
Encapsulated Ferritin-like Proteins: A Structural Perspective


Abstract
1. Introduction
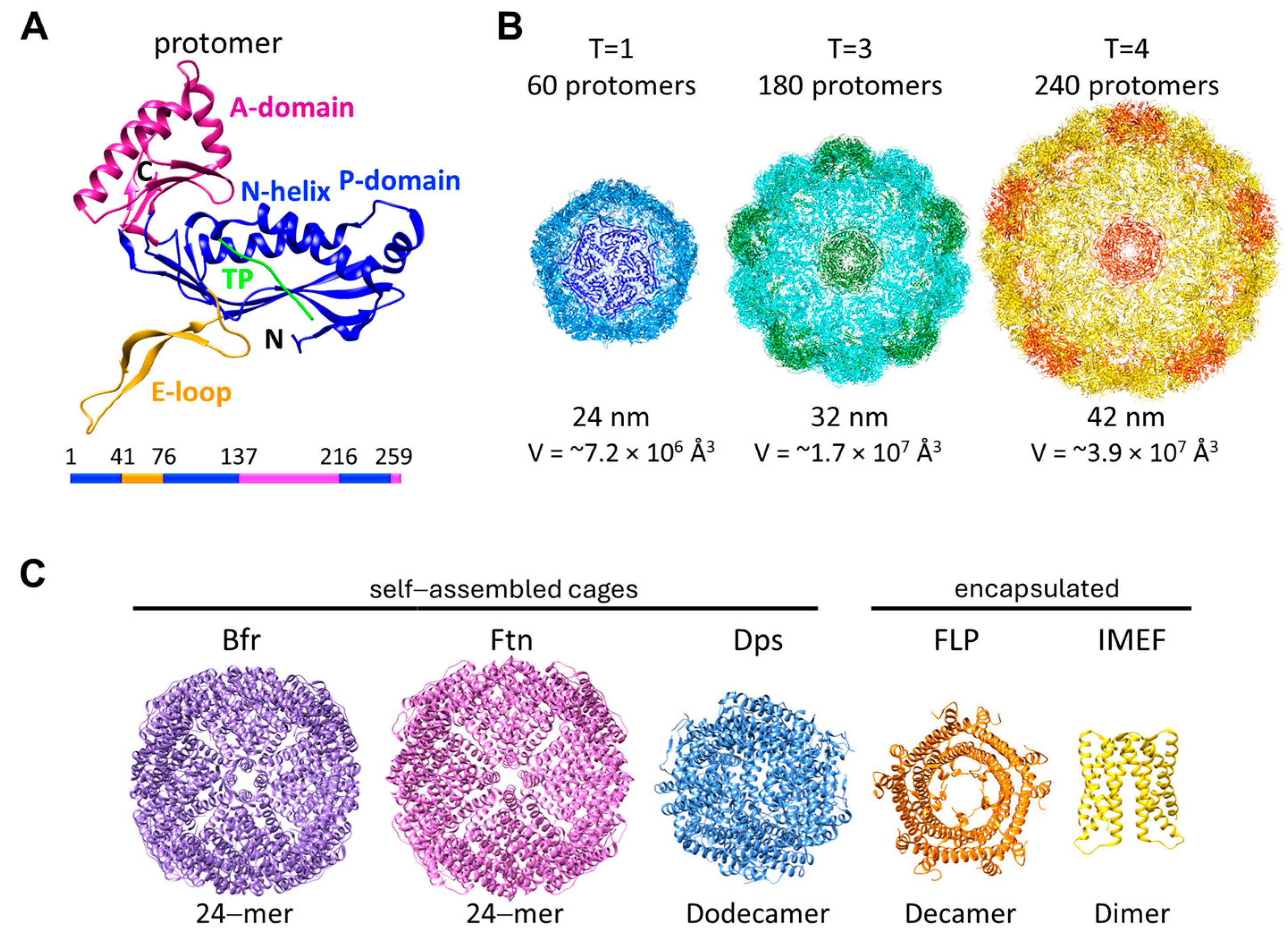
2. Encapsulins
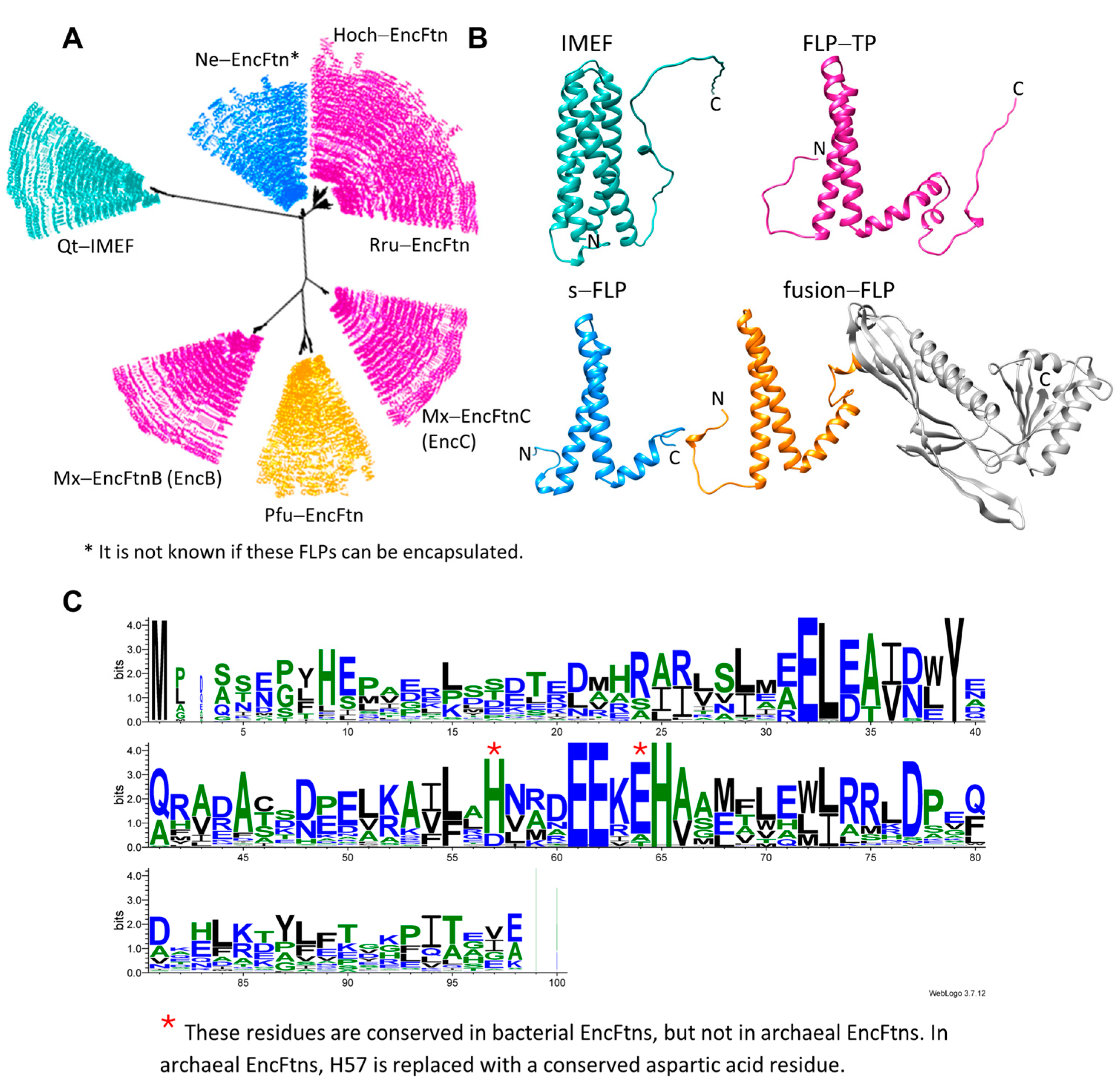
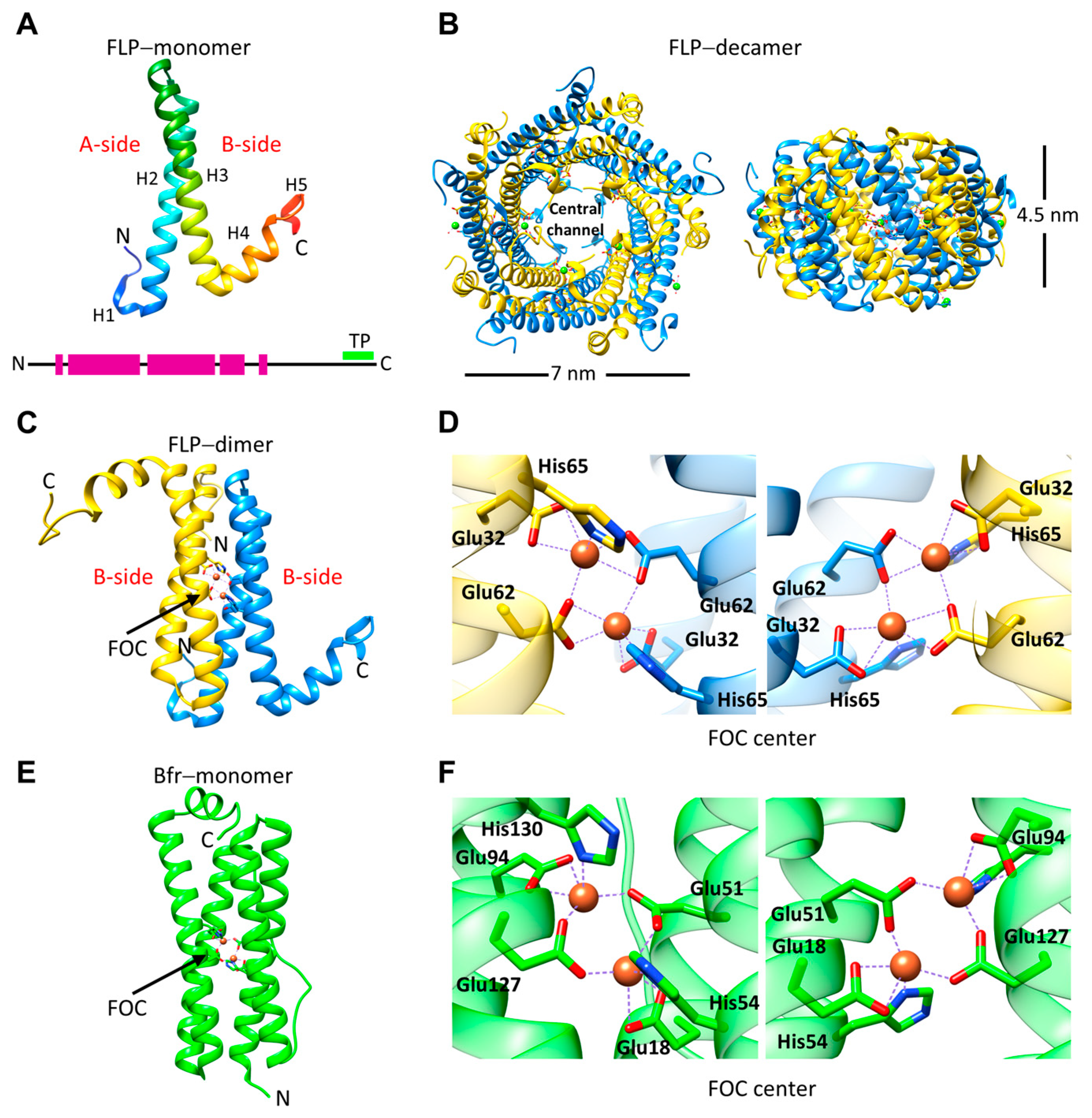
3. Encapsulated FLPs
4. Ferroxidase Activity of EncFtns and IMEF
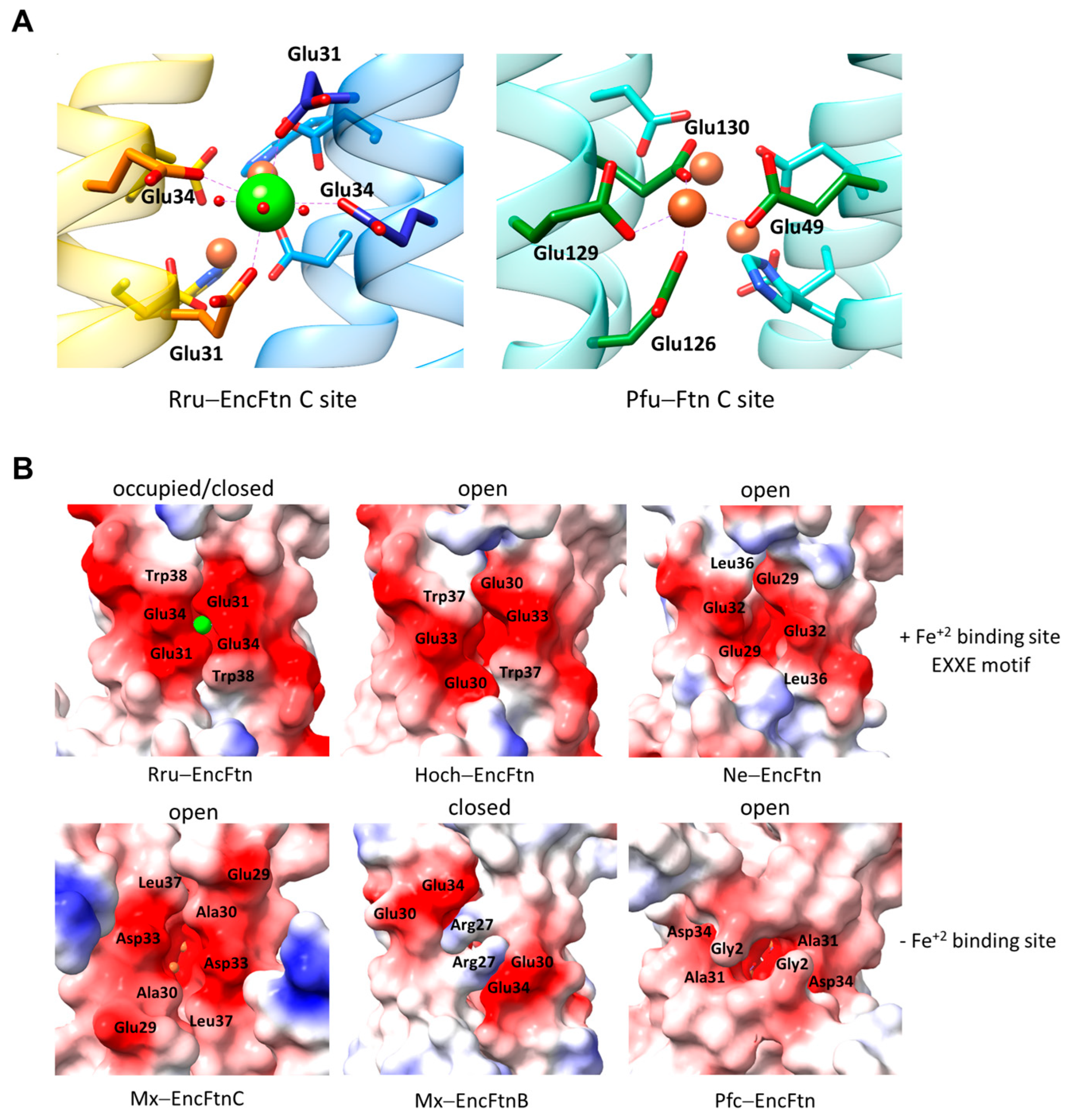
5. Additional Metal Binding Sites, Fe+2 Entry, and Fe+3 Exit Routes
6. Iron Storage
7. Iron Release
8. Implications of Unusual EncFtn and Encapsulin Systems
9. Summary
10. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Enriquez, R.P.H.; Do, T.N. Bioavailability of Metal Ions and Evolutionary Adaptation. Life 2012, 2, 274–285. [Google Scholar] [CrossRef]
- Abe, Y. Physical state of the very early Earth. Lithos 1993, 30, 223–235. [Google Scholar] [CrossRef]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R Soc. Lond. B Biol. Sci. 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.P.; Frausto da Silva, J.J.R. The Chemistry of Evolution; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Lee, J.A.; Yeung, L.Y.; McKenzie, N.R.; Yokoyama, Y.; Ozaki, K.; Lenardic, A. Two-step rise of atmospheric oxygen linked to the growth of continents. Nat. Geosci. 2016, 9, 417–424. [Google Scholar] [CrossRef]
- Andrews, S.C.; Robinson, A.K.; Rodriguez-Quinones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–911. [Google Scholar] [CrossRef]
- Luo, Y.; Henle, E.S.; Linn, S. Oxidative damage to DNA constituents by iron-mediated fenton reactions. The deoxycytidine family. J. Biol. Chem. 1996, 271, 21167–21176. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Rensing, C.; Helmann, J.D. Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 2017, 15, 338–350. [Google Scholar] [CrossRef]
- Semsey, S.; Andersson, A.M.; Krishna, S.; Jensen, M.H.; Masse, E.; Sneppen, K. Genetic regulation of fluxes: Iron homeostasis of Escherichia coli. Nucleic Acids Res. 2006, 34, 4960–4967. [Google Scholar] [CrossRef]
- Hider, R.; Aviles, M.V.; Chen, Y.L.; Latunde-Dada, G.O. The Role of GSH in Intracellular Iron Trafficking. Int. J. Mol. Sci. 2021, 22, 1278. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Kiley, P.J. Iron-containing transcription factors and their roles as sensors. Curr. Opin. Chem. Biol. 2011, 15, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.M.; Voskuil, M.I.; Gold, B.; Schoolnik, G.K.; Smith, I. ideR, An essential gene in Mycobacterium tuberculosis: Role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 2002, 70, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Sritharan, M. Iron Homeostasis in Mycobacterium tuberculosis: Mechanistic Insights into Siderophore-Mediated Iron Uptake. J. Bacteriol. 2016, 198, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.D.; Sawers, G.; Rodionov, D.A.; Johnston, A.W. The Rhizobium leguminosarum regulator IrrA affects the transcription of a wide range of genes in response to Fe availability. Mol. Genet. Genom. 2006, 275, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sangwan, I.; Lindemann, A.; Hauser, F.; Hennecke, H.; Fischer, H.M.; O’Brian, M.R. Bradyrhizobium japonicum senses iron through the status of haem to regulate iron homeostasis and metabolism. Mol. Microbiol. 2006, 60, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Delany, I.; Rappuoli, R.; Scarlato, V. Fur functions as an activator and as a repressor of putative virulence genes in Neisseria meningitidis. Mol. Microbiol. 2004, 52, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Helmann, J.D. Specificity of metal sensing: Iron and manganese homeostasis in Bacillus subtilis. J. Biol. Chem. 2014, 289, 28112–28120. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.W.; Kim, D.; Latif, H.; O’Brien, E.J.; Szubin, R.; Palsson, B.O. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 2014, 5, 4910. [Google Scholar] [CrossRef]
- Yu, C.; Genco, C.A. Fur-mediated activation of gene transcription in the human pathogen Neisseria gonorrhoeae. J. Bacteriol. 2012, 194, 1730–1742. [Google Scholar] [CrossRef]
- Ollinger, J.; Song, K.B.; Antelmann, H.; Hecker, M.; Helmann, J.D. Role of the Fur regulon in iron transport in Bacillus subtilis. J. Bacteriol. 2006, 188, 3664–3673. [Google Scholar] [CrossRef]
- Smaldone, G.T.; Revelles, O.; Gaballa, A.; Sauer, U.; Antelmann, H.; Helmann, J.D. A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J. Bacteriol. 2012, 194, 2594–2605. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Pinochet-Barros, A.; Gaballa, A.; Patel, S.J.; Arguello, J.M.; Helmann, J.D. PfeT, a P1B4-type ATPase, effluxes ferrous iron and protects Bacillus subtilis against iron intoxication. Mol. Microbiol. 2015, 98, 787–803. [Google Scholar] [CrossRef]
- Andrews, S.C. The Ferritin-like superfamily: Evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim. Biophys. Acta 2010, 1800, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Kang, S.; Reichhardt, C.; Harlen, K.; Douglas, T. The ferritin superfamily: Supramolecular templates for materials synthesis. Biochim. Biophys. Acta 2010, 1800, 834–845. [Google Scholar] [CrossRef]
- Carrondo, M.A. Ferritins, iron uptake and storage from the bacterioferritin viewpoint. EMBO J. 2003, 22, 1959–1968. [Google Scholar] [CrossRef]
- He, D.; Piergentili, C.; Ross, J.; Tarrant, E.; Tuck, L.R.; Mackay, C.L.; McIver, Z.; Waldron, K.J.; Clarke, D.J.; Marles-Wright, J. Conservation of the structural and functional architecture of encapsulated ferritins in bacteria and archaea. Biochem. J. 2019, 476, 975–989. [Google Scholar] [CrossRef]
- Honarmand Ebrahimi, K.; Hagedoorn, P.L.; Hagen, W.R. Unity in the biochemistry of the iron-storage proteins ferritin and bacterioferritin. Chem. Rev. 2015, 115, 295–326. [Google Scholar] [CrossRef] [PubMed]
- Piergentili, C.; Ross, J.; He, D.; Gallagher, K.J.; Stanley, W.A.; Adam, L.; Mackay, C.L.; Basle, A.; Waldron, K.J.; Clarke, D.J.; et al. Dissecting the structural and functional roles of a putative metal entry site in encapsulated ferritins. J. Biol. Chem. 2020, 295, 15511–15526. [Google Scholar] [CrossRef]
- Rivera, M. Bacterioferritin: Structure, Dynamics, and Protein-Protein Interactions at Play in Iron Storage and Mobilization. Acc. Chem. Res. 2017, 50, 331–340. [Google Scholar] [CrossRef]
- Williams, S.M.; Chatterji, D. Dps Functions as a Key Player in Bacterial Iron Homeostasis. ACS Omega 2023, 8, 34299–34309. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Roberts, J.N.; Hardiman, E.M.; Singh, R.; Eltis, L.D.; Bugg, T.D. Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 2011, 50, 5096–5107. [Google Scholar] [CrossRef]
- Contreras, H.; Joens, M.S.; McMath, L.M.; Le, V.P.; Tullius, M.V.; Kimmey, J.M.; Bionghi, N.; Horwitz, M.A.; Fitzpatrick, J.A.; Goulding, C.W. Characterization of a Mycobacterium tuberculosis nanocompartment and its potential cargo proteins. J. Biol. Chem. 2014, 289, 18279–18289. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Fontana, J.; Nemecek, D.; Cheng, N.; Aksyuk, A.A.; Heymann, J.B.; Winkler, D.C.; Lam, A.S.; Wall, J.S.; Steven, A.C.; et al. A virus capsid-like nanocompartment that stores iron and protects bacteria from oxidative stress. EMBO J. 2014, 33, 1896–1911. [Google Scholar] [CrossRef]
- Nichols, R.J.; LaFrance, B.; Phillips, N.R.; Radford, D.R.; Oltrogge, L.M.; Valentin-Alvarado, L.E.; Bischoff, A.J.; Nogales, E.; Savage, D.F. Discovery and characterization of a novel family of prokaryotic nanocompartments involved in sulfur metabolism. Elife 2021, 10, e59288. [Google Scholar] [CrossRef]
- Rahmanpour, R.; Bugg, T.D. Assembly in vitro of Rhodococcus jostii RHA1 encapsulin and peroxidase DypB to form a nanocompartment. FEBS J. 2013, 280, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.H.; Grimes, J.M.; Stuart, D.I. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 2005, 15, 655–663. [Google Scholar] [CrossRef]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef]
- Pietila, M.K.; Laurinmaki, P.; Russell, D.A.; Ko, C.C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc. Natl. Acad. Sci. USA 2013, 110, 10604–10609. [Google Scholar] [CrossRef]
- Sutter, M.; Boehringer, D.; Gutmann, S.; Gunther, S.; Prangishvili, D.; Loessner, M.J.; Stetter, K.O.; Weber-Ban, E.; Ban, N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 2008, 15, 939–947. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Duda, R.L.; Teschke, C.M. The amazing HK97 fold: Versatile results of modest differences. Curr. Opin. Virol. 2019, 36, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Akita, F.; Chong, K.T.; Tanaka, H.; Yamashita, E.; Miyazaki, N.; Nakaishi, Y.; Suzuki, M.; Namba, K.; Ono, Y.; Tsukihara, T.; et al. The crystal structure of a virus-like particle from the hyperthermophilic archaeon Pyrococcus furiosus provides insight into the evolution of viruses. J. Mol. Biol. 2007, 368, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Orlando, B.J.; Verdegaal, A.A.; Chambers, M.G.; Gardener, J.; Bell, D.C.; Birrane, G.; Liao, M.; Silver, P.A. Large protein organelles form a new iron sequestration system with high storage capacity. Elife 2019, 8, e46070. [Google Scholar] [CrossRef] [PubMed]
- Loncar, N.; Rozeboom, H.J.; Franken, L.E.; Stuart, M.C.A.; Fraaije, M.W. Structure of a robust bacterial protein cage and its application as a versatile biocatalytic platform through enzyme encapsulation. Biochem. Biophys. Res. Commun. 2020, 529, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Wiryaman, T.; Toor, N. Cryo-EM structure of a thermostable bacterial nanocompartment. IUCrJ 2021, 8, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Wang, B.; Winkler, D.C.; Watts, N.R.; Steven, A.C.; Wingfield, P.T. Structural characterization of the Myxococcus xanthus encapsulin and ferritin-like cargo system gives insight into its iron storage mechanism. Structure 2022, 30, 551–563.e554. [Google Scholar] [CrossRef]
- Giessen, T.W. Encapsulins. Annu. Rev. Biochem. 2022, 91, 353–380. [Google Scholar] [CrossRef] [PubMed]
- Altenburg, W.J.; Rollins, N.; Silver, P.A.; Giessen, T.W. Exploring targeting peptide-shell interactions in encapsulin nanocompartments. Sci. Rep. 2021, 11, 4951. [Google Scholar] [CrossRef]
- Cassidy-Amstutz, C.; Oltrogge, L.; Going, C.C.; Lee, A.; Teng, P.; Quintanilla, D.; East-Seletsky, A.; Williams, E.R.; Savage, D.F. Identification of a Minimal Peptide Tag for in Vivo and in Vitro Loading of Encapsulin. Biochemistry 2016, 55, 3461–3468. [Google Scholar] [CrossRef]
- Almeida, A.V.; Carvalho, A.J.; Pereira, A.S. Encapsulin nanocages: Protein encapsulation and iron sequestration. Coord. Chem. Rev. 2021, 448, 214188. [Google Scholar] [CrossRef]
- Andreas, M.P.; Giessen, T.W. Large-scale computational discovery and analysis of virus-derived microbial nanocompartments. Nat. Commun. 2021, 12, 4748. [Google Scholar] [CrossRef]
- Kashif-Khan, N.; Savva, R.; Frank, S. Mining metagenomics data for novel bacterial nanocompartments. NAR Genom. Bioinform. 2024, 6, lqae025. [Google Scholar] [CrossRef]
- He, D.; Hughes, S.; Vanden-Hehir, S.; Georgiev, A.; Altenbach, K.; Tarrant, E.; Mackay, C.L.; Waldron, K.J.; Clarke, D.J.; Marles-Wright, J. Structural characterization of encapsulated ferritin provides insight into iron storage in bacterial nanocompartments. Elife 2016, 5, e18972. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Lambert, T.; Piergentili, C.; He, D.; Waldron, K.J.; Mackay, C.L.; Marles-Wright, J.; Clarke, D.J. Mass spectrometry reveals the assembly pathway of encapsulated ferritins and highlights a dynamic ferroxidase interface. Chem. Commun. 2020, 56, 3417–3420. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. Widespread distribution of encapsulin nanocompartments reveals functional diversity. Nat. Microbiol. 2017, 2, 17029. [Google Scholar] [CrossRef]
- Nordlund, P.; Eklund, H. Di-iron-carboxylate proteins. Curr. Opin. Struct. Biol. 1995, 5, 758–766. [Google Scholar] [CrossRef]
- Orban, K.; Finkel, S.E. Dps Is a Universally Conserved Dual-Action DNA-Binding and Ferritin Protein. J. Bacteriol. 2022, 204, e0003622. [Google Scholar] [CrossRef] [PubMed]
- LaFrance, B.; Cassidy-Amstutz, C.; Nichols, R.J.; Oltrogge, L.M.; Nogales, E.; Savage, D.F. The encapsulin from Thermatoga maritima is a flavoprotein with a symmetry matched ferritin-like cargo protein. Sci. Rep. 2021, 11, 22810. [Google Scholar] [CrossRef]
- Bou-Abdallah, F. The iron redox and hydrolysis chemistry of the ferritins. Biochim. Biophys. Acta 2010, 1800, 719–731. [Google Scholar] [CrossRef]
- Bou-Abdallah, F.; McNally, J.; Liu, X.X.; Melman, A. Oxygen catalyzed mobilization of iron from ferritin by iron(III) chelate ligands. Chem. Commun. 2011, 47, 731–733. [Google Scholar] [CrossRef]
- Bradley, J.M.; Moore, G.R.; Le Brun, N.E. Mechanisms of iron mineralization in ferritins: One size does not fit all. J. Biol. Inorg. Chem. 2014, 19, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Melman, A.; Bou-Abdallah, F. Iron mineralization and core dissociation in mammalian homopolymeric H-ferritin: Current understanding and future perspectives. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129700. [Google Scholar] [CrossRef]
- Ebrahimi, K.H.; Bill, E.; Hagedoorn, P.L.; Hagen, W.R. Spectroscopic evidence for the role of a site of the di-iron catalytic center of ferritins in tuning the kinetics of Fe(ii) oxidation. Mol. Biosyst. 2016, 12, 3576–3588. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Santambrogio, P.; Levi, S.; Arosio, P.; Chasteen, N.D. Unique iron binding and oxidation properties of human mitochondrial ferritin: A comparative analysis with Human H-chain ferritin. J. Mol. Biol. 2005, 347, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Sato, D.; Yanagida, Y.; Aoki, E.; Fujiwara, K.; Yoshimura, H.; Ikeguchi, M. Morphological difference of Escherichia coli non-heme ferritin iron cores reconstituted in the presence and absence of inorganic phosphate. J. Biol. Inorg. Chem. 2022, 27, 583–594. [Google Scholar] [CrossRef]
- Carter-Fenk, K.; Liu, M.; Pujal, L.; Loipersberger, M.; Tsanai, M.; Vernon, R.M.; Forman-Kay, J.D.; Head-Gordon, M.; Heidar-Zadeh, F.; Head-Gordon, T. The Energetic Origins of Pi-Pi Contacts in Proteins. J. Am. Chem. Soc. 2023, 145, 24836–24851. [Google Scholar] [CrossRef]
- Tatur, J.; Hagen, W.R.; Matias, P.M. Crystal structure of the ferritin from the hyperthermophilic archaeal anaerobe Pyrococcus furiosus. J. Biol. Inorg. Chem. 2007, 12, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Rui, H.; Kumar, R.; Eshelman, K.; Lovell, S.; Battaile, K.P.; Im, W.; Rivera, M. Concerted motions networking pores and distant ferroxidase centers enable bacterioferritin function and iron traffic. Biochemistry 2015, 54, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.M.; Svistunenko, D.A.; Lawson, T.L.; Hemmings, A.M.; Moore, G.R.; Le Brun, N.E. Three Aromatic Residues are Required for Electron Transfer during Iron Mineralization in Bacterioferritin. Angew. Chem. Weinh. Bergstr. Ger. 2015, 127, 14976–14980. [Google Scholar] [CrossRef]
- Crow, A.; Lawson, T.L.; Lewin, A.; Moore, G.R.; Le Brun, N.E. Structural basis for iron mineralization by bacterioferritin. J. Am. Chem. Soc. 2009, 131, 6808–6813. [Google Scholar] [CrossRef] [PubMed]
- Waldo, G.S.; Ling, J.; Sanders-Loehr, J.; Theil, E.C. Formation of an Fe(III)-tyrosinate complex during biomineralization of H-subunit ferritin. Science 1993, 259, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.M.; Svistunenko, D.A.; Moore, G.R.; Le Brun, N.E. Tyr25, Tyr58 and Trp133 of Escherichia coli bacterioferritin transfer electrons between iron in the central cavity and the ferroxidase centre. Metallomics 2017, 9, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, K.H.; Hagedoorn, P.L.; Hagen, W.R. A conserved tyrosine in ferritin is a molecular capacitor. Chembiochem 2013, 14, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Honarmand Ebrahimi, K.; Bill, E.; Hagedoorn, P.L.; Hagen, W.R. Spectroscopic evidence for the presence of a high-valent Fe(IV) species in the ferroxidase reaction of an archaeal ferritin. FEBS Lett. 2017, 591, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Cassidy-Amstutz, C.; Chaijarasphong, T.; Savage, D.F. Encapsulins: Molecular biology of the shell. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; McIver, Z.; Lambert, T.; Piergentili, C.; Gallagher, K.J.; Bird, J.E.; Cruickshank, F.L.; Zarazúa-Arvizu, E.; Horsfall, L.E.; Waldron, K.J.; et al. Pore dynamics and asymmetric cargo loading in an encapsulin nanocompartment. Sci. Adv. 2021, 8, eabj4461. [Google Scholar] [CrossRef]
- Wiryaman, T.; Toor, N. Recent advances in the structural biology of encapsulin bacterial nanocompartments. J. Struct. Biol. X 2022, 6, 100062. [Google Scholar] [CrossRef]
- Xiong, X.; Sun, C.; Vago, F.S.; Klose, T.; Zhu, J.; Jiang, W. Cryo-EM Structure of Heterologous Protein Complex Loaded Thermotoga Maritima Encapsulin Capsid. Biomolecules 2020, 10, 1342. [Google Scholar] [CrossRef]
- Pozzi, C.; Di Pisa, F.; Lalli, D.; Rosa, C.; Theil, E.; Turano, P.; Mangani, S. Time-lapse anomalous X-ray diffraction shows how Fe(2+) substrate ions move through ferritin protein nanocages to oxidoreductase sites. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 941–953. [Google Scholar] [CrossRef]
- Khare, G.; Nangpal, P.; Tyagi, A.K. Differential Roles of Iron Storage Proteins in Maintaining the Iron Homeostasis in Mycobacterium tuberculosis. PLoS ONE 2017, 12, e0169545. [Google Scholar] [CrossRef] [PubMed]
- Frolow, F.; Kalb, A.J.; Yariv, J. Structure of a unique twofold symmetric haem-binding site. Nat. Struct. Biol. 1994, 1, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Pysz, M.A.; Conners, S.B.; Montero, C.I.; Shockley, K.R.; Johnson, M.R.; Ward, D.E.; Kelly, R.M. Transcriptional analysis of biofilm formation processes in the anaerobic, hyperthermophilic bacterium Thermotoga maritima. Appl. Environ. Microbiol. 2004, 70, 6098–6112. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Choi, J.; Lee, S.; Hyun, H.; Lee, K.; Cho, K. Mutants defective in the production of encapsulin show a tan-phase-locked phenotype in Myxococcus xanthus. J. Microbiol. 2019, 57, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Watts, N.R.; Conway, J.F.; Wingfield, P.T. Myxococcus xanthus encapsulin cargo protein EncD is a flavin-binding protein with ferric reductase activity. Proc. Natl. Acad. Sci. USA 2024, 121, e2400426121. [Google Scholar] [CrossRef] [PubMed]
- Lien, K.A.; Dinshaw, K.; Nichols, R.J.; Cassidy-Amstutz, C.; Knight, M.; Singh, R.; Eltis, L.D.; Savage, D.F.; Stanley, S.A. A nanocompartment system contributes to defense against oxidative stress in Mycobacterium tuberculosis. Elife 2021, 10, e74358. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.; Vidal, X.; Sunna, A.; Care, A. Bioengineering a Light-Responsive Encapsulin Nanoreactor: A Potential Tool for In Vitro Photodynamic Therapy. ACS Appl. Mater. Interfaces 2021, 13, 7977–7986. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. Converting a Natural Protein Compartment into a Nanofactory for the Size-Constrained Synthesis of Antimicrobial Silver Nanoparticles. ACS Synth. Biol. 2016, 5, 1497–1504. [Google Scholar] [CrossRef]
- Torra, J.; Lafaye, C.; Signor, L.; Aumonier, S.; Flors, C.; Shu, X.; Nonell, S.; Gotthard, G.; Royant, A. Tailing miniSOG: Structural bases of the complex photophysics of a flavin-binding singlet oxygen photosensitizing protein. Sci. Rep. 2019, 9, 2428. [Google Scholar] [CrossRef]
- Moon, H.; Lee, J.; Kim, H.; Heo, S.; Min, J.; Kang, S. Genetically engineering encapsulin protein cage nanoparticle as a SCC-7 cell targeting optical nanoprobe. Biomater. Res. 2014, 18, 21. [Google Scholar] [CrossRef]
- Khaleeq, S.; Sengupta, N.; Kumar, S.; Patel, U.R.; Rajmani, R.S.; Reddy, P.; Pandey, S.; Singh, R.; Dutta, S.; Ringe, R.P.; et al. Neutralizing Efficacy of Encapsulin Nanoparticles against SARS-CoV-2 Variants of Concern. Viruses 2023, 15, 346. [Google Scholar] [CrossRef] [PubMed]
- Piraner, D.I.; Farhadi, A.; Davis, H.C.; Wu, D.; Maresca, D.; Szablowski, J.O.; Shapiro, M.G. Going Deeper: Biomolecular Tools for Acoustic and Magnetic Imaging and Control of Cellular Function. Biochemistry 2017, 56, 5202–5209. [Google Scholar] [CrossRef] [PubMed]
- Sigmund, F.; Massner, C.; Erdmann, P.; Stelzl, A.; Rolbieski, H.; Desai, M.; Bricault, S.; Worner, T.P.; Snijder, J.; Geerlof, A.; et al. Bacterial encapsulins as orthogonal compartments for mammalian cell engineering. Nat. Commun. 2018, 9, 1990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Chu, C.; Zhou, Z.; Chen, B.; Pang, X.; Lin, G.; Lin, H.; Guo, Y.; Ren, E.; et al. Genetically engineered magnetic nanocages for cancer magneto-catalytic theranostics. Nat. Commun. 2020, 11, 5421. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Name | UniProt Reference | Organism | Type | Form | Encapsulin T-Number | Organism Type | Amino Acids |
|---|---|---|---|---|---|---|---|---|
| 5da5 | Rru-EncFtn, Rru_A0973 | Q2RVS1 | Rhodospirillum rubrum | FLP-TP | Fe-bound | 1 1 | Bacteria, anaerobic, photosynthetic | 140 |
| 3k6c | Ne-EncFtn * NE0167 | Q82XT5 | Nitrosomonas europaea | s-FLP | apo | ? 4 | Bacteria, anaerobic, nitrifying | 95 |
| 5n5e | Pfc-EncFtn PFC_05175 | Q8U1L4 | Pyrococcus furiosus | fusion–FLP | Fe-bound | 3 (? 4) | Archaea, anaerobic, hyperthermophile | 345 2 1–109 2 |
| 5n5f | Hoch-EncFtn Hoch_3836 | D0LZ73 | Haliangium ochraceum | FLP-TP | apo | 1 | Bacteria, aerobic, myxobacteria | 131 |
| 7s5c, 7s5k | Mx-EncFtnB * EncB, MXAN_3557 | Q1D6H3 | Myxococcus xanthus | FLP-TP | Fe-bound | 3 | Bacteria, aerobic, myxobacteria | 158 3 |
| 7s8t | Mx-EncFtnC * EncC, MXAN_4464 | Q1D3Y8 | Myxococcus xanthus | FLP-TP | Fe-bound | 3 | Bacteria, aerobic, myxobacteria | 130 |
| 6n63 | Qt-IMEF * Qs IMEF, Qs IMEF cargo, QY95_01593 | A0A0F5HNH9 | Quasibacillus thermotolerans | IMEF | Fe-bound | 4 | Bacteria, thermophilic, firmicute, facultative anaerobe | 192 |
| FLP | Conserved C–Terminal TP Sequences |
|---|---|
| FLP-TP | |
| R. rubrum | SLGIGSLK(R) |
| H.ochraceum | SLGIGSLK |
| M. xanthus (EncC) | RLTVGS(N)L(M) |
| M. xanthus (EncB) | PLTV(I)GS(T)L |
| T. maritima | GG(S)D(S)LG(D)I(L)R(G)K(S)L |
| Fusion–FLP | RSLLKHLP 1 |
| s-FLP | LFTD(E/N)KPIAHE 2 |
| IMEF | G(S/T)F(L)TVGSLI(L) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eren, E.; Watts, N.R.; Montecinos, F.; Wingfield, P.T. Encapsulated Ferritin-like Proteins: A Structural Perspective. Biomolecules 2024, 14, 624. https://doi.org/10.3390/biom14060624
Eren E, Watts NR, Montecinos F, Wingfield PT. Encapsulated Ferritin-like Proteins: A Structural Perspective. Biomolecules. 2024; 14(6):624. https://doi.org/10.3390/biom14060624
Chicago/Turabian StyleEren, Elif, Norman R. Watts, Felipe Montecinos, and Paul T. Wingfield. 2024. "Encapsulated Ferritin-like Proteins: A Structural Perspective" Biomolecules 14, no. 6: 624. https://doi.org/10.3390/biom14060624
APA StyleEren, E., Watts, N. R., Montecinos, F., & Wingfield, P. T. (2024). Encapsulated Ferritin-like Proteins: A Structural Perspective. Biomolecules, 14(6), 624. https://doi.org/10.3390/biom14060624