


Tissue Resident and Infiltrating Immune Cells: Their Influence on the Demise of Beta Cells in Type 1 Diabetes

Abstract
1. Introduction
2. The Immune Landscape of the Exocrine Pancreas
3. The Role of Intra-Islet Macrophages in the Context of T1D
4. Intrinsic Beta Cell Mechanisms and Visibility
5. The Insulitis Trajectory and Immune Networks
6. Components of Pancreatic Insulitis in T1D
6.1. Neutrophils
6.2. Macrophages and Dendritic Cells (DCs)
6.3. Natural Killer (NK) Cells
6.4. B Lymphocytes
6.5. T Lymphocytes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CODEX | CO-Detection by indEXing |
| DC | Dendritic cell |
| HLA | Human leukocyte antigen |
| IAA | Insulin autoantibody |
| IMC | Imaging mass cytometry |
| MHC | Major Histocompatibility Complex |
| NET | Neutrophil extracellular trap |
| NOD | Non-obese diabetic mouse |
| pDCs | Plasmacytoid dendritic cells |
| PLN | Pancreatic lymph node |
| PPI | Preproinsulin |
| T1D | Type 1 diabetes |
| TCR | T-cell receptor |
| TLS | Tertiary lymphoid structure |
| TRM | Resident-memory T-cells |
References
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.R.; Kronenberg, D.; Zhao, M.; Huang, G.C.; Eichmann, M.; Bulek, A.; Wooldridge, L.; Cole, D.K.; Sewell, A.K.; Peakman, M.; et al. Human beta-cell killing by autoreactive preproinsulin-specific CD8 T cells is predominantly granule-mediated with the potency dependent upon T-cell receptor avidity. Diabetes 2013, 62, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Gepts, W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965, 14, 619–633. [Google Scholar] [CrossRef]
- Foster, T.P.; Bruggeman, B.; Campbell-Thompson, M.; Atkinson, M.A.; Haller, M.J.; Schatz, D.A. Exocrine Pancreas Dysfunction in Type 1 Diabetes. Endocr. Pract. 2020, 26, 1505–1513. [Google Scholar] [CrossRef]
- Foulis, A.K.; Liddle, C.N.; Farquharson, M.A.; Richmond, J.A.; Weir, R.S. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: A 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia 1986, 29, 267–274. [Google Scholar] [CrossRef]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.; Rodriguez-Calvo, T.; Battaglia, M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr. Diab Rep. 2015, 15, 79. [Google Scholar] [CrossRef]
- Radenkovic, M.; Uvebrant, K.; Skog, O.; Sarmiento, L.; Avartsson, J.; Storm, P.; Vickman, P.; Bertilsson, P.A.; Fex, M.; Korgsgren, O.; et al. Characterization of resident lymphocytes in human pancreatic islets. Clin. Exp. Immunol. 2017, 187, 418–427. [Google Scholar] [CrossRef]
- Bender, C.; Rodriguez-Calvo, T.; Amirian, N.; Coppieters, K.T.; von Herrath, M.G. The healthy exocrine pancreas contains preproinsulin-specific CD8 T cells that attack islets in type 1 diabetes. Sci. Adv. 2020, 6, eabc5586. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Carpenter, D.J.; Chait, M.; Dogra, P.; Gartrell-Corrado, R.D.; Chen, A.X.; Campbell, S.; Liu, W.; Saraf, P.; Snyder, M.E.; et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep. 2019, 29, 3916–3932.e3915. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.R.; Jacques-Silva, C.; Qadir, M.M.F.; Umland, O.; Pereira, E.; Qureshi, F.; Tamayo, A.; Dominguez-Bendala, J.; Rodriguez-Diaz, R.; Almaca, J.; et al. Secretory Functions of Macrophages in the Human Pancreatic Islet Are Regulated by Endogenous Purinergic Signaling. Diabetes 2020, 69, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Calderon, B.; Carrero, J.A.; Ferris, S.T.; Sojka, D.K.; Moore, L.; Epelman, S.; Murphy, K.M.; Yokoyama, W.M.; Randolph, G.J.; Unanue, E.R. The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med. 2015, 212, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; Coomans de Brachène, A.; Paula, F.M.M.; Op de Beeck, A.; Castela, A.; et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 2018, 36, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Dong, C.; Lin, L.; Du, J. Characteristics and sources of tissue-resident memory T cells in psoriasis relapse. Curr. Res. Immunol. 2023, 4, 100067. [Google Scholar] [CrossRef]
- Muñoz García, A.; Juksar, J.; Groen, N.; Zaldumbide, A.; de Koning, E.; Carlotti, F. Single-cell transcriptomics reveals a role for pancreatic duct cells as potential mediators of inflammation in diabetes mellitus. Front. Immunol. 2024, 15, 1381319. [Google Scholar] [CrossRef]
- Fasolino, M.; Schwartz, G.W.; Patil, A.R.; Mongia, A.; Golson, M.L.; Wang, Y.J.; Morgan, A.; Liu, C.; Schug, J.; Liu, J.; et al. Single-cell multi-omics analysis of human pancreatic islets reveals novel cellular states in type 1 diabetes. Nat. Metab. 2022, 4, 284–299. [Google Scholar] [CrossRef]
- Vomund, A.N.; Zinselmeyer, B.H.; Hughes, J.; Calderon, B.; Valderrama, C.; Ferris, S.T.; Wan, X.; Kanekura, K.; Carrero, J.A.; Urano, F.; et al. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5496–E5502. [Google Scholar] [CrossRef]
- Zirpel, H.; Roep, B.O. Islet-Resident Dendritic Cells and Macrophages in Type 1 Diabetes: In Search of Bigfoot’s Print. Front. Endocrinol. 2021, 12, 666795. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.T.; Zakharov, P.N.; Wan, X.; Calderon, B.; Artyomov, M.N.; Unanue, E.R.; Carrero, J.A. The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J. Exp. Med. 2017, 214, 2369–2385. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; McCarthy, D.P.; Ferris, S.T.; Wan, X.; Hu, H.; Zinselmeyer, B.H.; Vomund, A.N.; Unanue, E.R. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc. Natl. Acad. Sci. USA 2017, 114, E10418–E10427. [Google Scholar] [CrossRef] [PubMed]
- Arnush, M.; Heitmeier, M.R.; Scarim, A.L.; Marino, M.H.; Manning, P.T.; Corbett, J.A. IL-1 produced and released endogenously within human islets inhibits beta cell function. J. Clin. Investig. 1998, 102, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Thornley, T.B.; Agarwal, K.A.; Kyriazis, P.; Ma, L.; Chipashvili, V.; Aker, J.E.; Korniotis, S.; Csizmadia, E.; Strom, T.B.; Koulmanda, M. Contrasting Roles of Islet Resident Immunoregulatory Macrophages and Dendritic Cells in Experimental Autoimmune Type 1 Diabetes. PLoS ONE 2016, 11, e0150792. [Google Scholar] [CrossRef]
- Srivastava, N.; Hu, H.; Peterson, O.J.; Vomund, A.N.; Stremska, M.; Zaman, M.; Giri, S.; Li, T.; Lichti, C.F.; Zakharov, P.N.; et al. CXCL16-dependent scavenging of oxidized lipids by islet macrophages promotes differentiation of pathogenic CD8(+) T cells in diabetic autoimmunity. Immunity 2024, 57, 1629–1647.e1628. [Google Scholar] [CrossRef]
- Avrahami, D.; Klochendler, A.; Dor, Y.; Glaser, B. Beta cell heterogeneity: An evolving concept. Diabetologia 2017, 60, 1363–1369. [Google Scholar] [CrossRef]
- Miranda, M.A.; Macias-Velasco, J.F.; Lawson, H.A. Pancreatic β-cell heterogeneity in health and diabetes: Classes, sources, and subtypes. Am. J. Physiol.-Endocrinol. Metab. 2021, 320, E716–E731. [Google Scholar] [CrossRef]
- Benninger, R.K.P.; Dorrell, C.; Hodson, D.J.; Rutter, G.A. The Impact of Pancreatic Beta Cell Heterogeneity on Type 1 Diabetes Pathogenesis. Curr. Diab Rep. 2018, 18, 112. [Google Scholar] [CrossRef]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef]
- Bottazzo, G.F.; Dean, B.M.; McNally, J.M.; MacKay, E.H.; Swift, P.G.; Gamble, D.R. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N. Engl. J. Med. 1985, 313, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Szymczak, F.; Mallone, R. Why does the immune system destroy pancreatic β-cells but not α-cells in type 1 diabetes? Nat. Rev. Endocrinol. 2023, 19, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Op de Beeck, A.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus—why the beta cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Deng, S.; Perdigoto, A.L.; Ponath, G.; Kursawe, R.; Lawlor, N.; Sumida, T.; Levine-Ritterman, M.; Stitzel, M.L.; Pitt, D.; et al. Tet2 Controls the Responses of β cells to Inflammation in Autoimmune Diabetes. Nat. Commun. 2021, 12, 5074. [Google Scholar] [CrossRef]
- Frigerio, S.; Junt, T.; Lu, B.; Gerard, C.; Zumsteg, U.; Holländer, G.A.; Piali, L. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat. Med. 2002, 8, 1414–1420. [Google Scholar] [CrossRef]
- Schulthess, F.T.; Paroni, F.; Sauter, N.S.; Shu, L.; Ribaux, P.; Haataja, L.; Strieter, R.M.; Oberholzer, J.; King, C.C.; Maedler, K. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009, 9, 125–139. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- Chen, M.C.; Proost, P.; Gysemans, C.; Mathieu, C.; Eizirik, D.L. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia 2001, 44, 325–332. [Google Scholar] [CrossRef]
- Piemonti, L.; Leone, B.E.; Nano, R.; Saccani, A.; Monti, P.; Maffi, P.; Bianchi, G.; Sica, A.; Peri, G.; Melzi, R.; et al. Human pancreatic islets produce and secrete MCP-1/CCL2: Relevance in human islet transplantation. Diabetes 2002, 51, 55–65. [Google Scholar] [CrossRef]
- Martin, A.P.; Rankin, S.; Pitchford, S.; Charo, I.F.; Furtado, G.C.; Lira, S.A. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes 2008, 57, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, M.A.; Rathinam, C.; Flavell, R.A. Pancreatic islet expression of chemokine CCL2 suppresses autoimmune diabetes via tolerogenic CD11c+ CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Scherm, M.G.; Wyatt, R.C.; Serr, I.; Anz, D.; Richardson, S.J.; Daniel, C. Beta cell and immune cell interactions in autoimmune type 1 diabetes: How they meet and talk to each other. Mol. Metab. 2022, 64, 101565. [Google Scholar] [CrossRef] [PubMed]
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634. [Google Scholar] [CrossRef]
- Huber, M.K.; Drotar, D.M.; Hiller, H.; Beery, M.L.; Joseph, P.; Kusmartseva, I.; Speier, S.; Atkinson, M.A.; Mathews, C.E.; Phelps, E.A. Observing Islet Function and Islet-Immune Cell Interactions in Live Pancreatic Tissue Slices. J. Vis. Exp. 2021. [Google Scholar] [CrossRef]
- Damond, N.; Engler, S.; Zanotelli, V.R.T.; Schapiro, D.; Wasserfall, C.H.; Kusmartseva, I.; Nick, H.S.; Thorel, F.; Herrera, P.L.; Atkinson, M.A.; et al. A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab. 2019, 29, 755–768.e755. [Google Scholar] [CrossRef]
- Wang, Y.J.; Traum, D.; Schug, J.; Gao, L.; Liu, C.; Consortium, H.; Atkinson, M.A.; Powers, A.C.; Feldman, M.D.; Naji, A.; et al. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 2019, 29, 769–783.e764. [Google Scholar] [CrossRef]
- Barlow, G.L.; Schürch, C.M.; Bhate, S.S.; Phillips, D.; Young, A.; Dong, S.; Martinez, H.A.; Kaber, G.; Nagy, N.; Ramachandran, S.; et al. The Extra-Islet Pancreas Supports Autoimmunity in Human Type 1 Diabetes. eLife, 2024; preprint. [Google Scholar] [CrossRef]
- Mariño, E.; Tan, B.; Binge, L.; Mackay, C.R.; Grey, S.T. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes 2012, 61, 2893–2905. [Google Scholar] [CrossRef]
- Serreze, D.V.; Fleming, S.A.; Chapman, H.D.; Richard, S.D.; Leiter, E.H.; Tisch, R.M. B Lymphocytes Are Critical Antigen-Presenting Cells for the Initiation of T Cell-Mediated Autoimmune Diabetes in Nonobese Diabetic Mice. J. Immunol. 1998, 161, 3912–3918. [Google Scholar] [CrossRef]
- Leete, P.; Willcox, A.; Krogvold, L.; Dahl-Jørgensen, K.; Foulis, A.K.; Richardson, S.J.; Morgan, N.G. Differential Insulitic Profiles Determine the Extent of β-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 2016, 65, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, F.; Lo Buono, N.; Stabilini, A.; Nigi, L.; Dufort, M.J.; Geyer, S.; Rancoita, P.M.; Cugnata, F.; Mandelli, A.; Valle, A.; et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight 2018, 3, e122146. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, Y.; Martens, P.J.; Sassi, G.; Viaene, M.; Wasserfall, C.H.; Mathieu, C.; Gysemans, C. Footprint of pancreas infiltrating and circulating immune cells throughout type 1 diabetes development. Front. Endocrinol. 2023, 14, 1275316. [Google Scholar] [CrossRef] [PubMed]
- Dufort, M.J.; Greenbaum, C.J.; Speake, C.; Linsley, P.S. Cell type-specific immune phenotypes predict loss of insulin secretion in new-onset type 1 diabetes. JCI Insight 2019, 4, e125556. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zakharov, P.N.; Peterson, O.J.; Unanue, E.R. Cytocidal macrophages in symbiosis with CD4 and CD8 T cells cause acute diabetes following checkpoint blockade of PD-1 in NOD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 31319–31330. [Google Scholar] [CrossRef]
- Poirot, L.; Benoist, C.; Mathis, D. Natural killer cells distinguish innocuous and destructive forms of pancreatic islet autoimmunity. Proc. Natl. Acad. Sci. USA 2004, 101, 8102–8107. [Google Scholar] [CrossRef]
- Saksida, T.; Paunović, V.; Koprivica, I.; Mićanović, D.; Jevtić, B.; Jonić, N.; Stojanović, I.; Pejnović, N. Development of Type 1 Diabetes in Mice Is Associated with a Decrease in IL-2-Producing ILC3 and FoxP3(+) Treg in the Small Intestine. Molecules 2023, 28, 3366. [Google Scholar] [CrossRef]
- Miani, M.; Le Naour, J.; Waeckel-Enée, E.; Verma, S.c.; Straube, M.; Emond, P.; Ryffel, B.; van Endert, P.; Sokol, H.; Diana, J. Gut Microbiota-Stimulated Innate Lymphoid Cells Support β-Defensin 14 Expression in Pancreatic Endocrine Cells, Preventing Autoimmune Diabetes. Cell Metab. 2018, 28, 557–572.e556. [Google Scholar] [CrossRef]
- Shi, S.; Ye, L.; Jin, K.; Xiao, Z.; Yu, X.; Wu, W. Innate Lymphoid Cells: Emerging Players in Pancreatic Disease. Int. J. Mol. Sci. 2022, 23, 3748. [Google Scholar] [CrossRef]
- Huang, J.; Peng, J.; Pearson, J.A.; Efthimiou, G.; Hu, Y.; Tai, N.; Xing, Y.; Zhang, L.; Gu, J.; Jiang, J.; et al. Toll-like receptor 7 deficiency suppresses type 1 diabetes development by modulating B-cell differentiation and function. Cell Mol. Immunol. 2021, 18, 328–338. [Google Scholar] [CrossRef]
- Boldison, J.; Hopkinson, J.R.; Davies, J.; Pearson, J.A.; Leete, P.; Richardson, S.; Morgan, N.G.; Wong, F.S. Gene expression profiling in NOD mice reveals that B cells are highly educated by the pancreatic environment during autoimmune diabetes. Diabetologia 2023, 66, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Korpos, É.; Kadri, N.; Loismann, S.; Findeisen, C.R.; Arfuso, F.; Burke, G.W.; Richardson, S.J.; Morgan, N.G.; Bogdani, M.; Pugliese, A.; et al. Identification and characterisation of tertiary lymphoid organs in human type 1 diabetes. Diabetologia 2021, 64, 1626–1641. [Google Scholar] [CrossRef] [PubMed]
- Smeets, S.; Staels, W.; Stangé, G.; Gillard, P.; De Leu, N.; in’t Veld, P. Insulitis and lymphoid structures in the islets of Langerhans of a 66-year-old patient with long-standing type 1 diabetes. Virchows Arch. 2021, 478, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, P.N.; Hu, H.; Wan, X.; Unanue, E.R. Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J. Exp. Med. 2020, 217, e20192362. [Google Scholar] [CrossRef]
- Obeagu, E.I.; Obeagu, G.U. Type 1 diabetes mellitus: Roles of neutrophils in the pathogenesis. Medicine 2023, 102, e36245. [Google Scholar] [CrossRef]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in chronic inflammatory diseases. Cell Mol. Immunol. 2022, 19, 177–191. [Google Scholar] [CrossRef]
- Petrelli, A.; Popp, S.K.; Fukuda, R.; Parish, C.R.; Bosi, E.; Simeonovic, C.J. The Contribution of Neutrophils and NETs to the Development of Type 1 Diabetes. Front. Immunol. 2022, 13, 930553. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, Y.; Xu, A.; Zhou, Z. Neutrophils in type 1 diabetes. J. Diabetes Investig. 2016, 7, 652–663. [Google Scholar] [CrossRef]
- Salami, F.; Lee, H.S.; Freyhult, E.; Elding Larsson, H.; Lernmark, Å.; Törn, C. Reduction in White Blood Cell, Neutrophil, and Red Blood Cell Counts Related to Sex, HLA, and Islet Autoantibodies in Swedish TEDDY Children at Increased Risk for Type 1 Diabetes. Diabetes 2018, 67, 2329–2336. [Google Scholar] [CrossRef]
- Klocperk, A.; Petruzelkova, L.; Pavlikova, M.; Rataj, M.; Kayserova, J.; Pruhova, S.; Kolouskova, S.; Sklenarova, J.; Parackova, Z.; Sediva, A.; et al. Changes in innate and adaptive immunity over the first year after the onset of type 1 diabetes. Acta Diabetol. 2020, 57, 297–307. [Google Scholar] [CrossRef]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Korsgren, S.; Molin, Y.; Salmela, K.; Lundgren, T.; Melhus, A.; Korsgren, O. On the etiology of type 1 diabetes: A new animal model signifying a decisive role for bacteria eliciting an adverse innate immunity response. Am. J. Pathol. 2012, 181, 1735–1748. [Google Scholar] [CrossRef] [PubMed]
- Popp, S.K.; Vecchio, F.; Brown, D.J.; Fukuda, R.; Suzuki, Y.; Takeda, Y.; Wakamatsu, R.; Sarma, M.A.; Garrett, J.; Giovenzana, A.; et al. Circulating platelet-neutrophil aggregates characterize the development of type 1 diabetes in humans and NOD mice. JCI Insight 2022, 7, e153993. [Google Scholar] [CrossRef]
- Nikolic, T.; Geutskens, S.B.; van Rooijen, N.; Drexhage, H.A.; Leenen, P.J. Dendritic cells and macrophages are essential for the retention of lymphocytes in (peri)-insulitis of the nonobese diabetic mouse: A phagocyte depletion study. Lab. Investig. 2005, 85, 487–501. [Google Scholar] [CrossRef]
- Ferris, S.T.; Carrero, J.A.; Mohan, J.F.; Calderon, B.; Murphy, K.M.; Unanue, E.R. A minor subset of Batf3-dependent antigen-presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity 2014, 41, 657–669. [Google Scholar] [CrossRef]
- Calderon, B.; Suri, A.; Unanue, E.R. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: Studies from an acute model. Am. J. Pathol. 2006, 169, 2137–2147. [Google Scholar] [CrossRef]
- Carrero, J.A.; Ferris, S.T.; Unanue, E.R. Macrophages and dendritic cells in islets of Langerhans in diabetic autoimmunity: A lesson on cell interactions in a mini-organ. Curr. Opin. Immunol. 2016, 43, 54–59. [Google Scholar] [CrossRef]
- Rodriguez-Fernandez, S.; Murillo, M.; Villalba, A.; Perna-Barrull, D.; Cano-Sarabia, M.; Gomez-Muñoz, L.; Aguilera, E.; Maspoch, D.; Vazquez, F.; Bel, J.; et al. Impaired Phagocytosis in Dendritic Cells From Pediatric Patients With Type 1 Diabetes Does Not Hamper Their Tolerogenic Potential. Front. Immunol. 2019, 10, 2811. [Google Scholar] [CrossRef]
- Gomez-Muñoz, L.; Perna-Barrull, D.; Caroz-Armayones, J.M.; Murillo, M.; Rodriguez-Fernandez, S.; Valls, A.; Vazquez, F.; Perez, J.; Corripio, R.; Castaño, L.; et al. Candidate Biomarkers for the Prediction and Monitoring of Partial Remission in Pediatric Type 1 Diabetes. Front. Immunol. 2022, 13, 825426. [Google Scholar] [CrossRef]
- Uno, S.; Imagawa, A.; Okita, K.; Sayama, K.; Moriwaki, M.; Iwahashi, H.; Yamagata, K.; Tamura, S.; Matsuzawa, Y.; Hanafusa, T.; et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia 2007, 50, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, S.; Imagawa, A.; Tauriainen, S.; Iino, M.; Oikarinen, M.; Abiru, H.; Tamaki, K.; Seino, H.; Nishi, K.; Takase, I.; et al. Expression of Toll-like Receptors in the Pancreas of Recent-onset Fulminant Type 1 Diabetes. Endocr. J. 2010, 57, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Muñoz, L.; Perna-Barrull, D.; Villalba, A.; Rodriguez-Fernandez, S.; Ampudia, R.-M.; Teniente-Serra, A.; Vazquez, F.; Murillo, M.; Perez, J.; Corripio, R.; et al. NK Cell Subsets Changes in Partial Remission and Early Stages of Pediatric Type 1 Diabetes. Front. Immunol. 2021, 11, 153993. [Google Scholar] [CrossRef]
- Sabetkam, S.; Kalarestaghi, H.; Mazloumi, Z.; Dizaji Asl, K.; Norouzi, N.; Rafat, A. The dysfunction of natural killer cells is essential for the development of type 1 diabetes. Pathol.—Res. Pract. 2023, 247, 154556. [Google Scholar] [CrossRef]
- Gardner, G.; Fraker, C.A. Natural Killer Cells as Key Mediators in Type I Diabetes Immunopathology. Front. Immunol. 2021, 12, 722979. [Google Scholar] [CrossRef]
- Torabi, F.; Vadakekolathu, J.; Wyatt, R.; Leete, P.; Tombs, M.A.; Richardson, C.C.; Boocock, D.J.; Turner, M.D.; Morgan, N.G.; Richardson, S.J.; et al. Differential expression of genes controlling lymphocyte differentiation and migration in two distinct endotypes of type 1 diabetes. Diabet. Med. 2023, 40, e15155. [Google Scholar] [CrossRef]
- Wang, Y.-n.; Li, R.; Huang, Y.; Chen, H.; Nie, H.; Liu, L.; Zou, X.; Zhong, J.; Zheng, B.; Gong, Q. The role of B cells in the pathogenesis of type 1 diabetes. Front. Immunol. 2024, 15, 1450366. [Google Scholar] [CrossRef]
- Smith, M.J.; Boldison, J.; Wong, F.S. The Role of B Lymphocytes in Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2024. [Google Scholar] [CrossRef]
- Stensland, Z.C.; Magera, C.A.; Broncucia, H.; Gomez, B.D.; Rios-Guzman, N.M.; Wells, K.L.; Nicholas, C.A.; Rihanek, M.; Hunter, M.J.; Toole, K.P.; et al. Identification of an anergic BND cell-derived activated B cell population (BND2) in young-onset type 1 diabetes patients. J. Exp. Med. 2023, 220, e20221604. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougouin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541.e525. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Aitken, R.J.; Wilson, I.V.; Mortimer, G.L.M.; Long, A.E.; Williams, A.J.K.; Group, B.O.X.S.; Gillespie, K.M. The measurement of autoantibodies to insulin informs diagnosis of diabetes in a childhood population negative for other autoantibodies. Diabet. Med. 2022, 39, e14979. [Google Scholar] [CrossRef] [PubMed]
- Apaolaza, P.S.; Balcacean, D.; Zapardiel-Gonzalo, J.; Rodriguez-Calvo, T. The extent and magnitude of islet T cell infiltration as powerful tools to define the progression to type 1 diabetes. Diabetologia 2023, 66, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.H.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Michels, A.W.; Landry, L.G.; McDaniel, K.A.; Yu, L.; Campbell-Thompson, M.; Kwok, W.W.; Jones, K.L.; Gottlieb, P.A.; Kappler, J.W.; Tang, Q.; et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes 2017, 66, 722–734. [Google Scholar] [CrossRef]
- Anderson, A.M.; Landry, L.G.; Alkanani, A.A.; Pyle, L.; Powers, A.C.; Atkinson, M.A.; Mathews, C.E.; Roep, B.O.; Michels, A.W.; Nakayama, M. Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2021, 118, e2107208118. [Google Scholar] [CrossRef]
- Kuric, E.; Seiron, P.; Krogvold, L.; Edwin, B.; Buanes, T.; Hanssen, K.F.; Skog, O.; Dahl-Jørgensen, K.; Korsgren, O. Demonstration of Tissue Resident Memory CD8 T Cells in Insulitic Lesions in Adult Patients with Recent-Onset Type 1 Diabetes. Am. J. Pathol. 2017, 187, 581–588. [Google Scholar] [CrossRef]
- Huber, M.K.; Widener, A.E.; Cuaycal, A.E.; Smurlick, D.; Butterworth, E.A.; Lenchik, N.I.; Chen, J.; Beery, M.; Hiller, H.; Verney, E.; et al. Beta cell dysfunction occurs independently of insulitis in type 1 diabetes pathogenesis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chiou, J.; Geusz, R.J.; Okino, M.L.; Han, J.Y.; Miller, M.; Melton, R.; Beebe, E.; Benaglio, P.; Huang, S.; Korgaonkar, K.; et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 2021, 594, 398–402. [Google Scholar] [CrossRef]
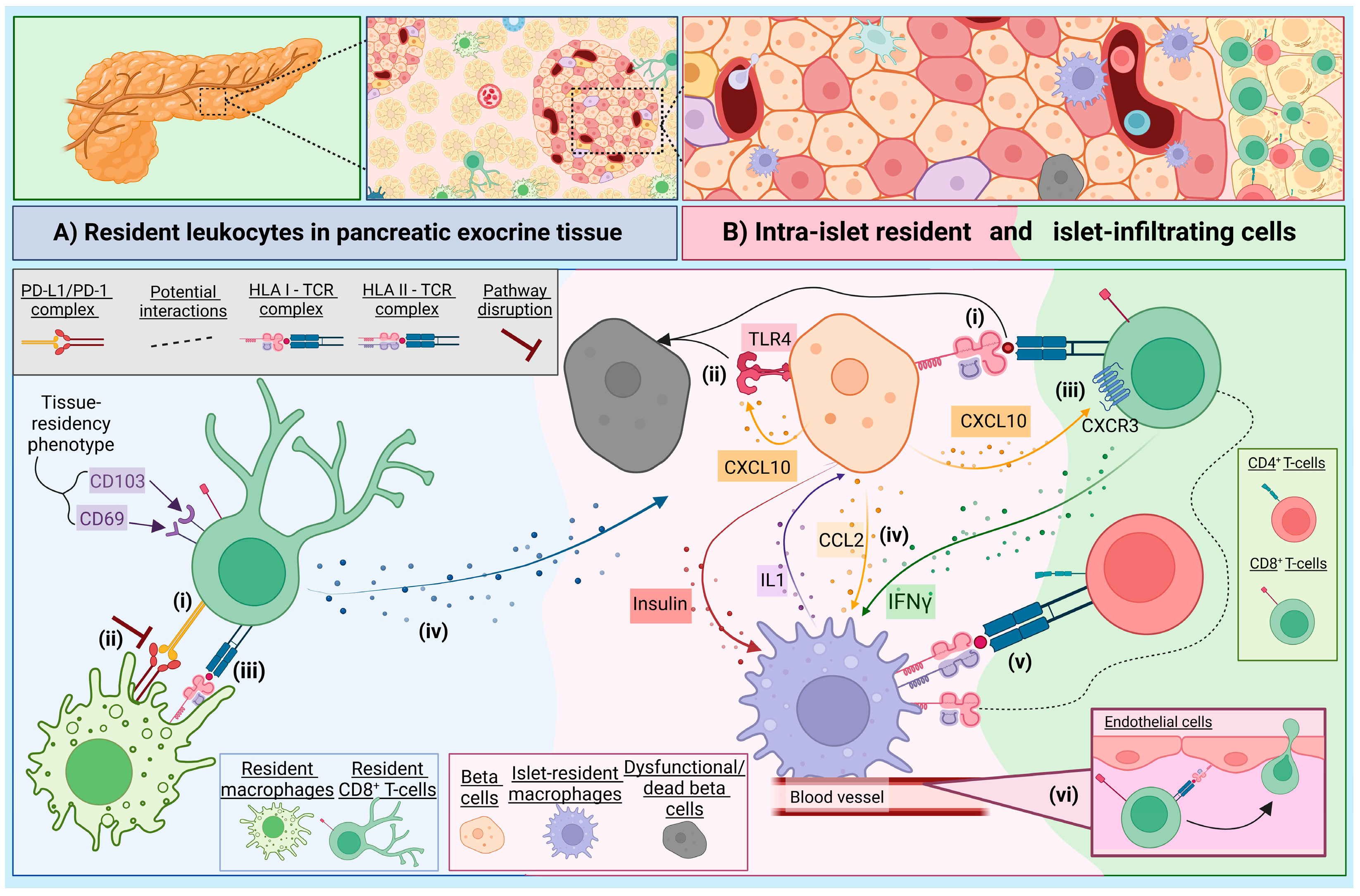
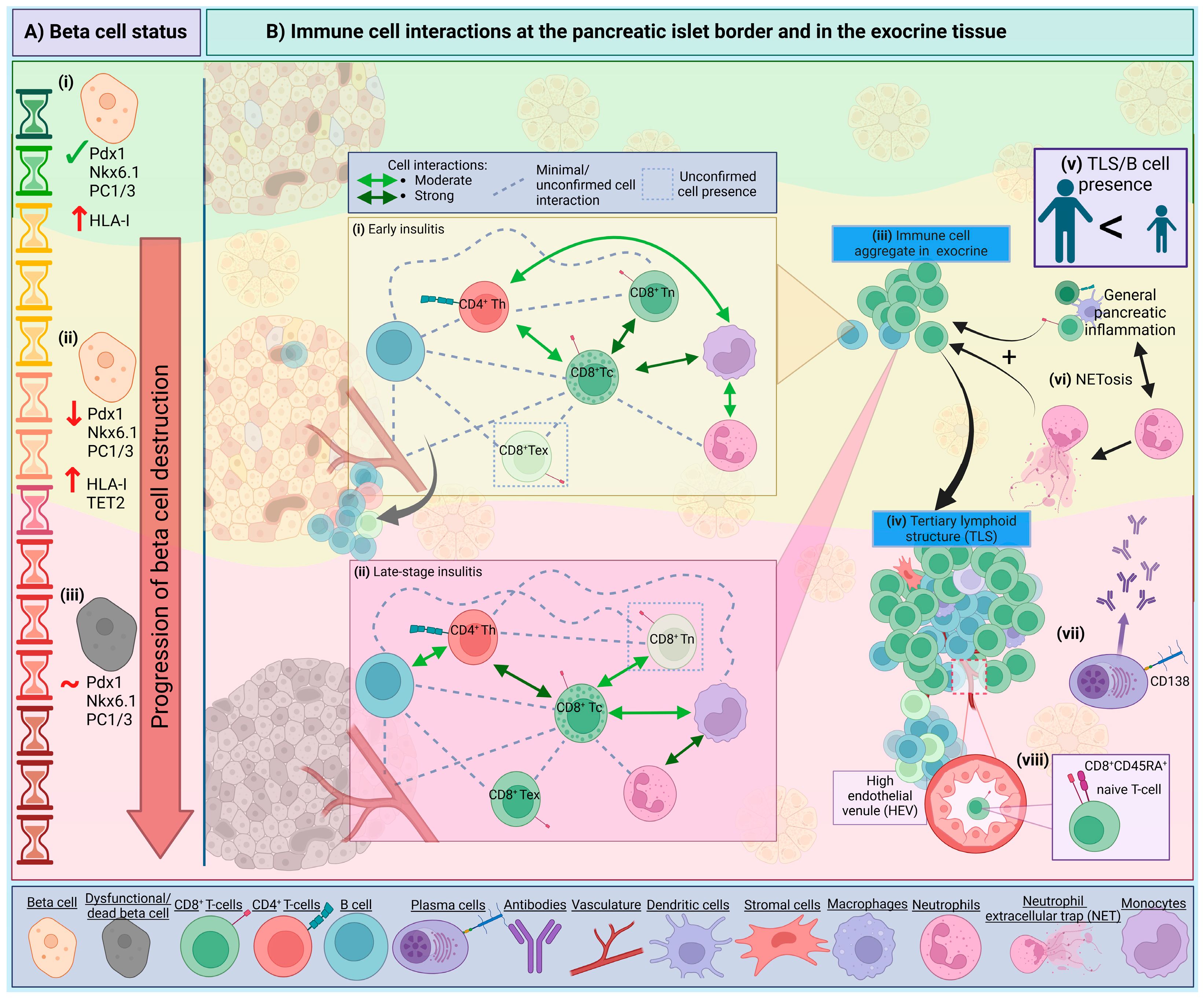

{kind=link}
{kind=link}
| Cell Type | Human or Mouse | Study Highlight | Suggested Role in Beta Cell Destruction/Targeting | References |
|---|---|---|---|---|
| Neutrophils | NOD mouse | Peak accumulation of neutrophils is at 8-weeks old, then a progressive decline is observed | Interact with CD8+ T-cells and monocytes at different disease stages resulting in enhanced T-cell responses and expansion | [53] |
| Human | Reduction in peripheral, circulating neutrophils correlates with the infiltration of neutrophils into the pancreatic tissues, particularly in the first year of diagnosis | [54] | ||
| Macrophages | NOD mouse | In response to IFN-γ from activated T-cells, macrophages become highly activated and attack beta cells via IL-1 and nitric oxide release | Release cytokines to effect beta cell health and interact with CD4+ T-cells via HLA-II TCR complexes | [55] |
| Human | Detected in early infiltrates, associated with both ICIs and IDIs, and not correlating with inflammatory clusters | [47,48] | ||
| Dendritic cells (DCs) | NOD mouse | Influx of pancreatic DCs is preceded by and increase in DC frequency in the pancreatic lymph node (PLN) | Presentation of islet antigens to result in further immune cell attack of beta cells, typically by CD8+ T-cells | [53] |
| Human | Relatively understudied in the human pancreas | [21] | ||
| Natural killer (NK) cells | Mouse | NK cell depletion was shown to significantly decrease the incidence of T1D | Due to the small sizes of the cell populations currently, the effect and interactions of other cells with NK cells is relatively unknown | [56] |
| Human | Increased presence in NK cells in pancreatic tissue donors with a diagnosis of under 2 years, although the populations are very small | [47] | ||
| Innate lymphoid cells (ILCs) | Mouse | Reduction in IL2-producing gut ILC3s and Tregs is associated with insulitis presence in the pancreatic tissues; IL-22 production induces islet-associated β defensin mBD14 which can protect NOD mice from autoimmune diabetes. | Modulate cytokine production in the gut to affect beta cell survival in the pancreas | [57,58] |
| Human | Data from T1D pancreatic tissues is limited | [59] | ||
| B lymphocytes | TLR7-knockout, and NOD mice | TLR7 knockout mice have a delayed onset and reduced incidence of type 1 diabetes development, with reduced B-cell function; TLR7 gene and protein expression is enriched in B-cell subsets | Present antigens to attract T-cells to increase beta cell destruction | [60,61] |
| Human | TLSs are associated with young age of onset; compartmentalised B- and T-cell zones were only present in a limited number of recent onset donors but also observed in a long-standing donor in an individual case-study | [62,63] | ||
| T lymphocytes | NOD mouse | CD4+ effector and regulatory T-cells present as early as 4-weeks of age, while CD8+ T-cells respond to local environment via functional-related changes | Direct cell-surface receptor interaction with beta cells to elicit beta cell destruction | [64] |
| Human | Substates of T-cells and the variable composition of CD8+ T-cells between islets in the same individual have been identified, in response to islet microenvironment | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, S.L.; Leete, P.; Boldison, J. Tissue Resident and Infiltrating Immune Cells: Their Influence on the Demise of Beta Cells in Type 1 Diabetes. Biomolecules 2025, 15, 441. https://doi.org/10.3390/biom15030441
Walker SL, Leete P, Boldison J. Tissue Resident and Infiltrating Immune Cells: Their Influence on the Demise of Beta Cells in Type 1 Diabetes. Biomolecules. 2025; 15(3):441. https://doi.org/10.3390/biom15030441
Chicago/Turabian StyleWalker, Sophie L., Pia Leete, and Joanne Boldison. 2025. "Tissue Resident and Infiltrating Immune Cells: Their Influence on the Demise of Beta Cells in Type 1 Diabetes" Biomolecules 15, no. 3: 441. https://doi.org/10.3390/biom15030441
APA StyleWalker, S. L., Leete, P., & Boldison, J. (2025). Tissue Resident and Infiltrating Immune Cells: Their Influence on the Demise of Beta Cells in Type 1 Diabetes. Biomolecules, 15(3), 441. https://doi.org/10.3390/biom15030441