
Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) in Keratinocytes by Endogenous Fatty Acids

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Keratinocyte Culture
2.2. HPLC Fractionation
2.3. Quantitative Polymerase Chain Reaction (qPCR)
2.4. ChIP Assays
2.5. HRAS Infection
2.6. Reporter Assays
2.7. Quantitative Western Blot Analyses
2.8. Flow Cytometry
2.9. Coulter Counting and BrdU Labeling of Keratinocytes
2.10. GC/MS
2.11. Topical Tetradecanoylphorbol-13-Acetate (TPA)Treatment in Wild-Type and Scd1-Null Mice
2.12. UV-Induced Skin Cancer Using Pparb/d-Null Crossed with SKH Mice
3. Results
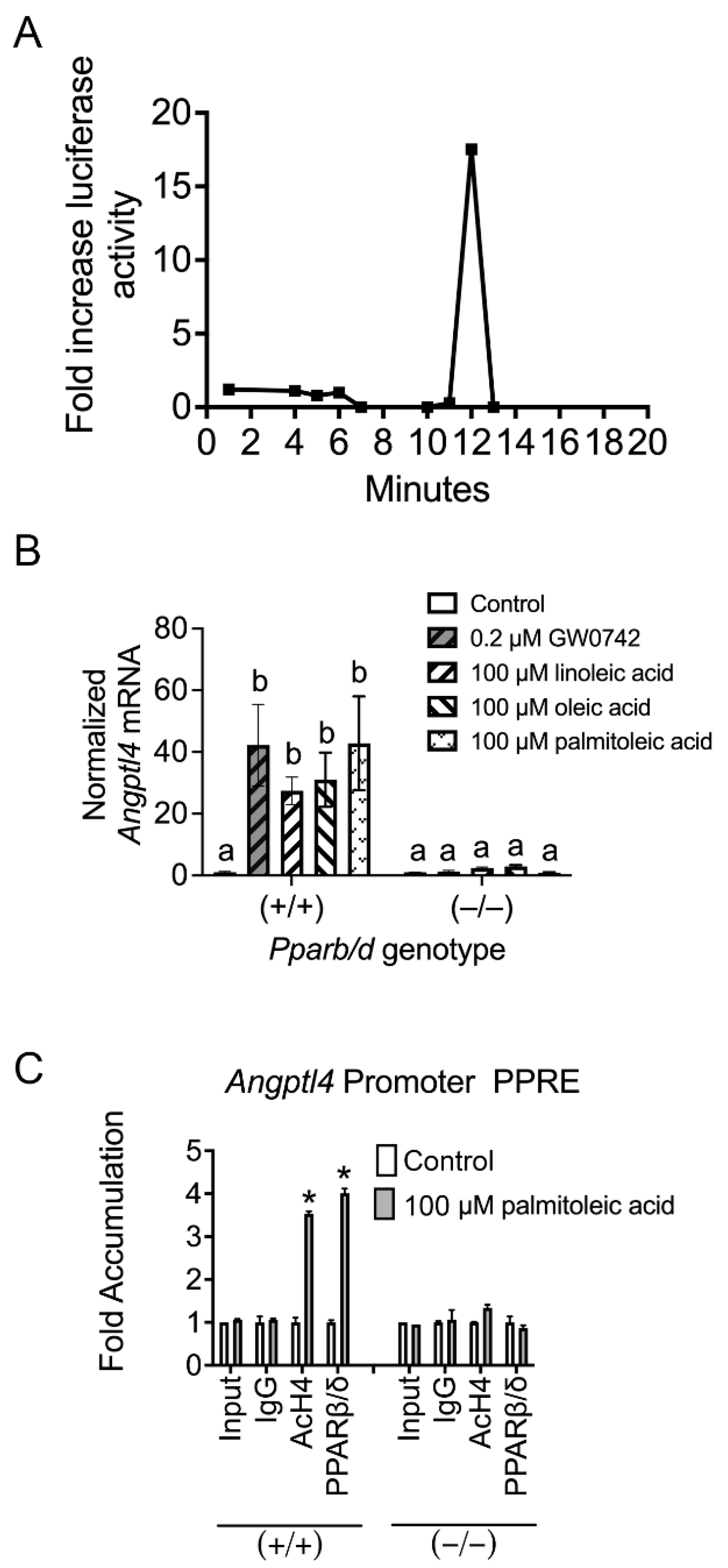
3.1. PPARβ/δ Activity Is Increased in the Cytosol of Keratinocytes Post-TPA Treatment
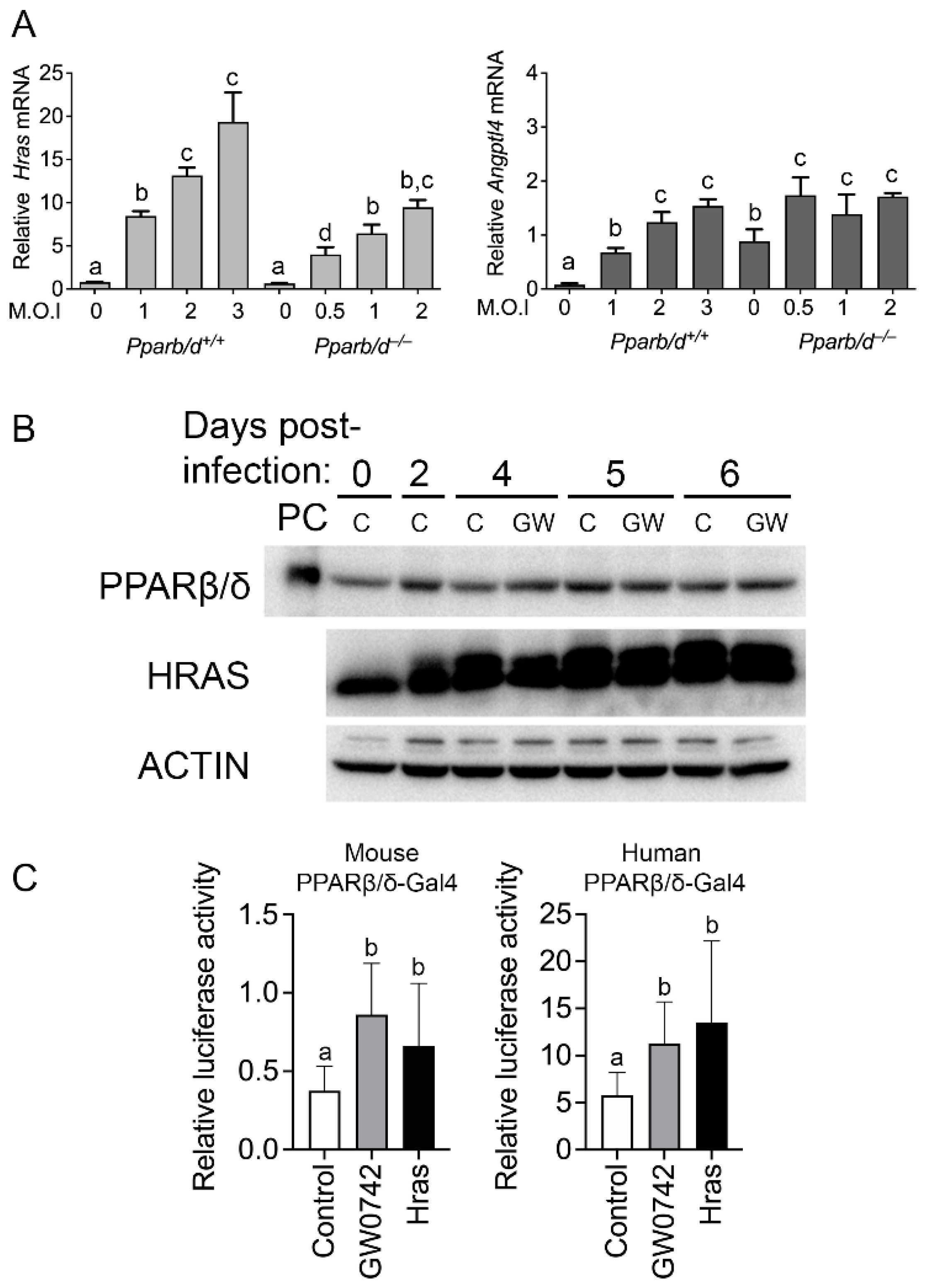
3.2. HRAS Activates PPARβ/δ
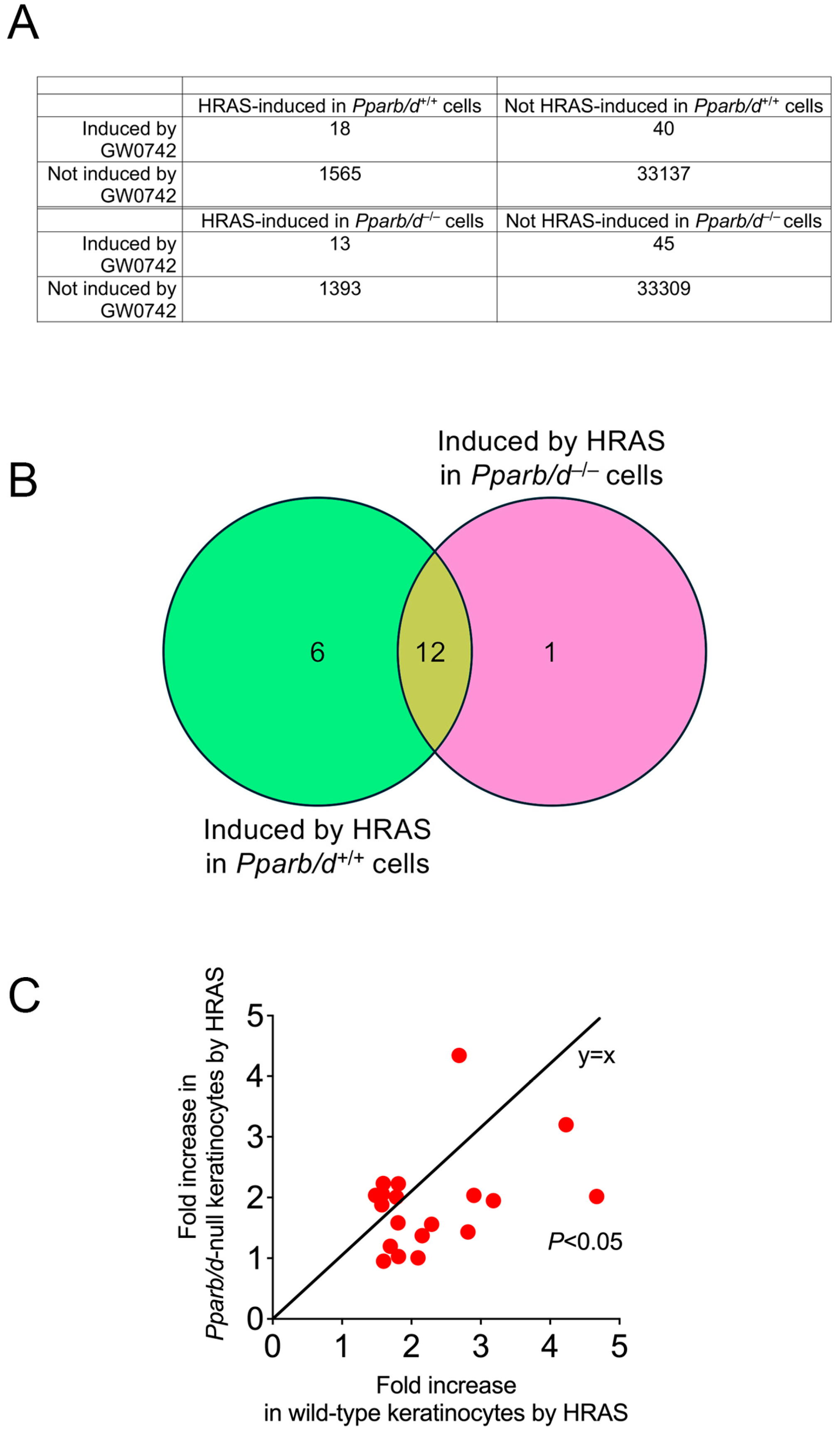
3.3. Bioinformatic Analyses Reveals Role of PPARβ/δ in Lipid Metabolism
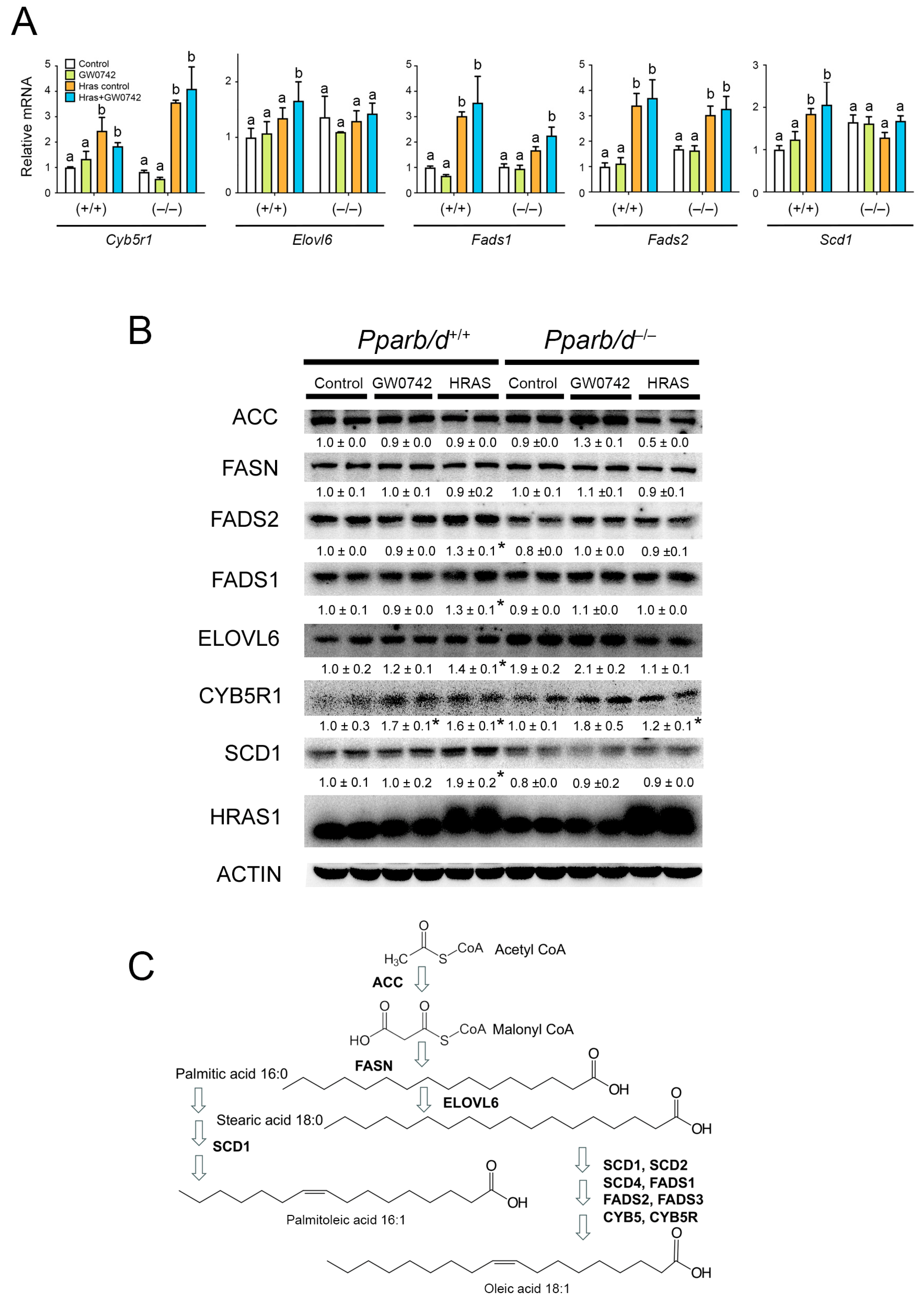
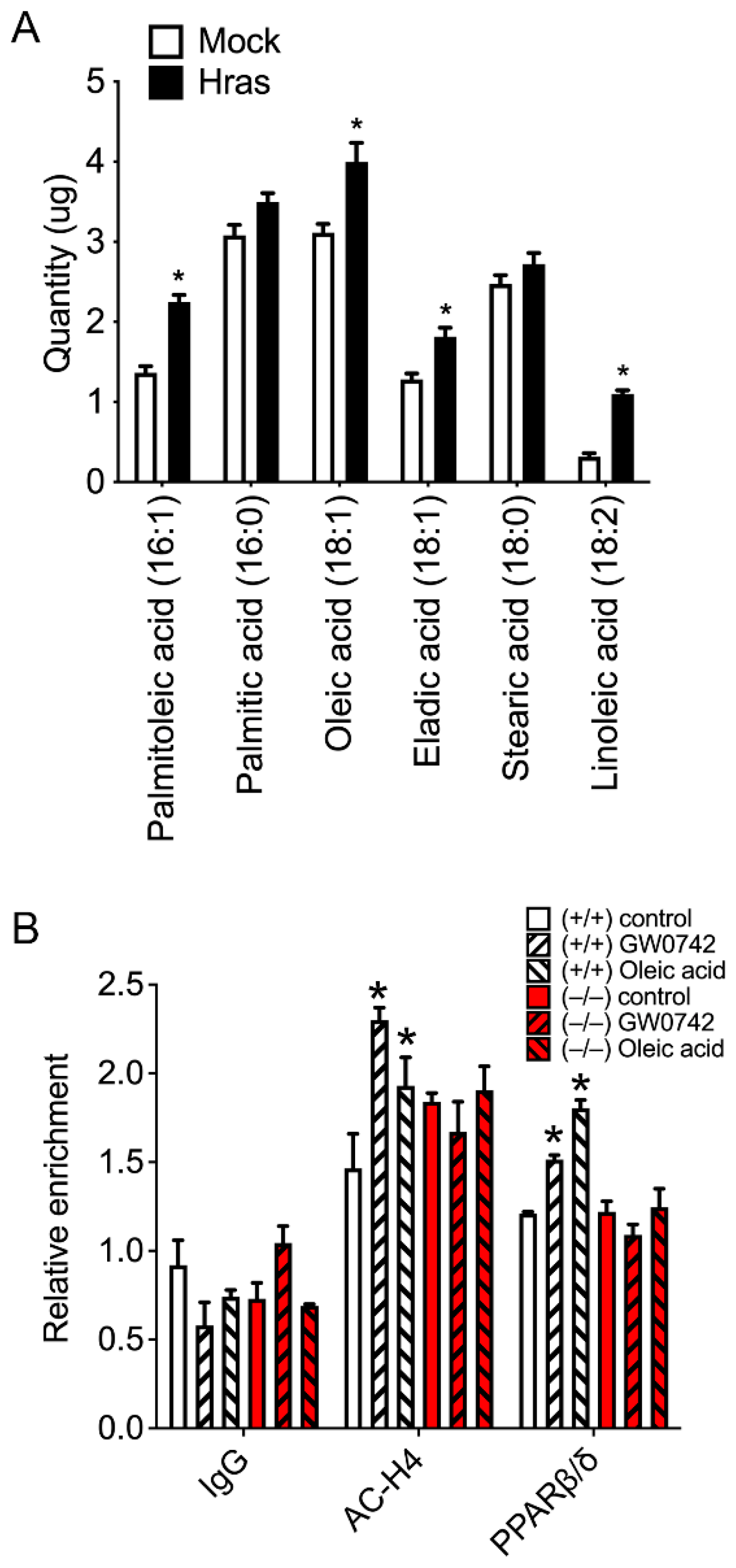
3.4. HRAS Increases Oleic and Palmitoleic Acid Levels in Primary Keratinocytes and Activates PPARβ/δ
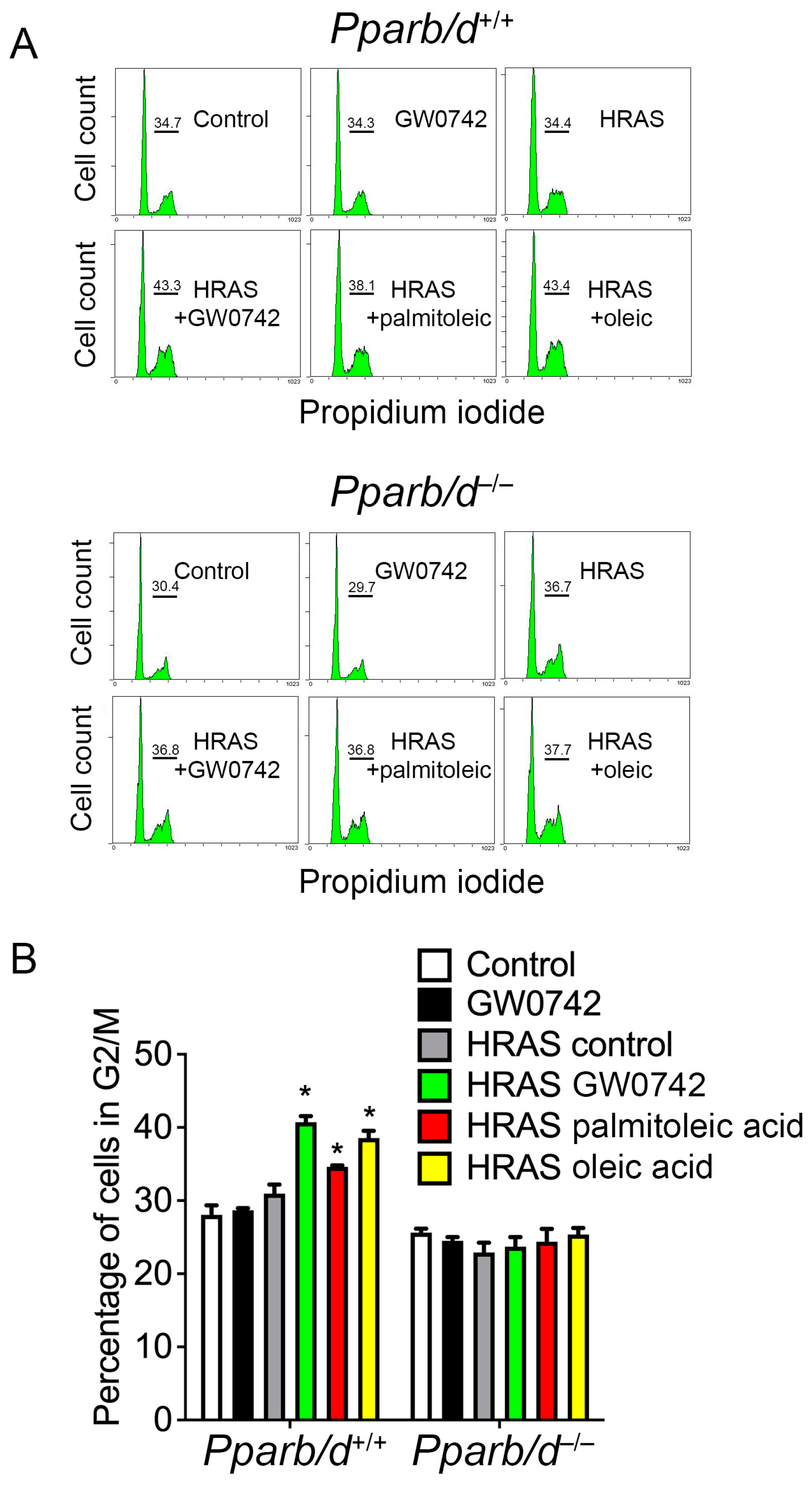
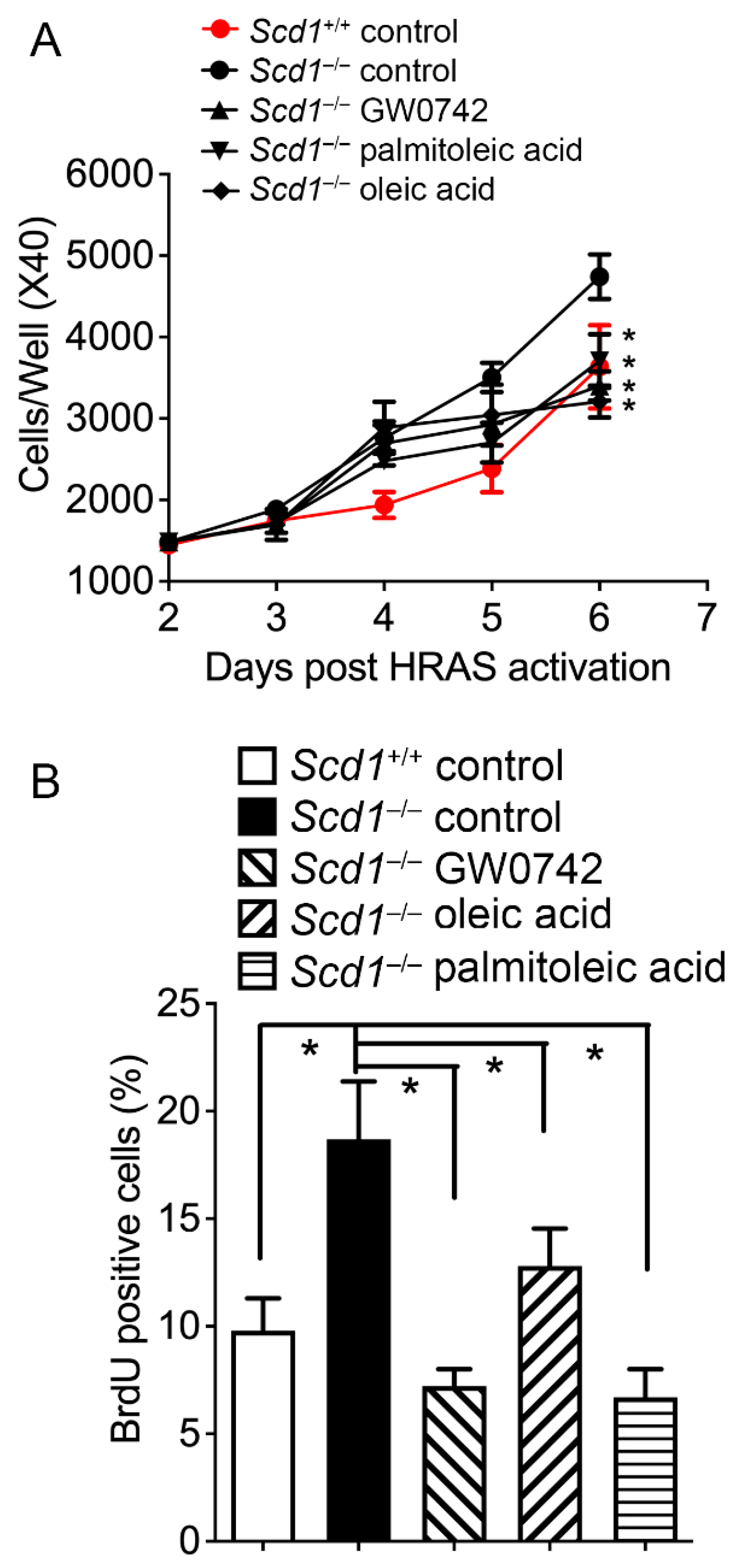
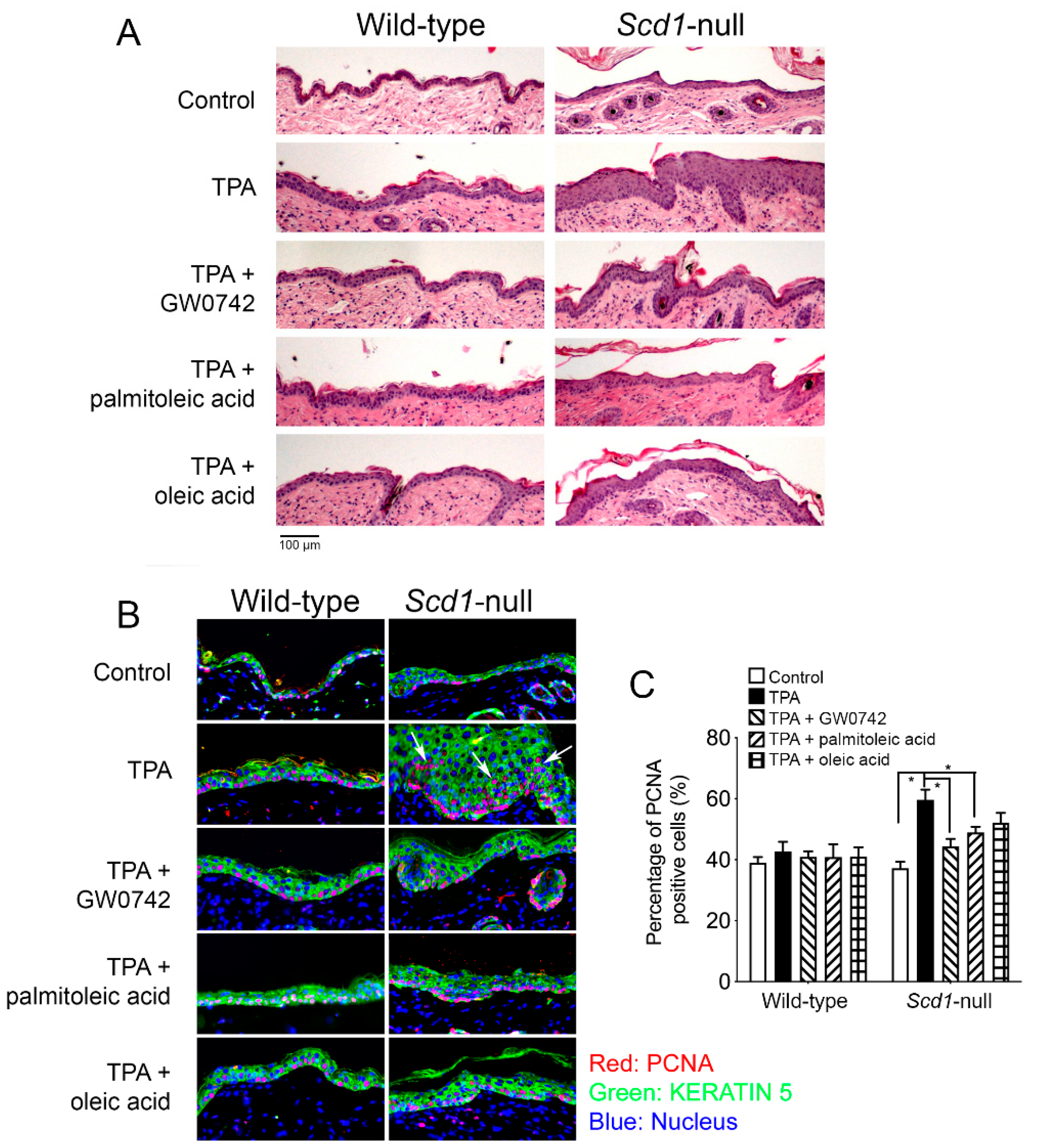
3.5. Oleic Acid and Palmitoleic Acid Inhibit Hyperplasia in Keratinocytes
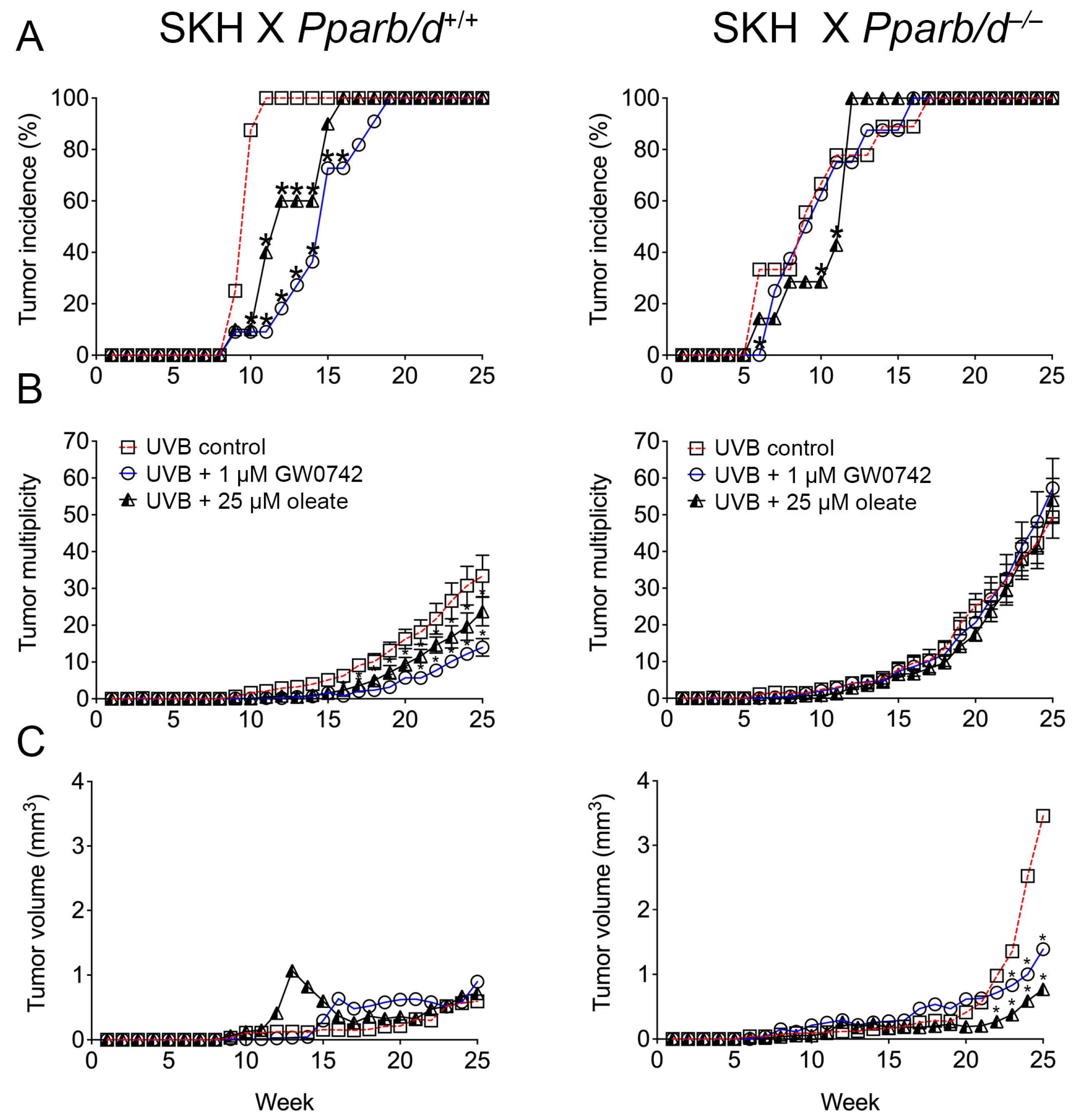
3.6. PPARβ/δ-Dependent Inhibition of UVB-Induced Skin Cancer with Oleic Acid and Synthetic Ligand
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Green, S.; Wahli, W. Peroxisome proliferator-activated receptors: Finding the orphan a home. Mol. Cell Endocrinol. 1994, 100, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Biddie, S.C.; Hager, G.L. Glucocorticoid receptor dynamics and gene regulation. Stress 2009, 12, 193–205. [Google Scholar] [CrossRef]
- Biddie, S.C.; John, S.; Hager, G.L. Genome-wide mechanisms of nuclear receptor action. Trends Endocrinol. Metab. 2010, 21, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hager, G.L.; Varticovski, L. Chromatin in time and space. Biochim. Biophys. Acta 2012, 1819, 631. [Google Scholar] [CrossRef] [PubMed]
- Khozoie, C.; Borland, M.G.; Zhu, B.; Baek, S.; John, S.; Hager, G.L.; Shah, Y.M.; Gonzalez, F.J.; Peters, J.M. Analysis of the peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) cistrome reveals novel co-regulatory role of ATF4. BMC Genom. 2012, 13, 665. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Girroir, E.E.; Hollingshead, H.E.; He, P.; Zhu, B.; Perdew, G.H.; Peters, J.M. Quantitative expression patterns of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) protein in mice. Biochem. Biophys. Res. Commun. 2008, 371, 456–461. [Google Scholar] [CrossRef]
- Modica, S.; Gofflot, F.; Murzilli, S.; D’Orazio, A.; Salvatore, L.; Pellegrini, F.; Nicolucci, A.; Tognoni, G.; Copetti, M.; Valanzano, R.; et al. The intestinal nuclear receptor signature with epithelial localization patterns and expression modulation in tumors. Gastroenterology 2010, 138, 636–648. [Google Scholar] [CrossRef]
- Montagner, A.; Wahli, W. Contributions of peroxisome proliferator-activated receptor β/δ to skin health and disease. Biomol. Concepts 2013, 4, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Kim, D.J.; Bility, M.T.; Borland, M.G.; Zhu, B.; Gonzalez, F.J. Regulatory mechanisms mediated by peroxisome proliferator-activated receptor-β/δ in skin cancer. Mol. Carcinog. 2019, 58, 1612–1622. [Google Scholar] [CrossRef]
- Bility, M.T.; Devlin-Durante, M.K.; Blazanin, N.; Glick, A.B.; Ward, J.M.; Kang, B.H.; Kennett, M.J.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits chemically-induced skin tumorigenesis. Carcinogenesis 2008, 29, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Bility, M.T.; Zhu, B.; Kang, B.H.; Gonzalez, F.J.; Peters, J.M. Ligand Activation of Peroxisome Proliferator-Activated Receptor-beta/delta and Inhibition of Cyclooxygenase-2 Enhances Inhibition of Skin Tumorigenesis. Toxicol. Sci. 2010, 113, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Akiyama, T.E.; Harman, F.S.; Burns, A.M.; Shan, W.; Ward, J.M.; Kennett, M.J.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor β (δ)-dependent regulation of ubiquitin C expression contributes to attenuation of skin carcinogenesis. J. Biol. Chem. 2004, 279, 23719–23727. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Khozoie, C.; Bility, M.T.; Ferry, C.H.; Blazanin, N.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor β/δ cross talks with E2F and attenuates mitosis in HRAS-expressing cells. Mol. Cell Biol. 2012, 32, 2065–2082. [Google Scholar] [CrossRef] [PubMed]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Michalik, L.; Noy, N.; Yasmin, R.; Pacot, C.; Heim, M.; Fluhmann, B.; Desvergne, B.; Wahli, W. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes. Dev. 2001, 15, 3263–3277. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ). Mol. Cell. Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar] [CrossRef]
- Dlugosz, A.A.; Glick, A.B.; Tennenbaum, T.; Weinberg, W.C.; Yuspa, S.H. Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol. 1995, 254, 3–20. [Google Scholar] [PubMed]
- Heinaniemi, M.; Uski, J.O.; Degenhardt, T.; Carlberg, C. Meta-analysis of primary target genes of peroxisome proliferator-activated receptors. Genome Biol. 2007, 8, R147. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Zandbergen, F.; Tan, N.S.; Escher, P.; Patsouris, D.; Koenig, W.; Kleemann, R.; Bakker, A.; Veenman, F.; Wahli, W.; et al. The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 2004, 279, 34411–34420. [Google Scholar] [CrossRef]
- Roop, D.R.; Lowy, D.R.; Tambourin, P.E.; Strickland, J.; Harper, J.R.; Balaschak, M.; Spangler, E.F.; Yuspa, S.H. An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature 1986, 323, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Bility, M.; Thompson, J.T.; McKee, R.H.; David, R.M.; Butala, J.H.; Vanden Heuvel, J.P.; Peters, J.M. Activation of mouse and human peroxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicol. Sci. 2004, 82, 170–182. [Google Scholar] [CrossRef]
- Borland, M.G.; Khozoie, C.; Albrecht, P.P.; Zhu, B.; Lee, C.; Lahoti, T.S.; Gonzalez, F.J.; Peters, J.M. Stable over-expression of PPARβ/δ and PPARγ to examine receptor signaling in human HaCaT keratinocytes. Cell Signal 2011, 23, 2039–2050. [Google Scholar] [CrossRef]
- Borland, M.G.; Foreman, J.E.; Girroir, E.E.; Zolfaghari, R.; Sharma, A.K.; Amin, S.M.; Gonzalez, F.J.; Ross, A.C.; Peters, J.M. Ligand Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) Inhibits Cell Proliferation in Human HaCaT Keratinocytes. Mol. Pharmacol. 2008, 74, 1429–1442. [Google Scholar] [CrossRef]
- He, P.; Borland, M.G.; Zhu, B.; Sharma, A.K.; Amin, S.; El-Bayoumy, K.; Gonzalez, F.J.; Peters, J.M. Effect of ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in human lung cancer cell lines. Toxicology 2008, 254, 112–117. [Google Scholar] [CrossRef]
- Zhu, B.; Ferry, C.H.; Markell, L.K.; Blazanin, N.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. The nuclear receptor peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) promotes oncogene-induced cellular senescence through repression of endoplasmic reticulum stress. J. Biol. Chem. 2014, 289, 20102–20119. [Google Scholar] [CrossRef]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef]
- Shearer, B.G.; Hoekstra, W.J. Recent advances in peroxisome proliferator-activated receptor science. Curr. Med. Chem. 2003, 10, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Li-Stiles, B.; Lo, H.H.; Fischer, S.M. Identification and characterization of several forms of phospholipase A2 in mouse epidermal keratinocytes. J. Lipid Res. 1998, 39, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Alexandropoulos, K.; Qureshi, S.A.; Foster, D.A. Ha-Ras functions downstream from protein kinase C in v-Fps-induced gene expression mediated by TPA response elements. Oncogene 1993, 8, 803–807. [Google Scholar] [PubMed]
- Clark, J.A.; Black, A.R.; Leontieva, O.V.; Frey, M.R.; Pysz, M.A.; Kunneva, L.; Woloszynska-Read, A.; Roy, D.; Black, J.D. Involvement of the ERK signaling cascade in protein kinase C-mediated cell cycle arrest in intestinal epithelial cells. J. Biol. Chem. 2004, 279, 9233–9247. [Google Scholar] [CrossRef] [PubMed]
- Kaddatz, K.; Adhikary, T.; Finkernagel, F.; Meissner, W.; Muller-Brusselbach, S.; Muller, R. Transcriptional profiling identifies functional interactions of TGFβ and PPARβ/δ signaling: Synergistic induction of ANGPTL4 transcription. J. Biol. Chem. 2010, 285, 29469–29479. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog. Lipid Res. 2004, 43, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Walter, V.; Patterson, A.D.; Gonzalez, F.J. Unraveling the role of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) expression in colon carcinogenesis. NPJ Precis. Oncol. 2019, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Bility, M.T.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peters, J.M. PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006, 13, 53–60. [Google Scholar] [CrossRef]
- Man, M.Q.; Barish, G.D.; Schmuth, M.; Crumrine, D.; Barak, Y.; Chang, S.; Jiang, Y.; Evans, R.M.; Elias, P.M.; Feingold, K.R. Deficiency of PPARβ/δ in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J. Investig. Dermatol. 2007, 128, 370–377. [Google Scholar] [CrossRef]
- Schmuth, M.; Haqq, C.M.; Cairns, W.J.; Holder, J.C.; Dorsam, S.; Chang, S.; Lau, P.; Fowler, A.J.; Chuang, G.; Moser, A.H.; et al. Peroxisome proliferator-activated receptor (PPAR)-β/δ stimulates differentiation and lipid accumulation in keratinocytes. J. Investig. Dermatol. 2004, 122, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Desvergne, B.; Tan, N.S.; Basu-Modak, S.; Escher, P.; Rieusset, J.; Peters, J.M.; Kaya, G.; Gonzalez, F.J.; Zakany, J.; et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. J. Cell Biol. 2001, 154, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Coleman, C.; Sen, T. Stearoyl coenzyme A desaturase-1: Multitasker in cancer, metabolism, and ferroptosis. Trends Cancer 2023, 9, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Jin, X.J.; Kim, Y.K.; Oh, I.K.; Kim, J.E.; Park, C.H.; Chung, J.H. UV decreases the synthesis of free fatty acids and triglycerides in the epidermis of human skin in vivo, contributing to development of skin photoaging. J. Dermatol. Sci. 2010, 57, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Fang, L.; Ni, R.; Zhang, H.; Pan, G. Changing trends in the disease burden of non-melanoma skin cancer globally from 1990 to 2019 and its predicted level in 25 years. BMC Cancer 2022, 22, 836. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Delgado, M.B.; Tallichet-Blanc, C.; Chan, J.S.; Sng, M.K.; Mottaz, H.; Degueurce, G.; Lippi, Y.; Moret, C.; Baruchet, M.; et al. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor β/δ in ultraviolet radiation-induced skin cancer. EMBO Mol. Med. 2014, 6, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.F.; Sidorova, J.M.; Nghiem, P.; Kawasumi, M. The 6-4 photoproduct is the trigger of UV-induced replication blockage and ATR activation. Proc. Natl. Acad. Sci. USA 2020, 117, 12806–12816. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, A.; Tachiwana, H.; Kagawa, W.; Horikoshi, N.; Matsumoto, S.; Hasegawa, M.; Matsumoto, N.; Toga, T.; Yamamoto, J.; Hanaoka, F.; et al. Structural basis of pyrimidine-pyrimidone (6-4) photoproduct recognition by UV-DDB in the nucleosome. Sci. Rep. 2015, 5, 16330. [Google Scholar] [CrossRef]
- Yokoyama, H.; Mizutani, R. Structural biology of DNA (6-4) photoproducts formed by ultraviolet radiation and interactions with their binding proteins. Int. J. Mol. Sci. 2014, 15, 20321–20338. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, B.; Zhu, X.; Borland, M.G.; Ralph, D.H.; Chiaro, C.R.; Krausz, K.W.; Ntambi, J.M.; Glick, A.B.; Patterson, A.D.; Perdew, G.H.; et al. Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) in Keratinocytes by Endogenous Fatty Acids. Biomolecules 2024, 14, 606. https://doi.org/10.3390/biom14060606
Zhu B, Zhu X, Borland MG, Ralph DH, Chiaro CR, Krausz KW, Ntambi JM, Glick AB, Patterson AD, Perdew GH, et al. Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) in Keratinocytes by Endogenous Fatty Acids. Biomolecules. 2024; 14(6):606. https://doi.org/10.3390/biom14060606
Chicago/Turabian StyleZhu, Bokai, Xiaoyang Zhu, Michael G. Borland, Douglas H. Ralph, Christopher R. Chiaro, Kristopher W. Krausz, James M. Ntambi, Adam B. Glick, Andrew D. Patterson, Gary H. Perdew, and et al. 2024. "Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) in Keratinocytes by Endogenous Fatty Acids" Biomolecules 14, no. 6: 606. https://doi.org/10.3390/biom14060606
APA StyleZhu, B., Zhu, X., Borland, M. G., Ralph, D. H., Chiaro, C. R., Krausz, K. W., Ntambi, J. M., Glick, A. B., Patterson, A. D., Perdew, G. H., Gonzalez, F. J., & Peters, J. M. (2024). Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) in Keratinocytes by Endogenous Fatty Acids. Biomolecules, 14(6), 606. https://doi.org/10.3390/biom14060606