 57, 475–483. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Sharma, N.; Arina, P.; Defilippi, P. Molecular Insights into the MAPK Cascade during Viral Infection: Potential Crosstalk between HCQ and HCQ Analogues. Biomed. Res. Int. 2020, 2020, 8827752. [Google Scholar] [CrossRef]
- Sinha, S.; Cheng, K.; Schaffer, A.A.; Aldape, K.; Schiff, E.; Ruppin, E. In vitro and in vivo identification of clinically approved drugs that modify ACE2 expression. Mol. Syst. Biol. 2020, 16, e9628. [Google Scholar] [CrossRef]
- Chen, X.; Yuan, S.; Mi, L.; Long, Y.; He, H. Pannexin1: Insight into inflammatory conditions and its potential involvement in multiple organ dysfunction syndrome. Front. Immunol. 2023, 14, 1217366. [Google Scholar] [CrossRef]
- Kozak, W.; Kluger, M.J.; Soszynski, D.; Conn, C.A.; Rudolph, K.; Leon, L.R.; Zheng, H. IL-6 and IL-1 beta in fever. Studies using cytokine-deficient (knockout) mice. Ann. N. Y. Acad. Sci. 1998, 856, 33–47. [Google Scholar] [CrossRef]
- Silverman, M.N.; Pearce, B.D.; Biron, C.A.; Miller, A.H. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005, 18, 41–78. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Jin, M.; Chen, H.; Wu, Z.; Yuan, L.; Liang, S.; Wang, X.; Memon, F.U.; Eldemery, F.; Si, H.; et al. Inflammasome activation by viral infection: Mechanisms of activation and regulation. Front. Microbiol. 2023, 14, 1247377. [Google Scholar] [CrossRef]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tang, W.; Zeng, H.; Peng, Y.; Yu, X.; Yan, F.; Cao, S. Probenecid-Blocked Pannexin-1 Channel Protects Against Early Brain Injury via Inhibiting Neuronal AIM2 Inflammasome Activation After Subarachnoid Hemorrhage. Front. Neurol. 2022, 13, 854671. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Wei, B.; Billman, Z.P.; Nozaki, K.; Goodridge, H.S.; Miao, E.A. NLRP3, NLRP6, and NLRP12 are inflammasomes with distinct expression patterns. Front. Immunol. 2024, 15, 1418290. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Rosli, S.; Kirby, F.J.; Lawlor, K.E.; Rainczuk, K.; Drummond, G.R.; Mansell, A.; Tate, M.D. Repurposing drugs targeting the P2X7 receptor to limit hyperinflammation and disease during influenza virus infection. Br. J. Pharmacol. 2019, 176, 3834–3844. [Google Scholar] [CrossRef]
- Wonnenberg, B.; Tschernig, T.; Voss, M.; Bischoff, M.; Meier, C.; Schirmer, S.H.; Langer, F.; Bals, R.; Beisswenger, C. Probenecid reduces infection and inflammation in acute Pseudomonas aeruginosa pneumonia. Int. J. Med. Microbiol. 2014, 304, 725–729. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Broz, P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur. J. Immunol. 2020, 50, 170–177. [Google Scholar] [CrossRef]
- Deryabin, P.I.; Shatrova, A.N.; Borodkina, A.V. Targeting Multiple Homeostasis-Maintaining Systems by Ionophore Nigericin Is a Novel Approach for Senolysis. Int. J. Mol. Sci. 2022, 23, 14251. [Google Scholar] [CrossRef]
- Rozario, P.; Pinilla, M.; Gorse, L.; Vind, A.C.; Robinson, K.S.; Toh, G.A.; Firdaus, M.J.; Martinez, J.F.; Kerk, S.K.; Lin, Z.; et al. Mechanistic basis for potassium efflux-driven activation of the human NLRP1 inflammasome. Proc. Natl. Acad. Sci. USA 2024, 121, e2309579121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jin, T.; Jian, S.; Han, X.; Song, H.; Zhou, Q.; Yu, X. A dominant pathogenic MEFV mutation causes atypical pyrin-associated periodic syndromes. JCI Insight 2023, 8, 172975. [Google Scholar] [CrossRef]
- Oh, S.; Lee, J.; Oh, J.; Yu, G.; Ryu, H.; Kim, D.; Lee, S. Integrated NLRP3, AIM2, NLRC4, Pyrin inflammasome activation and assembly drive PANoptosis. Cell. Mol. Immunol. 2023, 20, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Chen, W.; Monick, M.M.; Carter, A.B.; Hunninghake, G.W. Activation of ERK2 by respiratory syncytial virus in A549 cells is linked to the production of interleukin 8. Exp. Lung Res. 2000, 26, 13–26. [Google Scholar] [CrossRef]
- Manthey, C.L.; Wang, S.W.; Kinney, S.D.; Yao, Z. SB202190, a selective inhibitor of p38 mitogen-activated protein kinase, is a powerful regulator of LPS-induced mRNAs in monocytes. J. Leukoc. Biol. 1998, 64, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Borgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef]
- Lazcano, P.; Schmidtke, M.W.; Onu, C.J.; Greenberg, M.L. Phosphatidic acid inhibits inositol synthesis by inducing nuclear translocation of kinase IP6K1 and repression of myo-inositol-3-P synthase. J. Biol. Chem. 2022, 298, 102363. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Hu, Z.; Xu, X.; Dai, X.; Liu, Z. Key signal transduction pathways and crosstalk in cancer: Biological and therapeutic opportunities. Transl. Oncol. 2022, 26, 101510. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Meng, F.; Lowell, C.A. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 1997, 185, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.; Olona, A.; Leishman, S.; MacDonald-Ramsahai, K.; Cockcroft, S.; Larrouy-Maumus, G.; Anand, P.K. NLRP3 Inflammasome Priming and Activation Are Regulated by a Phosphatidylinositol-Dependent Mechanism. Immunohorizons 2022, 6, 642–659. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6. [Google Scholar] [CrossRef]
- Solt, L.A.; May, M.J. The IkappaB kinase complex: Master regulator of NF-kappaB signaling. Immunol. Res. 2008, 42, 3–18. [Google Scholar] [CrossRef]
- Mankan, A.K.; Dau, T.; Jenne, D.; Hornung, V. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur. J. Immunol. 2012, 42, 710–715. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, M.E.; Dubyak, G.R.; Abbott, D.W. Post-translational control of NLRP3 inflammasome signaling. J. Biol. Chem. 2024, 300, 107386. [Google Scholar] [CrossRef] [PubMed]
- Mazzarda, F.; Chittams-Miles, A.E.; Pittaluga, J.; Sozer, E.B.; Vernier, P.T.; Muratori, C. Inflammasome Activation and IL-1beta Release Triggered by Nanosecond Pulsed Electric Fields in Murine Innate Immune Cells and Skin. J. Immunol. 2024, 212, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kitanaka, T.; Nakano, R.; Kitanaka, N.; Kimura, T.; Okabayashi, K.; Narita, T.; Sugiya, H. JNK activation is essential for activation of MEK/ERK signaling in IL-1beta-induced COX-2 expression in synovial fibroblasts. Sci. Rep. 2017, 7, 39914. [Google Scholar] [CrossRef]
- Onodi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Toth, V.E.; Koncsos, G.; Novak, J.; Brenner, G.B.; Makkos, A.; Baranyai, T.; et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 2021, 117, 2639–2651. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, L.P.; Martin, D.E.; Murray, J.; Sancilio, F.; Tripp, R.A. Probenecid Inhibits NLRP3 Inflammasome Activity and Mitogen-Activated Protein Kinases (MAPKs). Biomolecules 2025, 15, 511. https://doi.org/10.3390/biom15040511
Jones LP, Martin DE, Murray J, Sancilio F, Tripp RA. Probenecid Inhibits NLRP3 Inflammasome Activity and Mitogen-Activated Protein Kinases (MAPKs). Biomolecules. 2025; 15(4):511. https://doi.org/10.3390/biom15040511
Chicago/Turabian StyleJones, Les P., David E. Martin, Jackelyn Murray, Fred Sancilio, and Ralph A. Tripp. 2025. "Probenecid Inhibits NLRP3 Inflammasome Activity and Mitogen-Activated Protein Kinases (MAPKs)" Biomolecules 15, no. 4: 511. https://doi.org/10.3390/biom15040511
APA StyleJones, L. P., Martin, D. E., Murray, J., Sancilio, F., & Tripp, R. A. (2025). Probenecid Inhibits NLRP3 Inflammasome Activity and Mitogen-Activated Protein Kinases (MAPKs). Biomolecules, 15(4), 511. https://doi.org/10.3390/biom15040511