


Genetic Analysis and Predictive Modeling of COVID-19 Severity in a Hospital-Based Patient Cohort


 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
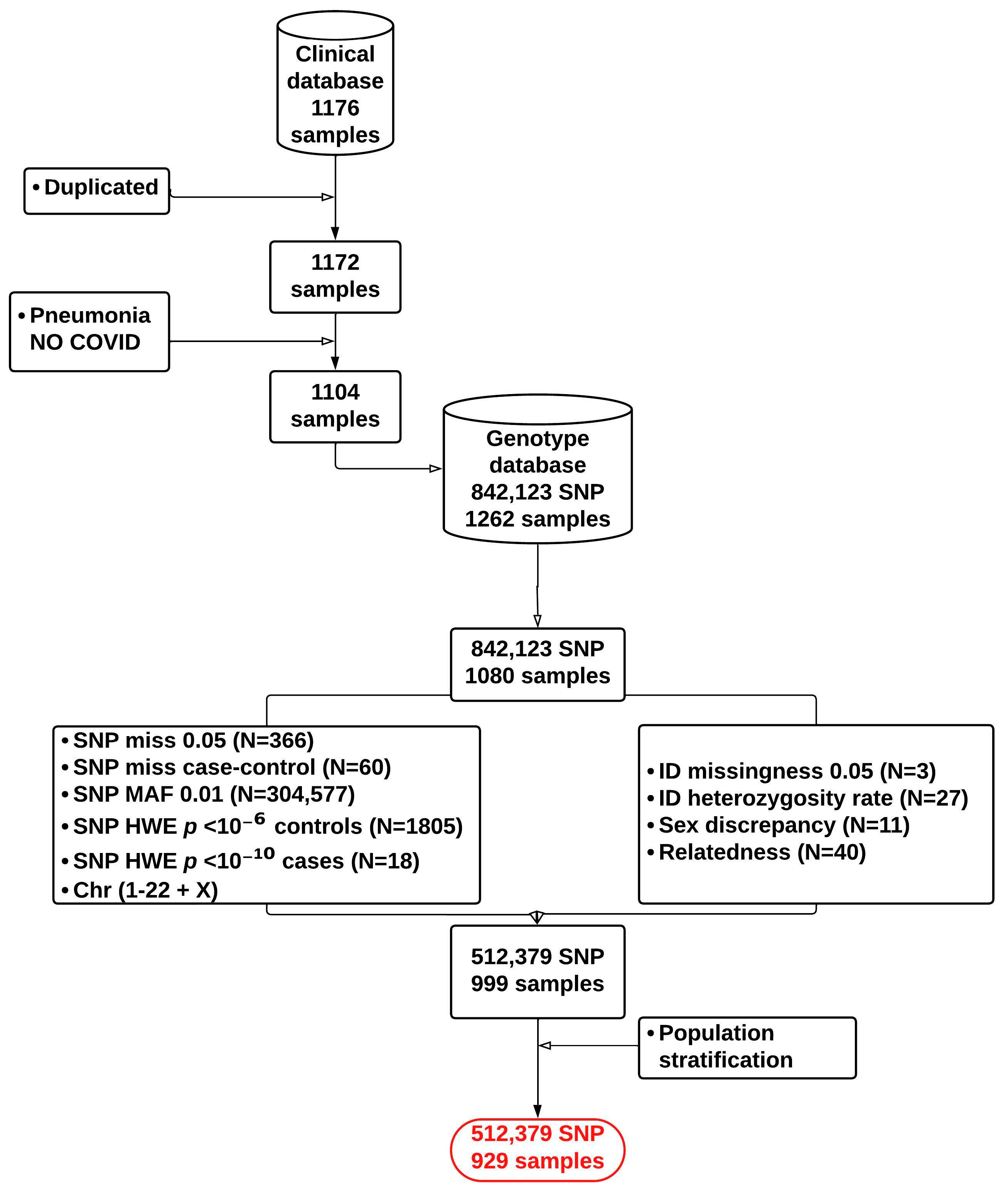
2.1. Patient Cohort
2.2. Genomic DNA Preparation and Genotyping
2.3. Genotyping Data Quality Control
2.4. Genome-Wide Association Analysis
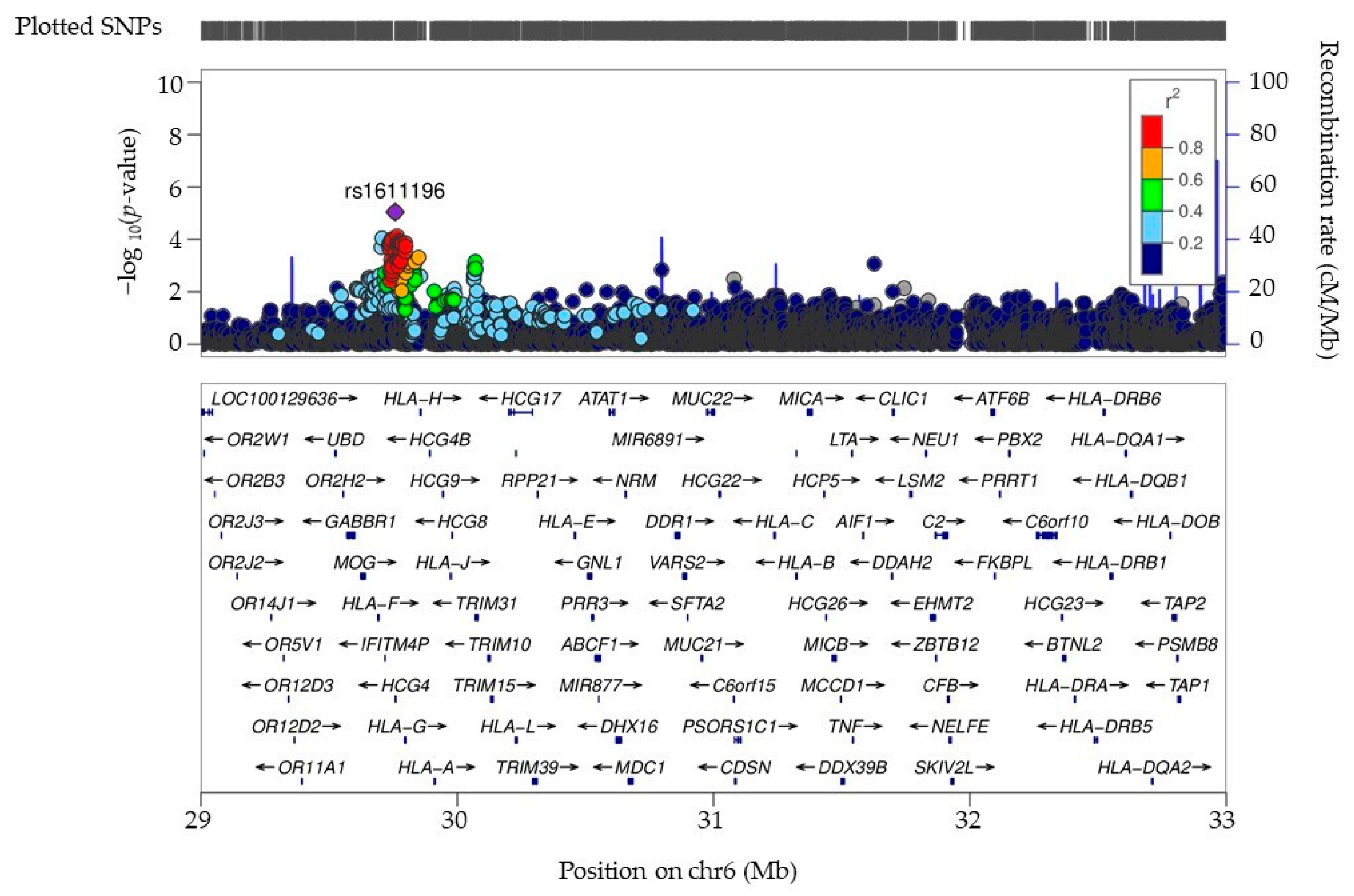
2.5. Analysis of the COVID-19 3p21.31 Locus
2.6. Analysis of HLA Allele Association
2.7. SNP Identification for PRS Replication Analysis
2.8. Polygenic Risk Scoring and Model Estimation: Univariable and Multivariable Method
3. Results
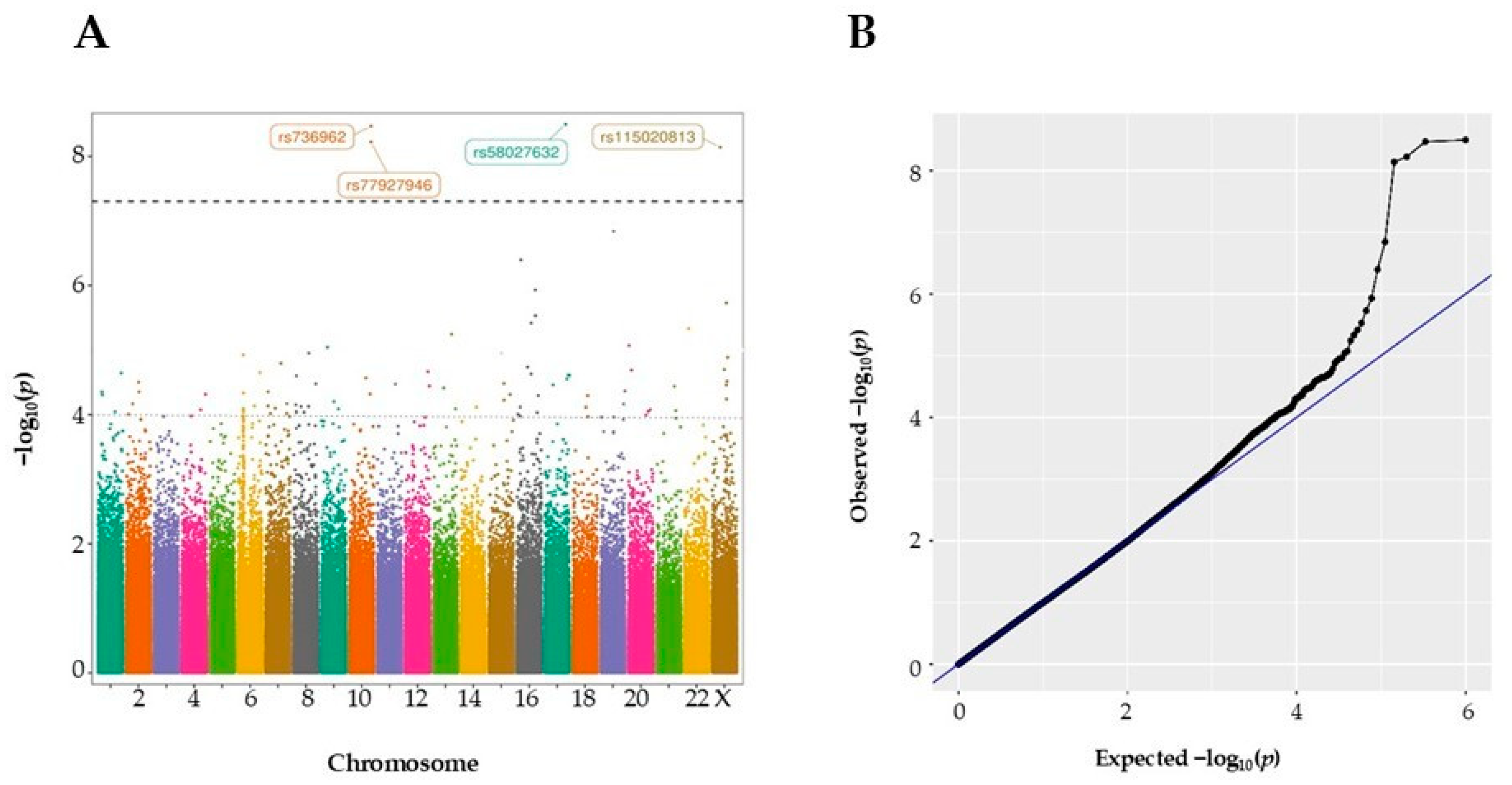
3.1. Identification and Exploration of GWAS Variants in a COVID-19 Hospital-Based Patient Cohort
3.2. Association of HLA Alleles with COVID-19 Severity
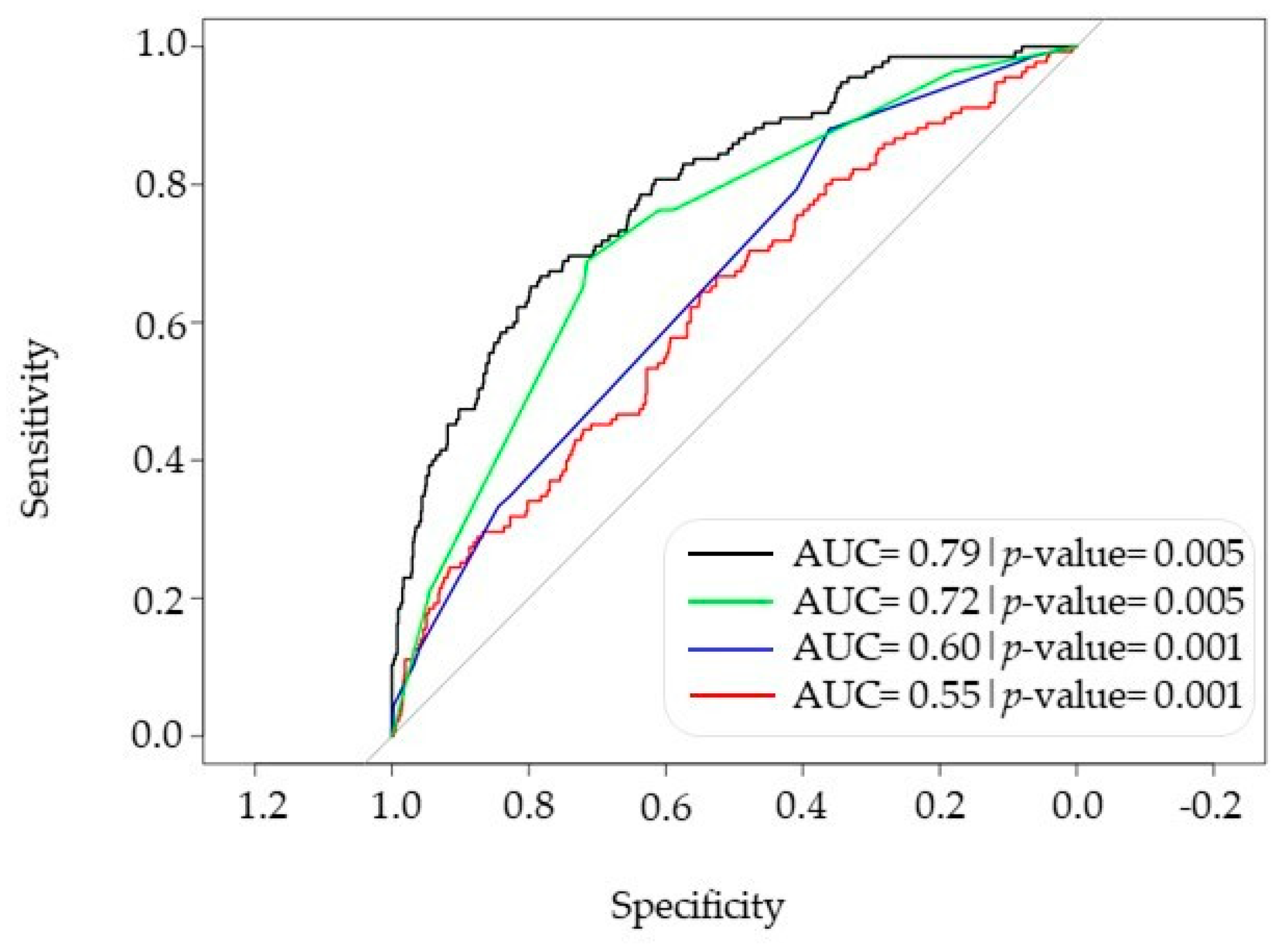
3.3. Comprehensive Model Combining PRS, HLA and Phenotype for Predicting Severe COVID-19 Risk
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Oran, D.P.; Topol, E.J. The proportion of SARS-CoV-2 infections that are asymptomatic: A systematic review. Ann. Intern. Med. 2021, 174, 655–662. [Google Scholar] [CrossRef]
- Bellani, G.; Grasselli, G.; Cecconi, M.; Antolini, L.; Borelli, M.; De Giacomi, F.; Bosio, G.; Latronico, N.; Filippini, M.; Gemma, M.; et al. Noninvasive ventilatory support of patients with COVID-19 outside the intensive care units (WARd-COVID). Ann. Am. Thorac. Soc. 2021, 18, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, Q.; Chi, J.; Dong, B.; Lv, W.; Shen, L.; Wang, Y. Comorbidities and the risk of severe or fatal outcomes associated with coronavirus disease 2019: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 99, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Goncheva, M.I.; Gibson, R.M.; Shouldice, A.C.; Dikeakos, J.D.; Heinrichs, D.E. The Staphylococcus aureus protein IsdA increases SARS CoV-2 replication by modulating JAK-STAT signaling. iScience 2023, 26, 105975. [Google Scholar] [CrossRef]
- Brogna, C.; Cristoni, S.; Brogna, B.; Bisaccia, D.R.; Marino, G.; Viduto, V.; Montano, L.; Piscopo, M. Toxin-like peptides from the bacterial cultures derived from gut microbiome infected by SARS-CoV-2—New data for a possible role in the long COVID pattern. Biomedicines 2022, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Host Genetics Initiative. The COVID-19 host genetics initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020, 28, 715–718. [Google Scholar] [CrossRef]
- Kanai, M.; Andrews, S.J.; Cordioli, M.; Stevens, C.; Neale, B.M.; Daly, M.; Ganna, A.; Pathak, G.A.; Iwasaki, A.; Karjalainen, J.; et al. A second update on mapping the human genetic architecture of COVID-19. Nature 2023, 621, E7–E26. [Google Scholar]
- Severe COVID-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide association study of severe COVID-19 with respiratory failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar]
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Yaghmouri, M.; Izadi, P. Role of the Neanderthal Genome in Genetic Susceptibility to COVID-19: 3p21. 31 Locus in the Spotlight. Biochem. Genet. 2024, 62, 4239–4263. [Google Scholar] [CrossRef] [PubMed]
- Uricchio, L.H. Evolutionary perspectives on polygenic selection, missing heritability, and GWAS. Hum. Genet. 2020, 139, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Horowitz, J.E.; Kosmicki, J.A.; Damask, A.; Sharma, D.; Roberts, G.H.; Justice, A.E.; Banerjee, N.; Coignet, M.V.; Yadav, A.; Leader, J.B.; et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat. Genet. 2022, 54, 382–392. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Marchini, J.; Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010, 11, 499–511. [Google Scholar] [CrossRef]
- van Rossum, B.; Kruijer, W. statgenGWAS: Genome Wide Association Studies. R Package Version 1.0.10.9000. 2024. Available online: https://github.com/Biometris/statgenGWAS/ (accessed on 17 December 2024).
- Brzyski, D.; Peterson, C.B.; Sobczyk, P.; Candès, E.J.; Bogdan, M.; Sabatti, C. Controlling the rate of GWAS false discoveries. Genetics 2017, 205, 61–75. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.; Müller, M. pROC: An open-source package for R and S to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Harrell, F.E. Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis; Springer: Berlin/Heidelberg, Germany, 2001; Volume 608. [Google Scholar]
- Słomian, D.; Szyda, J.; Dobosz, P.; Stojak, J.; Michalska-Foryszewska, A.; Sypniewski, M.; Liu, J.; Kotlarz, K.; Suchocki, T.; Mroczek, M.; et al. Better safe than sorry—Whole-genome sequencing indicates that missense variants are significant in susceptibility to COVID-19. PLoS ONE 2023, 18, e0279356. [Google Scholar] [CrossRef] [PubMed]
- Alyami, A.; Barnawi, F.B.; Christmas, S.; Alyafee, Y.; Awadalla, M.; Al-Bayati, Z.; Alshehri, A.A.; Saif, A.M.; Mansour, L. Relationships between Polymorphisms in HLA-G 3’UTR Region and COVID-19 Disease Severity. Biochem. Genet. 2024, 1–22. [Google Scholar] [CrossRef]
- Bozkurt, I.; Gözler, T.; Yüksel, I.; Ulucan, K.; Tarhan, K.N. Prognostic Value of (rs2069514 and rs762551) Polymorphisms in COVID-19 Patients. Balk. J. Med. Genet. 2023, 26, 35–42. [Google Scholar] [CrossRef]
- Zhang, J.; Kennedy, A.; de Melo Jorge, D.M.; Xing, L.; Reid, W.; Bui, S.; Joppich, J.; Rose, M.; Ercan, S.; Tang, Q. SARS-CoV-2 remodels the Golgi apparatus to facilitate viral assembly and secretion. bioRxiv 2024. [Google Scholar] [CrossRef]
- Abramczyk, U.; Nowaczyński, M.; Słomczyński, A.; Wojnicz, P.; Zatyka, P.; Kuzan, A. Consequences of COVID-19 for the Pancreas. Int. J. Mol. Sci. 2022, 23, 864. [Google Scholar] [CrossRef]
- Finsterer, J.; Scorza, F.A. The pituitary gland in SARS-CoV-2 infections, vaccinations, and post-COVID syndrome. Clinics 2023, 78, 100157. [Google Scholar] [CrossRef]
- Xie, J.; Mothe, B.; Alcalde Herraiz, M.; Li, C.; Xu, Y.; Jödicke, A.M.; Gao, Y.; Wang, Y.; Feng, S.; Wei, J.; et al. Relationship between HLA genetic variations, COVID-19 vaccine antibody response, and risk of breakthrough outcomes. Nat. Commun. 2024, 15, 4031. [Google Scholar] [CrossRef]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Allison, P.D. Measures of fit for logistic regression. In Proceedings of the SAS Global Forum 2014 Conference, Washington, DC, USA, 23–26 March 2014; SAS Institute Inc.: Cary, NC, USA, 2014; pp. 1–13. [Google Scholar]
- Gao, Z.; Xu, Y.; Sun, C.; Wang, X.; Guo, Y.; Qiu, S.; Ma, K. A systematic review of asymptomatic infections with COVID-19. J. Microbiol. Immunol. Infect. 2021, 54, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Oran, D.P.; Topol, E.J. Prevalence of asymptomatic SARS-CoV-2 infection: A narrative review. Ann. Intern. Med. 2020, 173, 362–367. [Google Scholar] [CrossRef]
- Van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of genetic variants among young men with severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Cobat, A.; Casanova, J.-L. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef]
- Robinot, R.; Hubert, M.; de Melo, G.D.; Lazarini, F.; Bruel, T.; Smith, N.; Levallois, S.; Larrous, F.; Fernandes, J.; Gellenoncourt, S. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat. Commun. 2021, 12, 4354. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Polak, P.; Uryasev, S. Classification and severity progression measure of COVID-19 patients using pairs of multi-omic factors. J. Appl. Stat. 2023, 50, 2473–2503. [Google Scholar] [CrossRef] [PubMed]
- Kotov, J.A.; Kotov, D.I.; Linehan, J.L.; Bardwell, V.J.; Gearhart, M.D.; Jenkins, M.K. BCL6 corepressor contributes to Th17 cell formation by inhibiting Th17 fate suppressors. J. Exp. Med. 2019, 216, 1450–1464. [Google Scholar] [CrossRef]
- Caillet-Saguy, C.; Durbesson, F.; Rezelj, V.V.; Gogl, G.; Tran, Q.D.; Twizere, J.; Vignuzzi, M.; Vincentelli, R.; Wolff, N. Host PDZ-containing proteins targeted by SARS-CoV-2. FEBS J. 2021, 288, 5148–5162. [Google Scholar] [CrossRef]
- Zarei, M.; Bose, D.; Ali Akbari Ghavimi, S.; Nouri-Vaskeh, M.; Mohammadi, M.; Sahebkar, A. Potential role of glycoprotein 340 in milder SARS-CoV-2 infection in children. Expert Rev. Anti Infect. Ther. 2021, 19, 675–677. [Google Scholar] [CrossRef]
- Chung, J.; Vig, V.; Sun, X.; Han, X.; O’Connor, G.T.; Chen, X.; DeAngelis, M.M.; Farrer, L.A.; Subramanian, M.L. Genome-wide pleiotropy study identifies association of PDGFB with age-related macular degeneration and COVID-19 infection outcomes. J. Clin. Med. 2022, 12, 109. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Zhang, Q.; Bugge, K.; Breslow, D.K.; Searby, C.C.; Nachury, M.V.; Sheffield, V.C. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011, 7, e1002358. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Crux, N.B.; Elahi, S. Human. leukocyte antigen (HLA) and immune regulation: How do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front. Immunol. 2017, 8, 832. [Google Scholar] [CrossRef]
- Suslova, T.A.; Vavilov, M.N.; Belyaeva, S.V.; Evdokimov, A.V.; Stashkevich, D.S.; Galkin, A.; Kofiadi, I.A. Distribution of HLA-A,-B,-C,-DRB1,-DQB1,-DPB1 allele frequencies in patients with COVID-19 bilateral pneumonia in Russians, living in the Chelyabinsk region (Russia). Hum. Immunol. 2022, 83, 547–550. [Google Scholar] [CrossRef]
- Kakodkar, P.; Dokouhaki, P.; Wu, F.; Shavadia, J.; Nair, R.; Webster, D.; Sawyer, T.; Huan, T.; Mostafa, A. The role of the HLA allelic repertoire on the clinical severity of COVID-19 in Canadians, living in the Saskatchewan province. Hum. Immunol. 2023, 84, 163–171. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Rawlik, K.; Bretherick, A.D.; Qi, T.; Wu, Y.; Nassiri, I.; McConkey, G.A.; Zechner, M.; Klaric, L.; Griffiths, F.; et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature 2023, 617, 764–768. [Google Scholar] [CrossRef]
- Thibord, F.; Chan, M.V.; Chen, M.H..; Johnson, A.D. A year of COVID-19 GWAS results from the GRASP portal reveals potential genetic risk factors. HGG Adv. 2022, 3, 10009. [Google Scholar] [CrossRef]
- Bian, S.; Guo, X.; Yang, X.; Wei, Y.; Yang, Z.; Cheng, S.; Yan, J.; Chen, Y.; Chen, G.B.; Du, X.; et al. Genetic determinants of IgG antibody response to COVID-19 vaccination. Am. J. Hum. Genet. 2024, 111, 181–199. [Google Scholar] [CrossRef]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; Aslibekyan, S.; Auton, A. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All | Non-ICU | ICU | p-Value | n |
|---|---|---|---|---|---|
| Gender (n, %) | 0.041 | 1104 | |||
| Male | 706 (63.9%) | 583 (82.6%) | 123 (17.4%) | --- | --- |
| Female | 398 (36.1%) | 348 (87.4%) | 50 (12.6%) | --- | --- |
| Age (median, IQR) | 62.0 [52.0;72.0] | 62.0 [52.0;72.0] | 63.0 [52.0;71.0] | 0.507 | 1104 |
| Weight (median, IQR) | 81.0 [70.5;91.5] | 80.0 [70.0;90.0] | 84.0 [75.0;95.0] | 0.004 | 663 |
| BMI (median, IQ) | 29.4 [26.1;32.4] | 29.1 [26.0;32.2] | 29.9 [27.0;32.9] | 0.075 | 461 |
| Oxygen therapy | |||||
| IMV (n, %) | 117 (10.6%) | 2 (1.71%) | 115 (98.3%) | <0.001 | 1102 |
| NIMV (n, %) | 334 (32.8%) | 192 (57.5%) | 142 (42.5%) | <0.001 | 1018 |
| Oxygen glasses (n, %) | 899 (81.4%) | 737 (82.0%) | 162 (18.0%) | <0.001 | 1104 |
| Venturi mask | 378 (34.2%) | 262 (69.3%) | 116 (30.7%) | <0.001 | 1104 |
| Comorbidities | |||||
| HTA (n, %) | 465 (42.2%) | 383 (82.4%) | 82 (17.6%) | 0.151 | 1103 |
| DM (n, %) | 209 (18.9%) | 163 (78.0%) | 46 (22.0%) | 0.007 | 1103 |
| Chronic cardiomyopathy (n, %) | 108 (9.79%) | 91 (84.3%) | 17 (15.7%) | 1.000 | 1103 |
| Cardiac arrhythmia (n, %) | 94 (8.52%) | 78 (83.0%) | 16 (17.0%) | 0.822 | 1103 |
| Valvulopathy (n, %) | 40 (3.63%) | 37 (92.5%) | 3 (7.50%) | 0.219 | 1103 |
| Cardiac ischemia (n, %) | 90 (8.16%) | 72 (80.0%) | 18 (20.0%) | 0.306 | 1103 |
| DM treatment (n, %) | 0.093 | 978 | |||
| No treatment | 812 (83.0%) | 715 (88.1%) | 97 (11.9%) | --- | --- |
| Insulin | 21 (2.15%) | 20 (95.2%) | 1 (4.76%) | --- | --- |
| Insulin + oral antidiabetics | 21 (2.15%) | 18 (85.7%) | 3 (14.3%) | --- | --- |
| Oral antidiabetics | 124 (12.7%) | 100 (80.6%) | 24 (19.4%) | --- | --- |
| Chr:pos(b38) | rsID | EA | OA | OR | ICU Frequency | Non-ICU Frequency | p-Value | Nearest Gene |
|---|---|---|---|---|---|---|---|---|
| 17:74332949 | rs58027632 | T | C | 1.26 | 0.106 | 0.031 | 3.19 × 10−9 | KIF19 |
| 10:122498480 | rs736962 | G | A | 1.49 | 0.053 | 0.007 | 3.04 × 10−9 | HTRA1 |
| 10:122521643 | rs77927946 | A | C | 1.50 | 0.049 | 0.007 | 5.98 × 10−9 | DMBT1 |
| X:39639346 | rs115020813 | T | G | 1.34 | 0.064 | 0.006 | 7.23 × 10−9 | LINC01283 |
| Cytogenetic Band | Gene | p-Value | Number of Variants with p < 10−4 |
|---|---|---|---|
| 1q43 | LINC01139 | <5 × 10−5 | 2 |
| 5q24.1 | CYP1A2; CPLX3 | <5 × 10−5 | 2 |
| 1q43 | LOC105373220; MIR4426 | <5 × 10−5 | 2 |
| 16q23.1 | WWOX | <10−5 | 2 |
| Xq21.33 | MIR548 | <5 × 10−5 | 2 |
| 6p22.1 | HLA-G | <10−4 | 10 |
| 7p21.3 | --- | <10−4 | 2 |
| 7q34 | CASP2; CLCN1 | <10−4 | 2 |
| 8p23.1 | CLDN23 | <10−4 | 2 |
| Allele | Adjusted p-Value | OR |
|---|---|---|
| A*01:01 | 0.002 | 0.431 |
| DRB3*03:01 | 0.007 | 2.124 |
| DPB1*10:01 | 0.022 | 2.193 |
| B*45:01 | 0.031 | 3.422 |
| C*02:02 | 0.036 | 0.432 |
| DRB1*13:03 | 0.043 | 3.765 |
| DQB1*05:02 | 0.043 | 0.128 |
| DQB1*06:09 | 0.045 | 3.604 |
| DRB4*01:01 | 0.049 | 1.446 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alloza-Moral, I.; Aldekoa-Etxabe, A.; Tulloch-Navarro, R.; Fiat-Arriola, A.; Mar, C.; Urrechaga, E.; Ponga, C.; Artiga-Folch, I.; Garcia-Bediaga, N.; Aspichueta, P.; et al. Genetic Analysis and Predictive Modeling of COVID-19 Severity in a Hospital-Based Patient Cohort. Biomolecules 2025, 15, 393. https://doi.org/10.3390/biom15030393
Alloza-Moral I, Aldekoa-Etxabe A, Tulloch-Navarro R, Fiat-Arriola A, Mar C, Urrechaga E, Ponga C, Artiga-Folch I, Garcia-Bediaga N, Aspichueta P, et al. Genetic Analysis and Predictive Modeling of COVID-19 Severity in a Hospital-Based Patient Cohort. Biomolecules. 2025; 15(3):393. https://doi.org/10.3390/biom15030393
Chicago/Turabian StyleAlloza-Moral, Iraide, Ane Aldekoa-Etxabe, Raquel Tulloch-Navarro, Ainhoa Fiat-Arriola, Carmen Mar, Eloisa Urrechaga, Cristina Ponga, Isabel Artiga-Folch, Naiara Garcia-Bediaga, Patricia Aspichueta, and et al. 2025. "Genetic Analysis and Predictive Modeling of COVID-19 Severity in a Hospital-Based Patient Cohort" Biomolecules 15, no. 3: 393. https://doi.org/10.3390/biom15030393
APA StyleAlloza-Moral, I., Aldekoa-Etxabe, A., Tulloch-Navarro, R., Fiat-Arriola, A., Mar, C., Urrechaga, E., Ponga, C., Artiga-Folch, I., Garcia-Bediaga, N., Aspichueta, P., Martin, C., Zarandona-Garai, A., Pérez-Fernández, S., Arana-Arri, E., Triviño, J.-C., Uranga, A., España, P.-P., & Vandenbroeck-van-Caeckenbergh, K. (2025). Genetic Analysis and Predictive Modeling of COVID-19 Severity in a Hospital-Based Patient Cohort. Biomolecules, 15(3), 393. https://doi.org/10.3390/biom15030393