
CSN-CRL Complexes: New Regulators of Adipogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
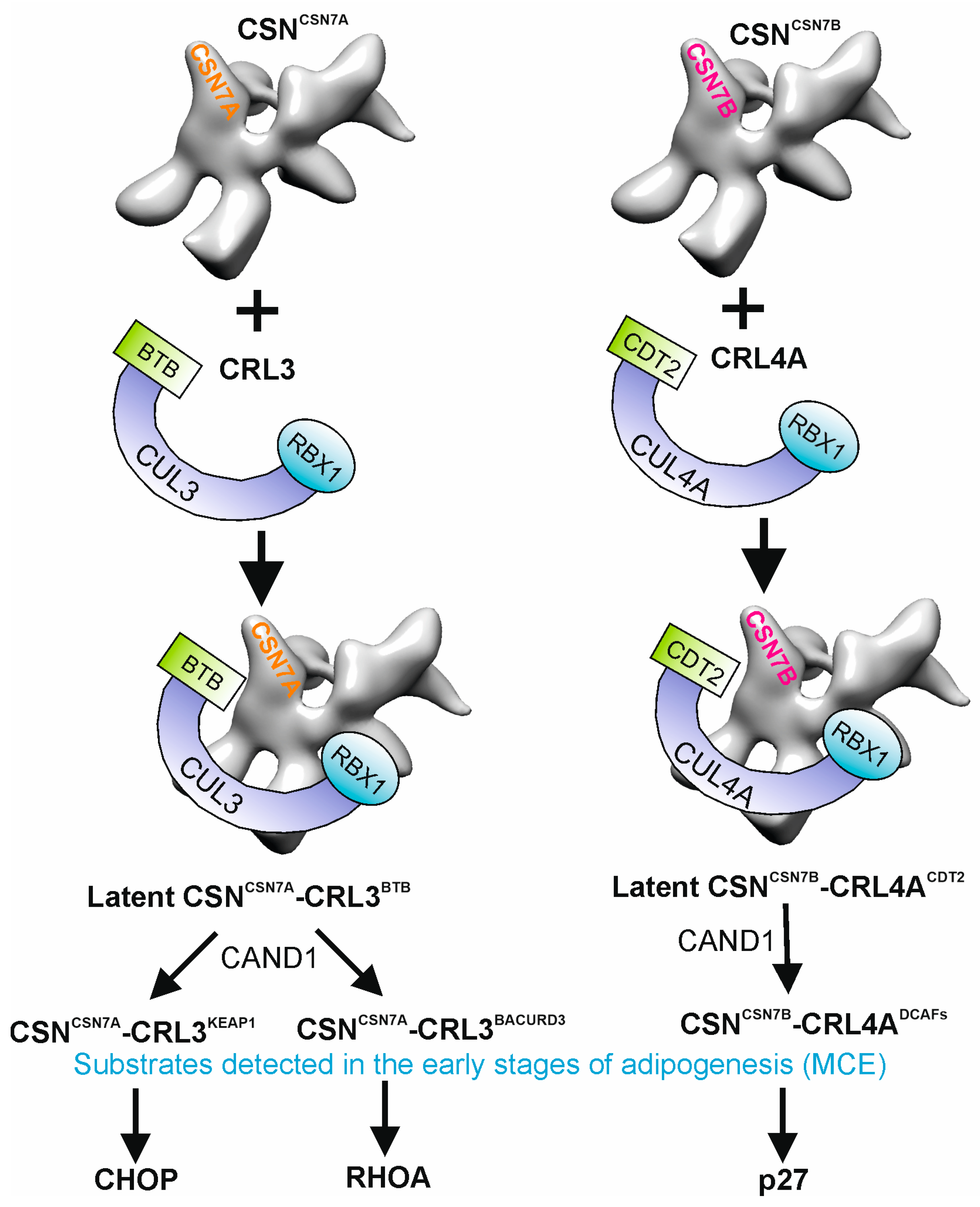
2. Composition of CSN-CRL Complexes
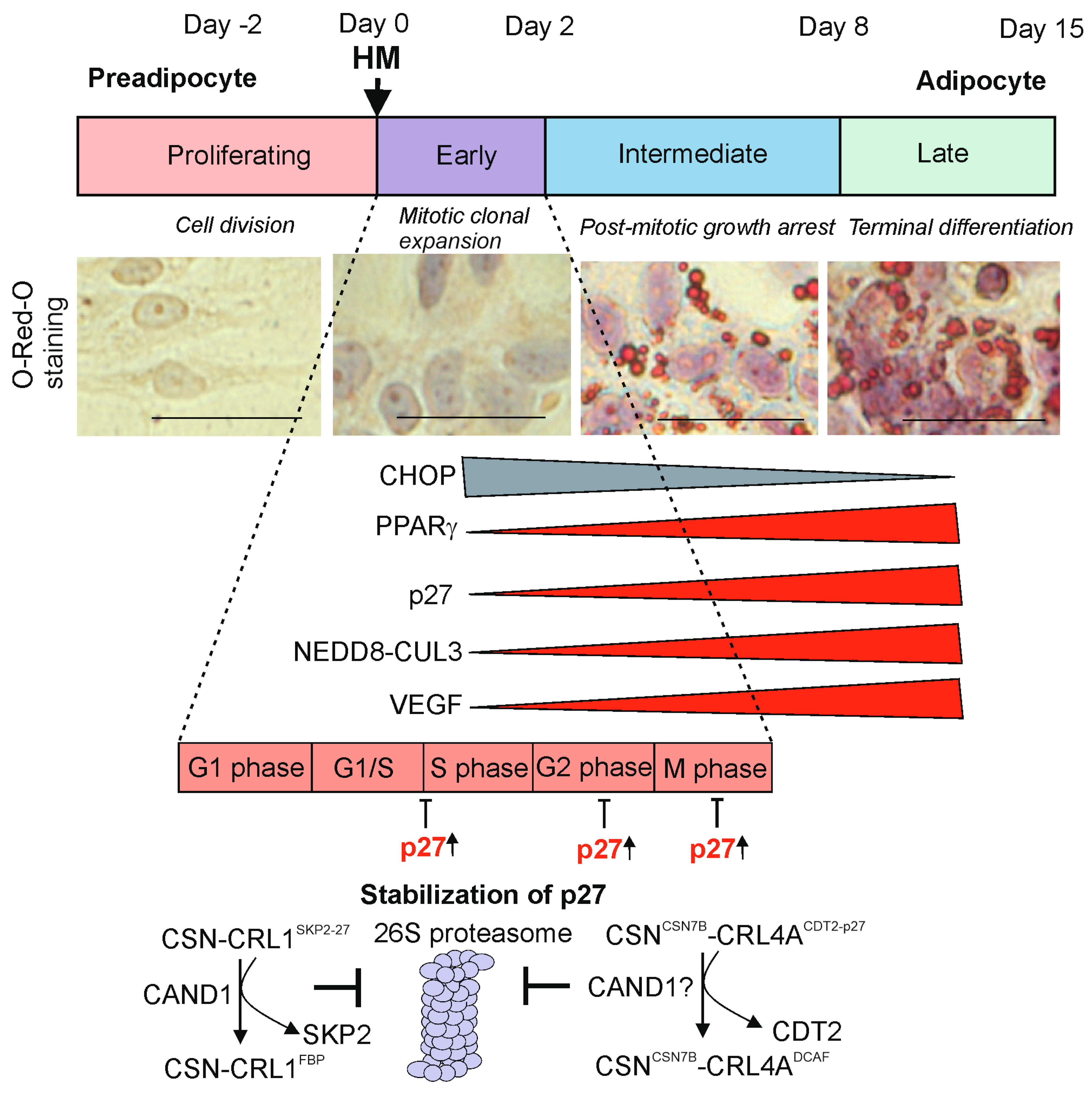
3. Functions of CSN-CRL Complexes in Mitotic Clonal Expansion
4. CSNCSN7A-CRL3BTB Is Recruited to Lipid Droplets by RAB18 During TAD
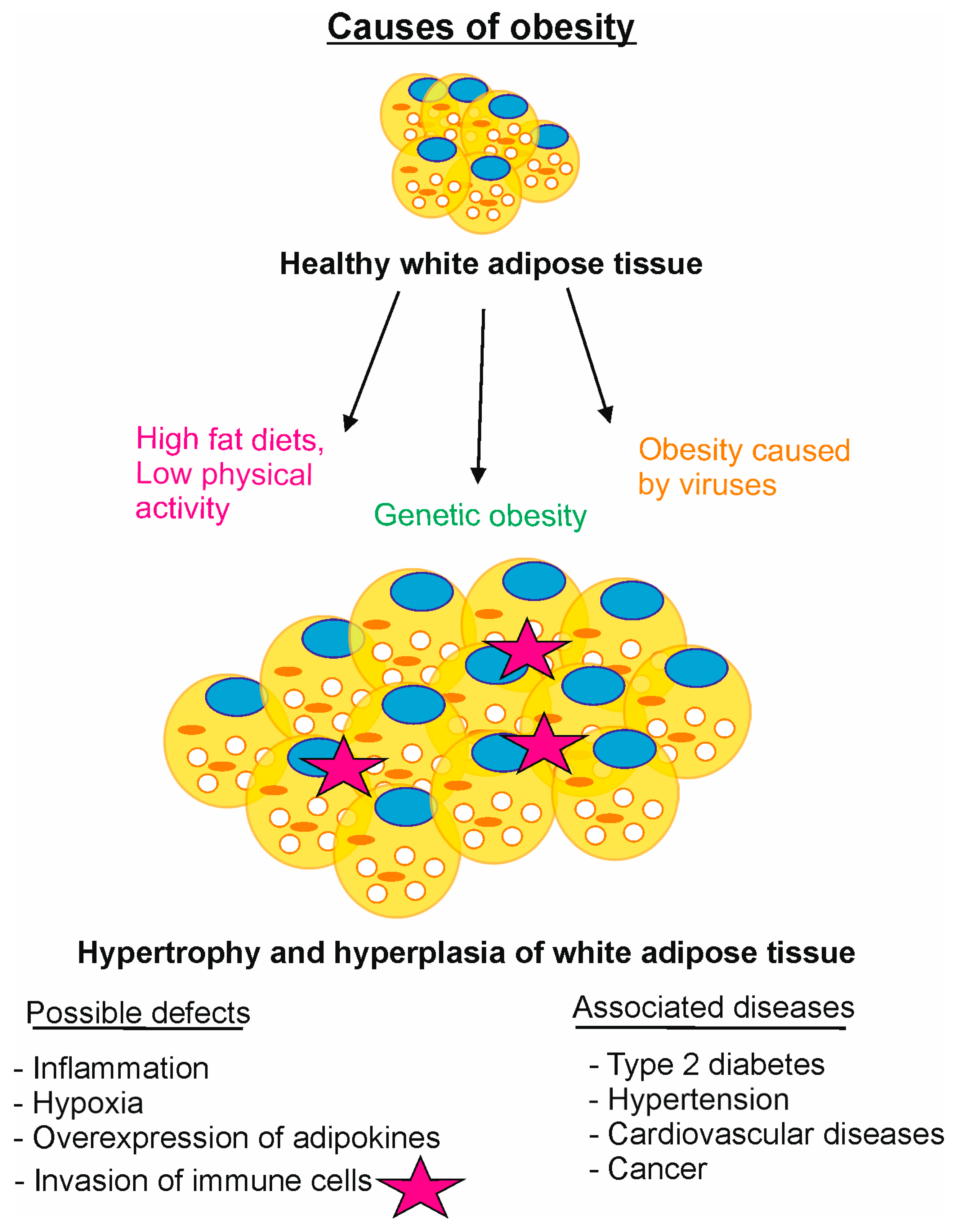
5. Selected Malfunctions of CSNCSN7A-CRL3BTB and CSNCSN7B-CRL4ADCAF Connected with Obesity
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dubiel, D.; Wang, J.; Hartig, R.; Chaithongyot, S.; Dubiel, W.; Naumann, M. Latent CSN-CRL complexes are crucial for curcumin-induced apoptosis and recruited during adipogenesis to lipid droplets via small GTPase RAB18. iScience 2023, 26, 106468. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Rush, J.; Gygi, S.P.; Harper, J.W. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 2010, 143, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, Y.; Cheng, S.; Li, X.; Wei, N. COP9 signalosome-mediated deneddylation of CULLIN1 is necessary for SCF(EBF1) assembly in Arabidopsis thaliana. Cell Rep. 2024, 43, 113638. [Google Scholar] [CrossRef] [PubMed]
- Diaz, S.; Wang, K.; Sjogren, B.; Liu, X. Roles of Cullin-RING Ubiquitin Ligases in Cardiovascular Diseases. Biomolecules 2022, 12, 416. [Google Scholar] [CrossRef]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Dubiel, D.; Bintig, W.; Kahne, T.; Dubiel, W.; Naumann, M. Cul3 neddylation is crucial for gradual lipid droplet formation during adipogenesis. Biochim. Biophys. Acta 2017, 1864, 1405–1412. [Google Scholar] [CrossRef]
- Zhou, H.; Patel, V.; Rice, R.; Lee, R.; Kim, H.W.; Weintraub, N.L.; Su, H.; Chen, W. Neddylation and Its Target Cullin 3 Are Essential for Adipocyte Differentiation. Cells 2024, 13, 1654. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Roberts, M.A.; Olzmann, J.A. Protein Quality Control and Lipid Droplet Metabolism. Annu. Rev. Cell Dev. Biol. 2020, 36, 115–139. [Google Scholar] [CrossRef]
- Farese, R.V., Jr.; Walther, T.C. Glycerolipid Synthesis and Lipid Droplet Formation in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2023, 15, a041246. [Google Scholar] [CrossRef]
- Goransson, O.; Kopietz, F.; Rider, M.H. Metabolic control by AMPK in white adipose tissue. Trends Endocrinol. Metab. 2023, 34, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Bruderlein, S.; Melzner, I.; Braun, M.; Mechtersheimer, G.; Moller, P. LiSa-2, a novel human liposarcoma cell line with a high capacity for terminal adipose differentiation. Int. J. Cancer 2000, 88, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef]
- Wu, Z.; Bucher, N.L.; Farmer, S.R. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol. Cell Biol. 1996, 16, 4128–4136. [Google Scholar] [CrossRef]
- Mishra, A.; Zhu, X.G.; Ge, K.; Cheng, S.Y. Adipogenesis is differentially impaired by thyroid hormone receptor mutant isoforms. J. Mol. Endocrinol. 2010, 44, 247–255. [Google Scholar] [CrossRef]
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341. [Google Scholar] [CrossRef]
- Hursting, S.D.; Hursting, M.J. Growth signals, inflammation, and vascular perturbations: Mechanistic links between obesity, metabolic syndrome, and cancer. Arter. Thromb. Vasc. Biol. 2012, 32, 1766–1770. [Google Scholar] [CrossRef] [PubMed]
- Vona-Davis, L.; Rose, D.P. Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor. Rev. 2009, 20, 193–201. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef]
- Jin, D.; Li, B.; Deng, X.W.; Wei, N. Plant COP9 Signalosome subunit 5, CSN5. Plant Sci. 2014, 224C, 54–61. [Google Scholar] [CrossRef]
- Schulze-Niemand, E.; Naumann, M. The COP9 signalosome: A versatile regulatory hub of Cullin-RING ligases. Trends Biochem. Sci. 2023, 48, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef] [PubMed]
- Milhollen, M.A.; Traore, T.; Adams-Duffy, J.; Thomas, M.P.; Berger, A.J.; Dang, L.; Dick, L.R.; Garnsey, J.J.; Koenig, E.; Langston, S.P.; et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood 2010, 116, 1515–1523. [Google Scholar] [CrossRef]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 Promotes Assembly of New SCF Complexes through Dynamic Exchange of F Box Proteins. Cell 2013, 153, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, J.M.; Liu, X.; Reichermeier, K.M.; Moradian, A.; Sweredoski, M.J.; Hess, S.; Deshaies, R.J. Composition and Regulation of the Cellular Repertoire of SCF Ubiquitin Ligases. Cell 2017, 171, 1326–1339.e14. [Google Scholar] [CrossRef]
- Dubiel, D.; Ordemann, J.; Pratschke, J.; Dubiel, W.; Naumann, M. CAND1 exchange factor promotes Keap1 integration into cullin 3-RING ubiquitin ligase during adipogenesis. Int. J. Biochem. Cell Biol. 2015, 66, 95–100. [Google Scholar] [CrossRef]
- Dubiel, W.; Chaithongyot, S.; Dubiel, D.; Naumann, M. The COP9 Signalosome: A Multi-DUB Complex. Biomolecules 2020, 10, 1082. [Google Scholar] [CrossRef]
- Chaithongyot, S.; Naumann, M. Helicobacter pylori-induced reactive oxygen species direct turnover of CSN-associated STAMBPL1 and augment apoptotic cell death. Cell Mol. Life Sci. 2022, 79, 86. [Google Scholar] [CrossRef]
- Jantaree, P.; Chaithongyot, S.; Sokolova, O.; Naumann, M. USP48 and A20 synergistically promote cell survival in Helicobacter pylori infection. Cell Mol. Life Sci. 2022, 79, 461. [Google Scholar] [CrossRef]
- Li, S.; Wang, D.; Zhao, J.; Weathington, N.M.; Shang, D.; Zhao, Y. The deubiquitinating enzyme USP48 stabilizes TRAF2 and reduces E-cadherin-mediated adherens junctions. FASEB J. 2018, 32, 230–242. [Google Scholar] [CrossRef]
- Li, B.; Jia, N.; Kapur, R.; Chun, K.T. Cul4A targets p27 for degradation and regulates proliferation, cell cycle exit, and differentiation during erythropoiesis. Blood 2006, 107, 4291–4299. [Google Scholar] [CrossRef] [PubMed]
- Audano, M.; Pedretti, S.; Caruso, D.; Crestani, M.; De Fabiani, E.; Mitro, N. Regulatory mechanisms of the early phase of white adipocyte differentiation: An overview. Cell Mol. Life Sci. 2022, 79, 139. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Li, M.; Wang, Z.; Wang, X.M.; Liu, S.Y.; Tian, S.; Wang, Z.X.; Cheng, X.; Hu, Y.; Zhang, P.; et al. Targeting peroxisome proliferator-activated receptor gamma proteasomal degradation by magnolol is a potential avenue for adipogenesis-mediated metabolic homeostasis. Obesity 2023, 31, 1584–1599. [Google Scholar] [CrossRef]
- Ferguson, B.S.; Nam, H.; Morrison, R.F. Curcumin Inhibits 3T3-L1 Preadipocyte Proliferation by Mechanisms Involving Post-transcriptional p27 Regulation. Biochem. Biophys. Rep. 2016, 5, 16–21. [Google Scholar] [CrossRef]
- Higa, L.A.; Yang, X.; Zheng, J.; Banks, D.; Wu, M.; Ghosh, P.; Sun, H.; Zhang, H. Involvement of CUL4 ubiquitin E3 ligases in regulating CDK inhibitors Dacapo/p27Kip1 and cyclin E degradation. Cell Cycle 2006, 5, 71–77. [Google Scholar] [CrossRef]
- Humphries, B.; Wang, Z.; Yang, C. Rho GTPases: Big Players in Breast Cancer Initiation, Metastasis and Therapeutic Responses. Cells 2020, 9, 2167. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Lane, M.D. Role of C/EBP homologous protein (CHOP-10) in the programmed activation of CCAAT/enhancer-binding protein-beta during adipogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 12446–12450. [Google Scholar] [CrossRef]
- Dubiel, W.; Dubiel, D.; Wolf, D.A.; Naumann, M. Cullin 3-Based Ubiquitin Ligases as Master Regulators of Mammalian Cell Differentiation. Trends Biochem. Sci. 2018, 43, 95–107. [Google Scholar] [CrossRef]
- Zhang, J.W.; Tang, Q.Q.; Vinson, C.; Lane, M.D. Dominant-negative C/EBP disrupts mitotic clonal expansion and differentiation of 3T3-L1 preadipocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Farese, R.V., Jr.; Walther, T.C. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cell Biol. 2016, 26, 535–546. [Google Scholar] [CrossRef]
- Chorlay, A.; Monticelli, L.; Verissimo Ferreira, J.; Ben M’barek, K.; Ajjaji, D.; Wang, S.; Johnson, E.; Beck, R.; Omrane, M.; Beller, M.; et al. Membrane Asymmetry Imposes Directionality on Lipid Droplet Emergence from the ER. Dev. Cell 2019, 50, 25–42. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Sztalryd, C.; Londos, C. Degradation of perilipin is mediated through ubiquitination-proteasome pathway. Biochim. Biophys. Acta 2006, 1761, 83–90. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shinoda, A.; Kamada, H.; Shimizu, M.; Inoue, J.; Sato, R. Perilipin2 plays a positive role in adipocytes during lipolysis by escaping proteasomal degradation. Sci. Rep. 2016, 6, 20975. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Richter, C.M.; Kopito, R.R. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 1345–1350. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; You, P.; Jiang, H.; Liu, Y.; Han, D.; Liu, M.; Yu, H.; Su, B. Mice with the Rab10 T73V mutation exhibit anxiety-like behavior and alteration of neuronal functions in the striatum. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166641. [Google Scholar] [CrossRef]
- Pfeffer, S.R. LRRK2 phosphorylation of Rab GTPases in Parkinson’s disease. FEBS Lett. 2023, 597, 811–818. [Google Scholar] [CrossRef]
- Malik, A.U.; Karapetsas, A.; Nirujogi, R.S.; Mathea, S.; Chatterjee, D.; Pal, P.; Lis, P.; Taylor, M.; Purlyte, E.; Gourlay, R.; et al. Deciphering the LRRK code: LRRK1 and LRRK2 phosphorylate distinct Rab proteins and are regulated by diverse mechanisms. Biochem. J. 2021, 478, 553–578. [Google Scholar] [CrossRef]
- Gschweitl, M.; Ulbricht, A.; Barnes, C.A.; Enchev, R.I.; Stoffel-Studer, I.; Meyer-Schaller, N.; Huotari, J.; Yamauchi, Y.; Greber, U.F.; Helenius, A.; et al. A SPOPL/Cullin-3 ubiquitin ligase complex regulates endocytic trafficking by targeting EPS15 at endosomes. eLife 2016, 5, e13841. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yoshida, K.; Iwamoto, S. Kbtbd11 gene expression in adipose tissue increases in response to feeding and affects adipocyte differentiation. J. Diabetes Investig. 2019, 10, 925–932. [Google Scholar] [CrossRef]
- Kusakabe, S.; Suzuki, T.; Sugiyama, Y.; Haga, S.; Horike, K.; Tokunaga, M.; Hirano, J.; Zhang, H.; Chen, D.V.; Ishiga, H.; et al. USP15 Participates in Hepatitis C Virus Propagation through Regulation of Viral RNA Translation and Lipid Droplet Formation. J. Virol. 2019, 93, 1–18. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Lee, C.S.; Mun, S.H.; Truong, N.T.; Park, S.K.; Hwang, C.S. N-terminal acetylation and the N-end rule pathway control degradation of the lipid droplet protein PLIN2. J. Biol. Chem. 2019, 294, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Stadiotti, I.; Di Bona, A.; Pilato, C.A.; Scalco, A.; Guarino, A.; Micheli, B.; Casella, M.; Tondo, C.; Rizzo, S.; Pilichou, K.; et al. Neuropeptide Y promotes adipogenesis of human cardiac mesenchymal stromal cells in arrhythmogenic cardiomyopathy. Int. J. Cardiol. 2021, 342, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.; Tokumaru, Y.; Angarita, F.A.; Lee, L.; Yan, L.; Matsuyama, R.; Endo, I.; Takabe, K. Adipogenesis in triple-negative breast cancer is associated with unfavorable tumor immune microenvironment and with worse survival. Sci. Rep. 2021, 11, 12541. [Google Scholar] [CrossRef]
- Pan, X.H.; Tan, B.; Chin, Y.H.; Lee, E.C.Z.; Kong, G.; Chong, B.; Kueh, M.; Khoo, C.M.; Mehta, A.; Majety, P.; et al. Efficacy and safety of tirzepatide, GLP-1 receptor agonists, and other weight loss drugs in overweight and obesity: A network meta-analysis. Obesity 2024, 32, 840–856. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Miyazawa, T.; Abe, C.; Ueno, T.; Suzuki, M.; Mizukami, M.; Kurihara, K.; Toda, M. Hypolipidemic and Anti-Inflammatory Effects of Curcuma longa-Derived Bisacurone in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2023, 24, 9366. [Google Scholar] [CrossRef]
- Weidlich, M.; Huang, X.; Pratschke, J.; Dubiel, W.; Ordemann, J. Bariatric Surgery Significantly Reduces Serum Concentration of Vascular Endothelial Growth Factor A and Increases Apelin-12 in Patients with Morbid Obesity. Bariatr. Surg. Pract. Patient Care 2017, 12, 16–20. [Google Scholar] [CrossRef]
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Elsea, S.H.; Mykytyn, K.; Ferrell, K.; Coulter, K.L.; Das, P.; Dubiel, W.; Patel, P.I.; Metherall, J.E. Hemizygosity for the COP9 signalosome subunit gene, SGN3, in the Smith-Magenis syndrome. Am. J. Med. Genet. 1999, 87, 342–348. [Google Scholar] [CrossRef]
- Shayota, B.J.; Elsea, S.H. Behavior and sleep disturbance in Smith-Magenis syndrome. Curr. Opin. Psychiatry 2019, 32, 73–78. [Google Scholar] [CrossRef]
- Chang, H.C.; Lee, Y.J.; Javed, S.; Haque, M.; Chang, Y.T.; Lin, Y.C.; Oram, C.; Huang, W.H. rAAV-CRISPRa therapy corrects Rai1 haploinsufficiency and rescues selective disease features in Smith-Magenis syndrome mice. J. Biol. Chem. 2023, 299, 102728. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Izumi, T. Analysis of RNA expression of normal and cancer tissues reveals high correlation of COP9 gene expression with respiratory chain complex components. BMC Genom. 2016, 17, 983. [Google Scholar] [CrossRef]
- Goldlust, I.S.; Hermetz, K.E.; Catalano, L.M.; Barfield, R.T.; Cozad, R.; Wynn, G.; Ozdemir, A.C.; Conneely, K.N.; Mulle, J.G.; Dharamrup, S.; et al. Mouse model implicates GNB3 duplication in a childhood obesity syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, 14990–14994. [Google Scholar] [CrossRef] [PubMed]
- Leroy, C.; Landais, E.; Briault, S.; David, A.; Tassy, O.; Gruchy, N.; Delobel, B.; Gregoire, M.J.; Leheup, B.; Taine, L.; et al. The 2q37-deletion syndrome: An update of the clinical spectrum including overweight, brachydactyly and behavioural features in 14 new patients. Eur. J. Hum. Genet. 2013, 21, 602–612. [Google Scholar] [CrossRef]
- Lin, H.; Yan, Y.; Luo, Y.; So, W.Y.; Wei, X.; Zhang, X.; Yang, X.; Zhang, J.; Su, Y.; Yang, X.; et al. IP6-assisted CSN-COP1 competition regulates a CRL4-ETV5 proteolytic checkpoint to safeguard glucose-induced insulin secretion. Nat. Commun. 2021, 12, 2461. [Google Scholar] [CrossRef]
- Scherer, P.C.; Ding, Y.; Liu, Z.; Xu, J.; Mao, H.; Barrow, J.C.; Wei, N.; Zheng, N.; Snyder, S.H.; Rao, F. Inositol hexakisphosphate (IP6) generated by IP5K mediates cullin-COP9 signalosome interactions and CRL function. Proc. Natl. Acad. Sci. USA 2016, 113, 3503–3508. [Google Scholar] [CrossRef]
- Qian, M.; Lin, S.; Tan, Y.; Chen, Q.; Wang, W.; Li, J.; Mu, C. A nonsense mutation in the CUL3 gene in a Chinese patient with autism spectrum disorder and epilepsy: A case report. Medicine 2023, 102, e33457. [Google Scholar] [CrossRef]
- Rapanelli, M.; Tan, T.; Wang, W.; Wang, X.; Wang, Z.J.; Zhong, P.; Frick, L.; Qin, L.; Ma, K.; Qu, J.; et al. Behavioral, circuitry, and molecular aberrations by region-specific deficiency of the high-risk autism gene Cul3. Mol. Psychiatry 2021, 26, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Siew, K.; Zhang, J.; Johnson, C.; Wood, N.; Cleary, S.E.; Al Maskari, R.S.; Ferryman, J.T.; Hardege, I.; Yasmin; et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol. Med. 2015, 7, 1285–1306. [Google Scholar] [CrossRef]
- Takahashi, D.; Mori, T.; Sohara, E.; Tanaka, M.; Chiga, M.; Inoue, Y.; Nomura, N.; Zeniya, M.; Ochi, H.; Takeda, S.; et al. WNK4 is an Adipogenic Factor and Its Deletion Reduces Diet-Induced Obesity in Mice. EBioMedicine 2017, 18, 118–127. [Google Scholar] [CrossRef]
- Reinstein, E.; Liberman, M.; Feingold-Zadok, M.; Tenne, T.; Graham, J.M., Jr. Terminal microdeletions of 13q34 chromosome region in patients with intellectual disability: Delineation of an emerging new microdeletion syndrome. Mol. Genet. Metab. 2016, 118, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. Pathogenic Role of the CRL4 Ubiquitin Ligase in Human Disease. Front. Oncol. 2012, 2, 21. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Liu, Z.; Wang, Q.; Wen, M.; Wang, Y.; Yuan, H.; Mao, J.H.; Wei, G. CUL4A overexpression enhances lung tumor growth and sensitizes lung cancer cells to erlotinib via transcriptional regulation of EGFR. Mol. Cancer 2014, 13, 252. [Google Scholar] [CrossRef]
- Groh, B.S.; Yan, F.; Smith, M.D.; Yu, Y.; Chen, X.; Xiong, Y. The antiobesity factor WDTC1 suppresses adipogenesis via the CRL4WDTC1 E3 ligase. EMBO Rep. 2016, 17, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Cancelier, A.C.L.; Schuelter-Trevisol, F.; Trevisol, D.J.; Atkinson, R.L. Adenovirus 36 infection and obesity risk: Current understanding and future therapeutic strategies. Expert. Rev. Endocrinol. Metab. 2022, 17, 143–152. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Cataldi, M. Findings from Studies Are Congruent with Obesity Having a Viral Origin, but What about Obesity-Related NAFLD? Viruses 2021, 13, 1285. [Google Scholar] [CrossRef]
- Barrera-Alcocer, J.; Garcia-Benavides, L.; Munoz-Valle, J.F.; de la Cruz-Mosso, U.; Gonzalez, R.A.; Luquin, S.; Alarcon-Romero, L.D.C.; Marino-Ortega, L.A.; Matia-Garcia, I.; Parra-Rojas, I. Presence of Adenovirus-36 DNA in Adipose Tissue of Women: Relationship with Adipocyte Morphology and the Expression of C/EBPbeta and HIF-1alpha. Diabetes Metab. Syndr. Obes. 2021, 14, 477–486. [Google Scholar] [CrossRef]
- Manriquez, V.; Brito, R.; Pavez, M.; Sapunar, J.; Fonseca, L.; Molina, V.; Ortiz, E.; Baeza, R.; Reimer, C.; Charles, M.; et al. Adenovirus 36 seropositivity is related to the expression of anti-adipogenic lncRNAs GAS5 and MEG3 in adipose tissue obtained from subjects with obesity. Int. J. Obes. 2024, 48, 1414–1420. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubiel, D.; Naumann, M.; Dubiel, W. CSN-CRL Complexes: New Regulators of Adipogenesis. Biomolecules 2025, 15, 372. https://doi.org/10.3390/biom15030372
Dubiel D, Naumann M, Dubiel W. CSN-CRL Complexes: New Regulators of Adipogenesis. Biomolecules. 2025; 15(3):372. https://doi.org/10.3390/biom15030372
Chicago/Turabian StyleDubiel, Dawadschargal, Michael Naumann, and Wolfgang Dubiel. 2025. "CSN-CRL Complexes: New Regulators of Adipogenesis" Biomolecules 15, no. 3: 372. https://doi.org/10.3390/biom15030372
APA StyleDubiel, D., Naumann, M., & Dubiel, W. (2025). CSN-CRL Complexes: New Regulators of Adipogenesis. Biomolecules, 15(3), 372. https://doi.org/10.3390/biom15030372