
Mitochondrial and Nuclear DNA Variants in Amyotrophic Lateral Sclerosis: Enrichment in the Mitochondrial Control Region and Sirtuin Pathway Genes in Spinal Cord Tissue

 , , ,
, , ,  and
and 
Abstract
1. Introduction
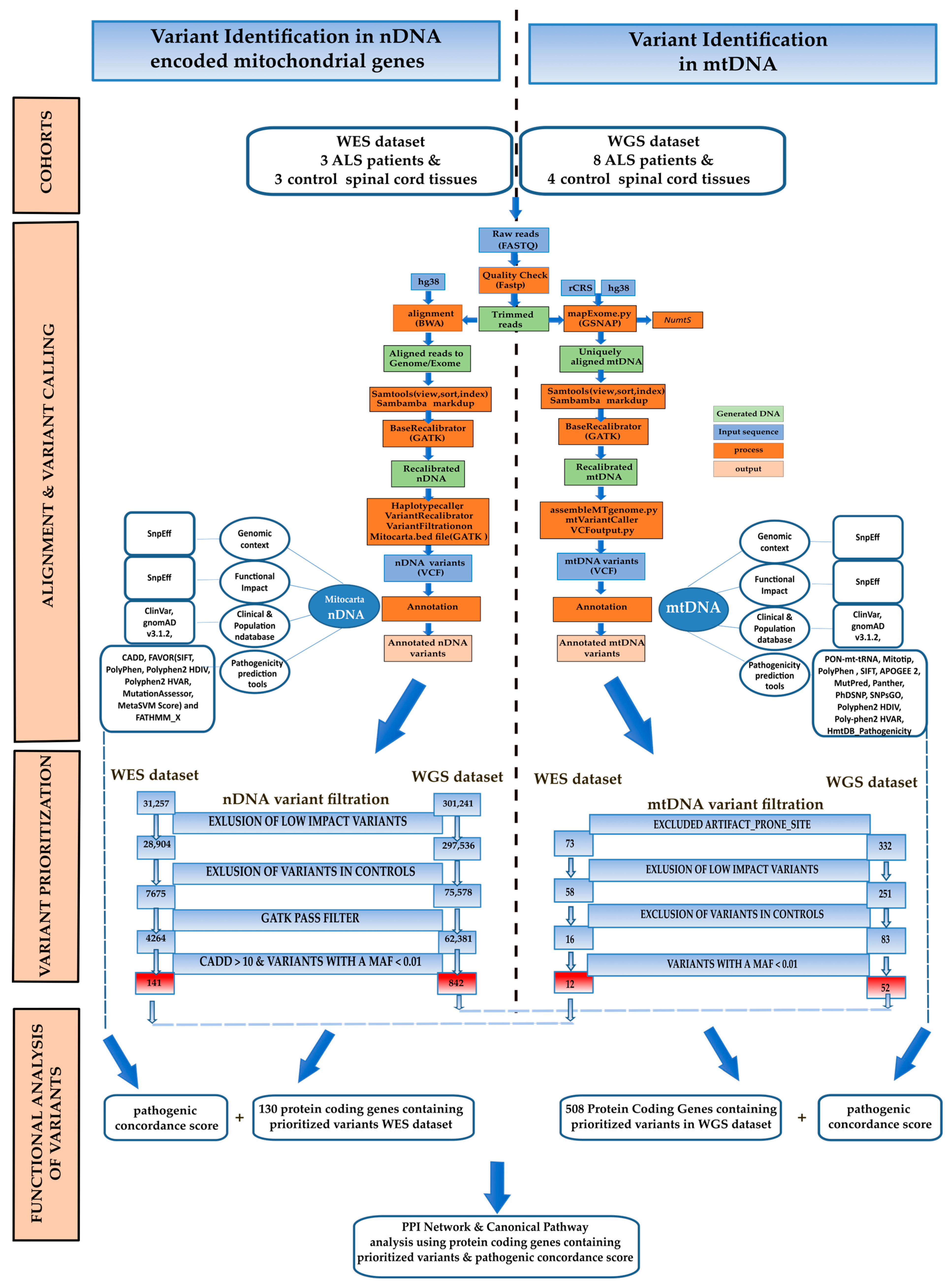
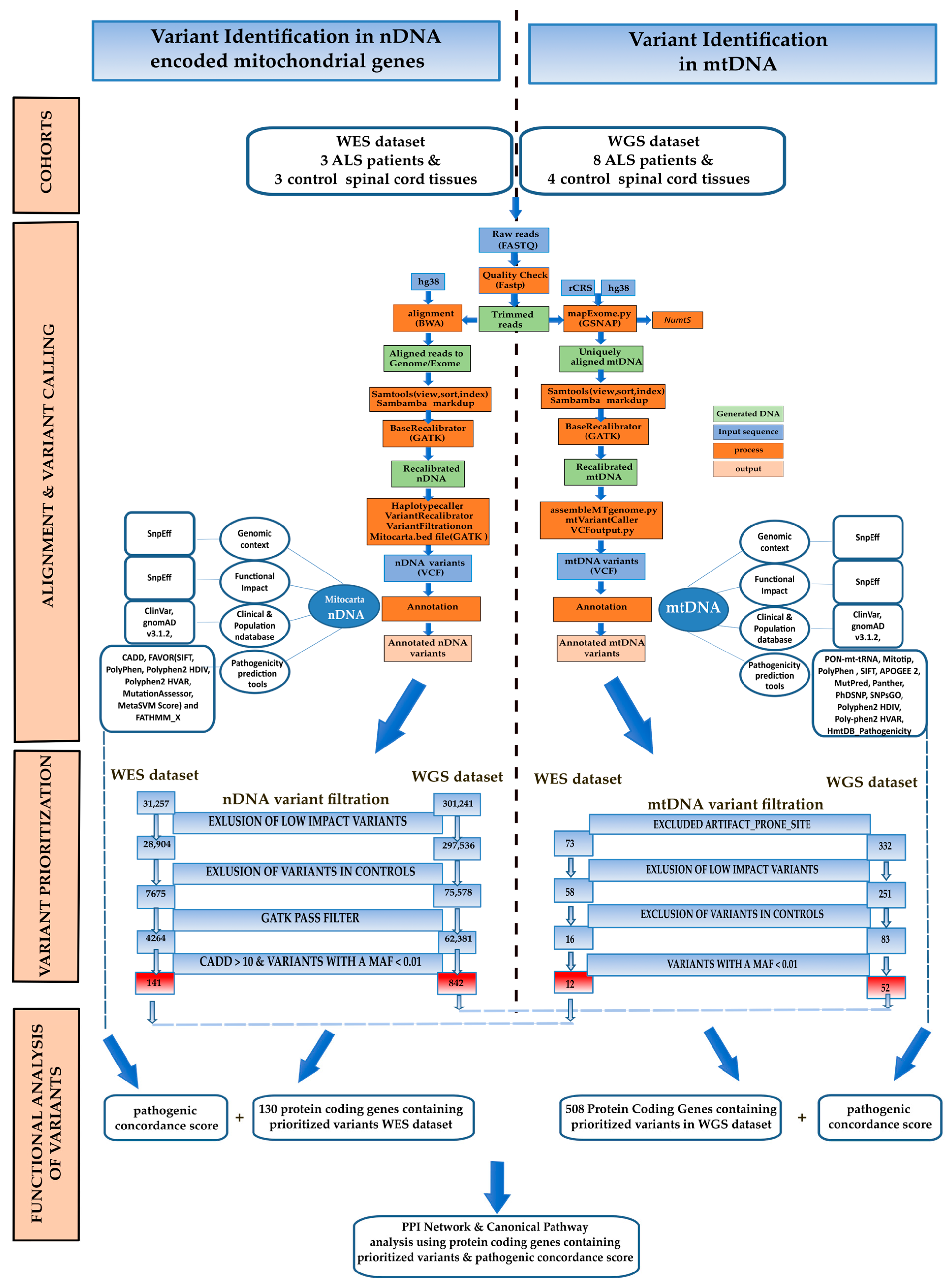
2. Materials and Methods
2.1. nDNA Alignment and nDNA Variant Calling
2.2. mtDNA Assembly and Variant Calling
2.3. nDNA and mtDNA Variant Annotation and Filtering
2.4. Disease Association Analysis
2.5. Structural Predictions
2.6. Network Analysis of Genes Containing Candidate Variants
2.7. mtDNA-CN Estimation
2.8. Statistical Analyses
3. Results
3.1. WES Dataset
3.1.1. nDNA Alignment, Variant Calling Filtration, and Prioritisation
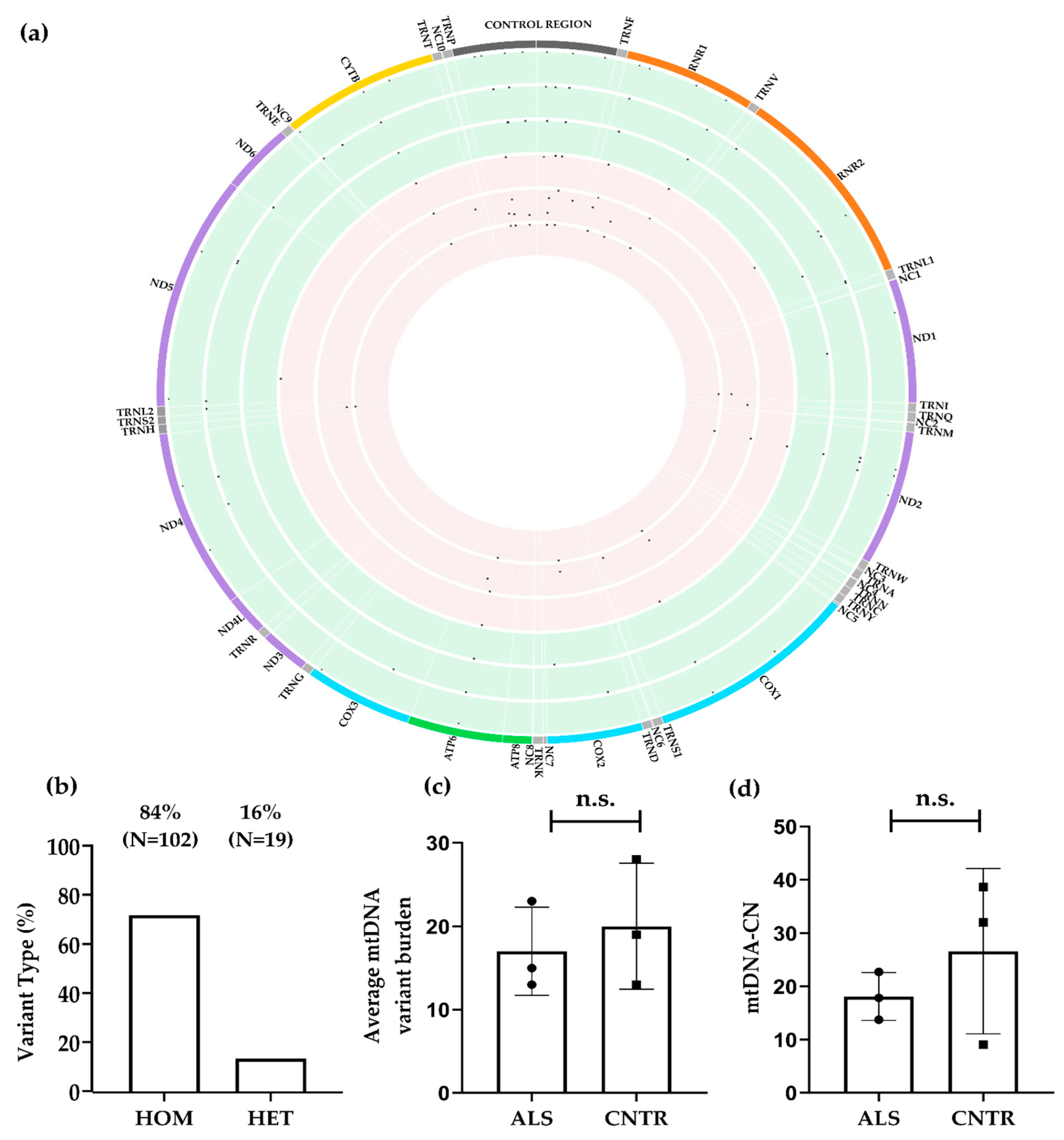
3.1.2. mtDNA Alignment, Variant Calling Filtration, and Proritisation
3.2. WGS Dataset
3.2.1. nDNA Alignment, Variant Calling Filtration, and Prioritisation
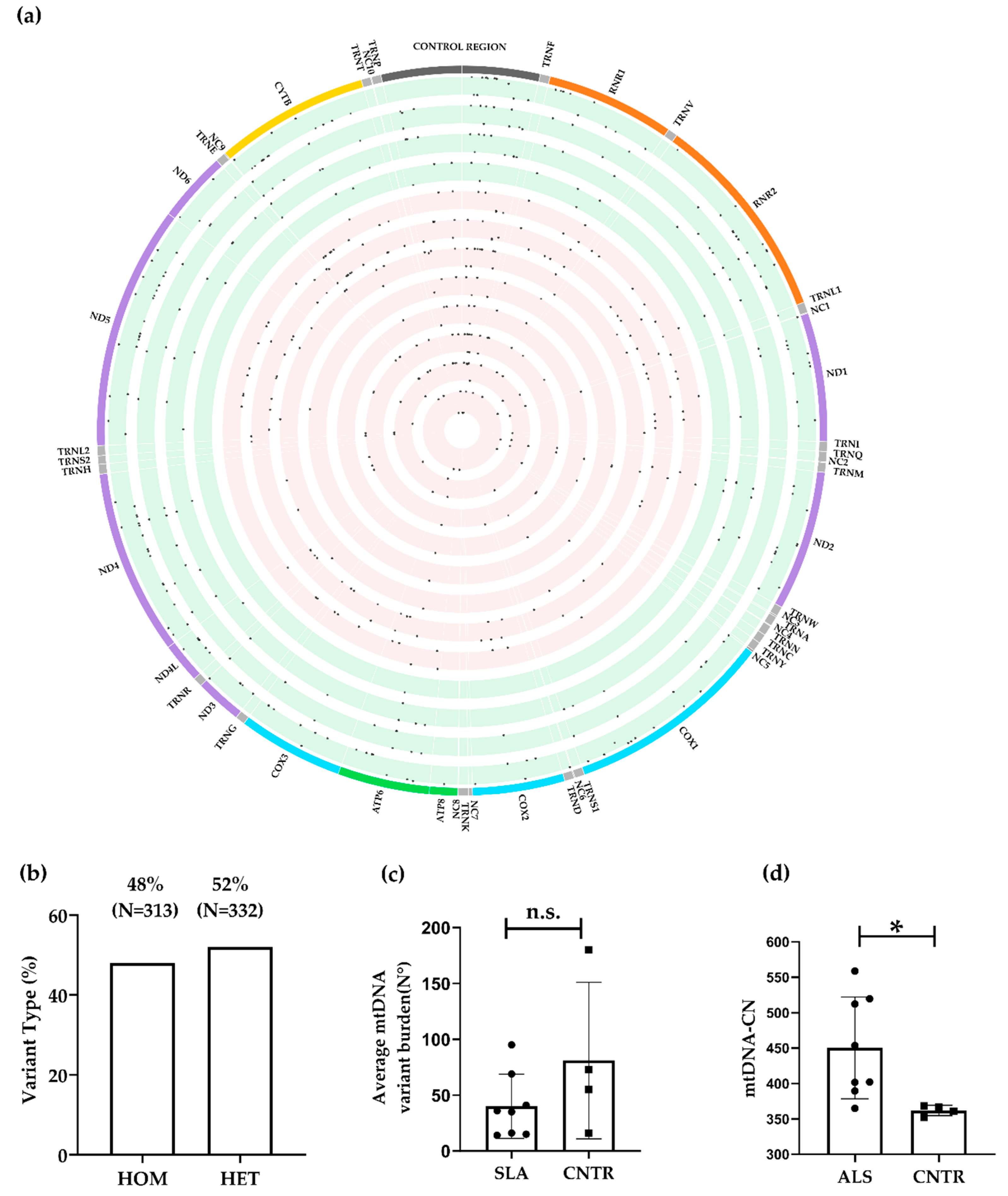
3.2.2. mtDNA Assembly, Variant Calling Filtration, and Prioritisation
3.2.3. mtDNA Copy Number
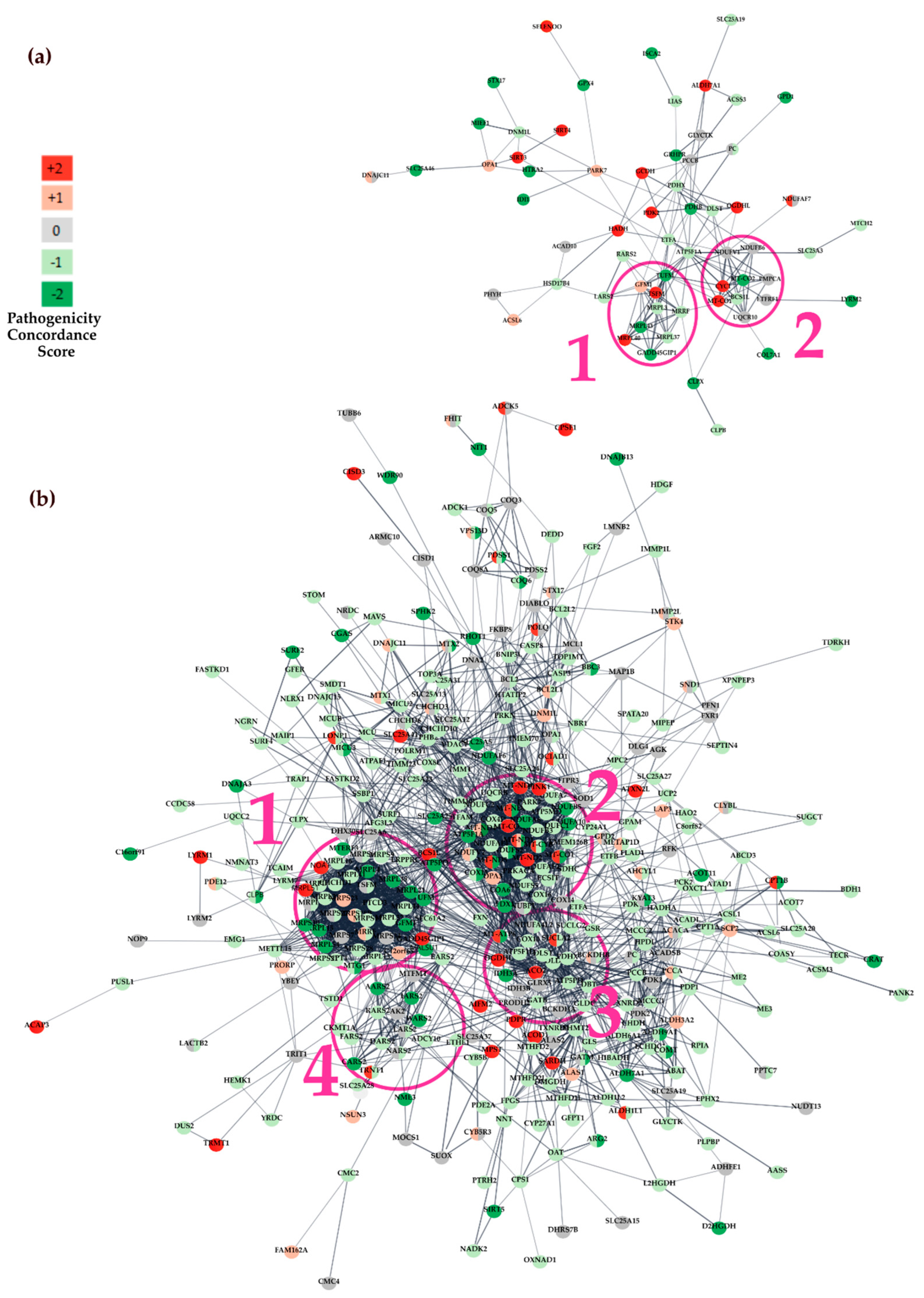
3.3. Functional Analysis of Genes Containing Variants
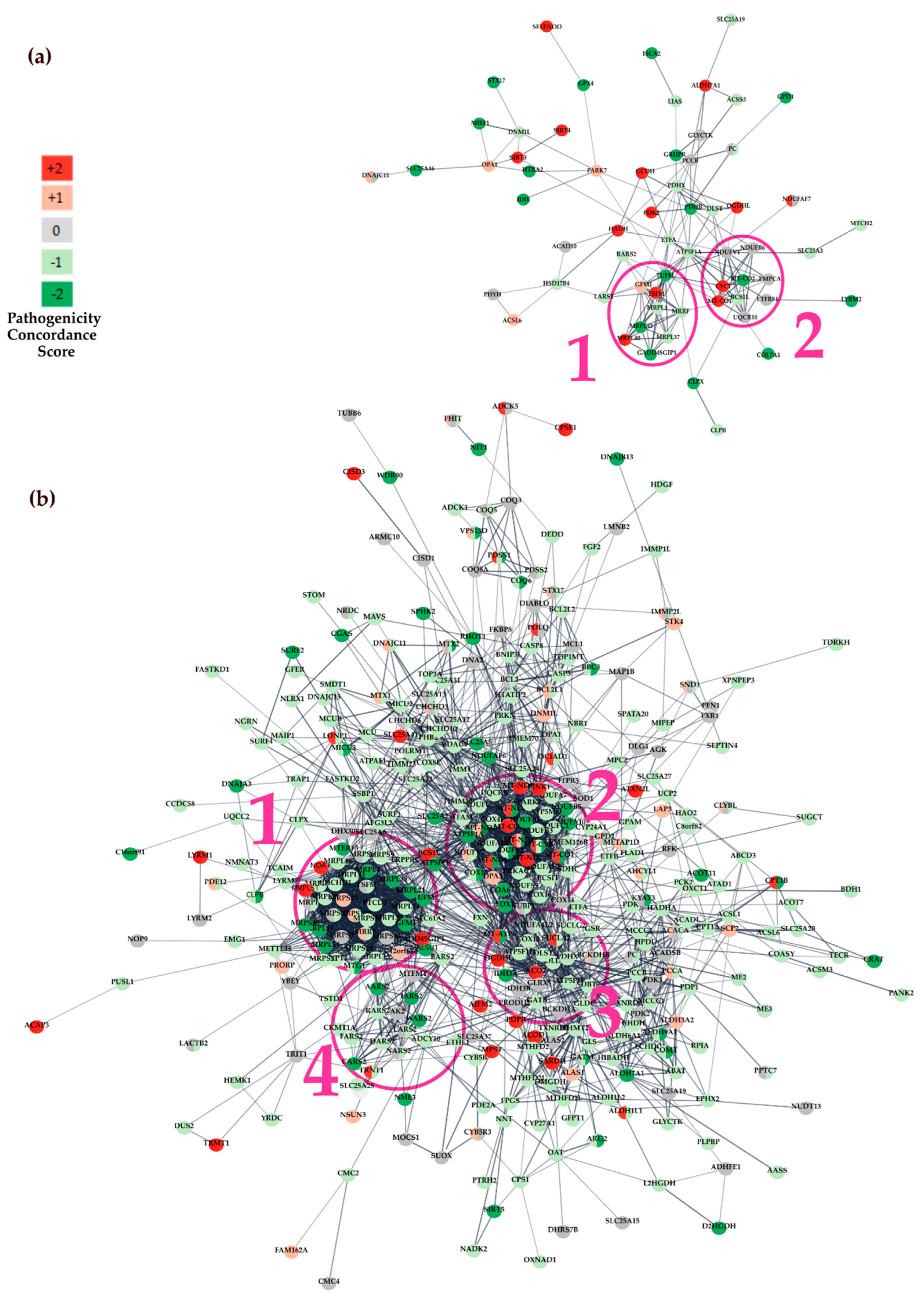
3.3.1. Protein-Protein Interaction (PPI) Network Analysis
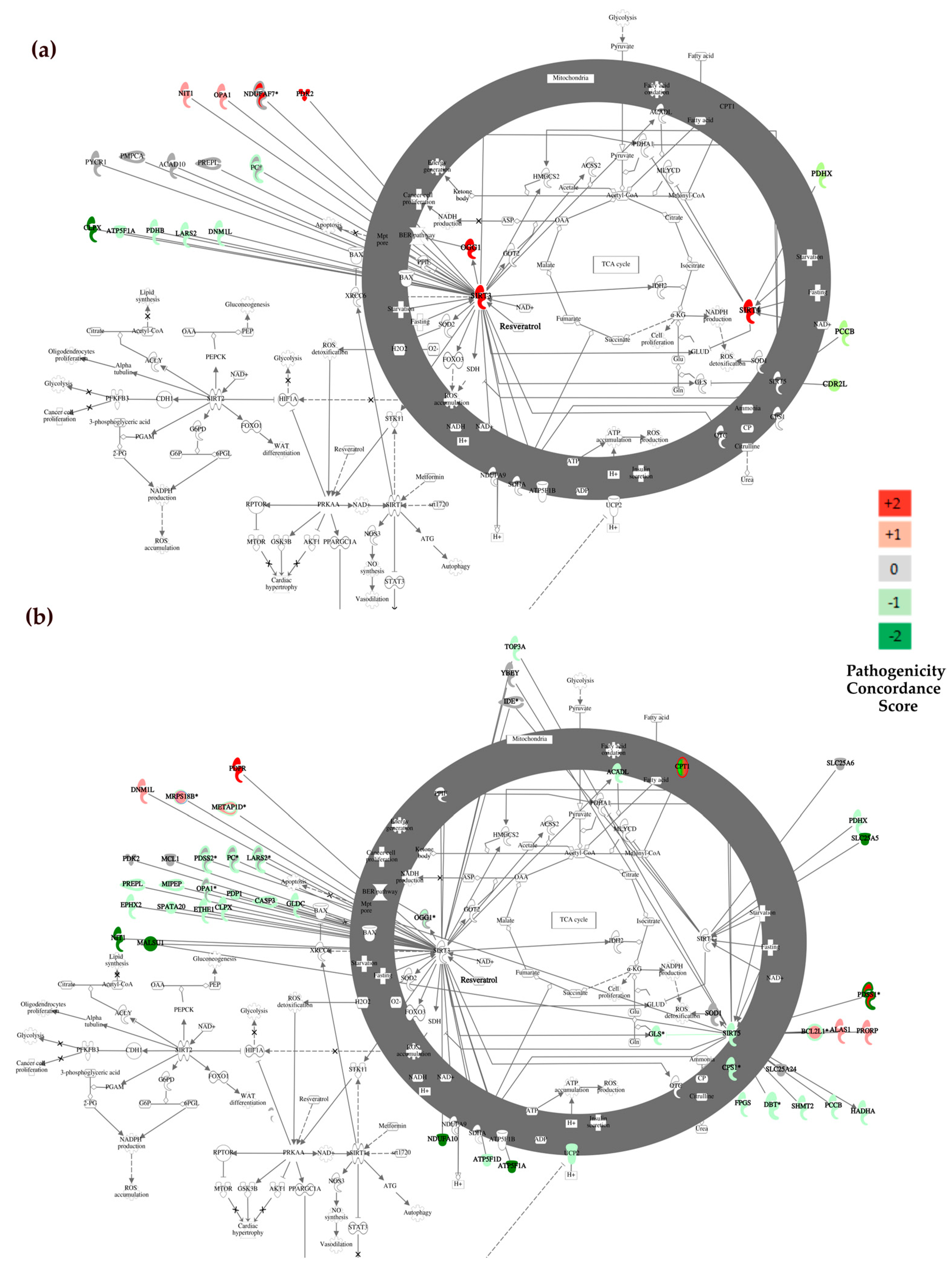
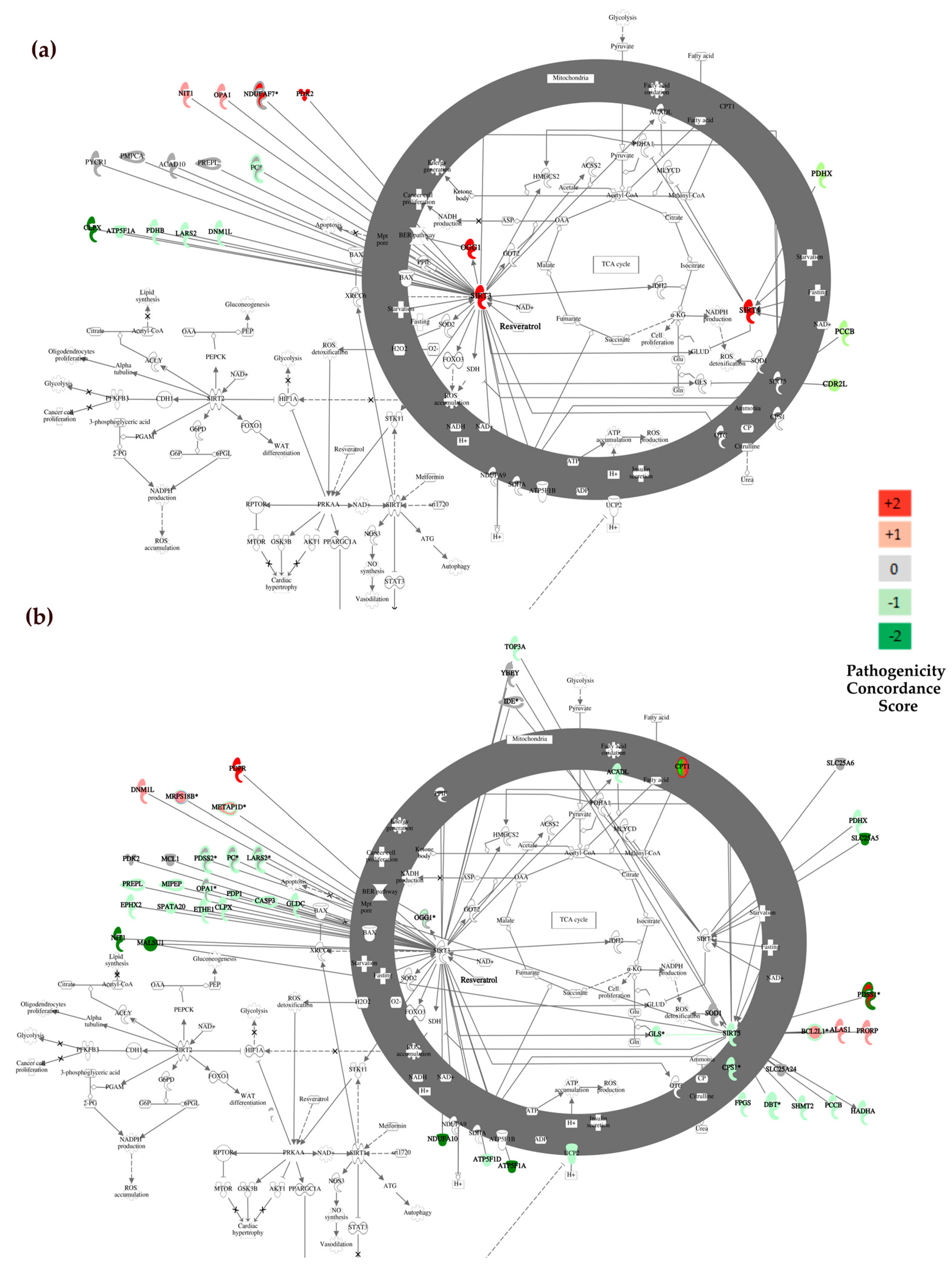
3.3.2. Canonical Pathway Analysis with Ingenuity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swinnen, B.; Robberecht, W. The Phenotypic Variability of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Saini, A.; Chawla, P.A. Breaking Barriers with Tofersen: Enhancing Therapeutic Opportunities in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2024, 31, e16140. [Google Scholar] [CrossRef]
- Alqallaf, A.; Cates, D.W.; Render, K.P.; Patel, K.A. Sodium Phenylbutyrate and Taurursodiol: A New Therapeutic Option for the Treatment of Amyotrophic Lateral Sclerosis. Ann. Pharmacother. 2023, 58, 165–173. [Google Scholar] [CrossRef]
- Fralick, M.; Sacks, C.A.; Kesselheim, A.S. Assessment of Use of Combined Dextromethorphan and Quinidine in Patients with Dementia or Parkinson Disease After US Food and Drug Administration Approval for Pseudobulbar Affect. JAMA Intern. Med. 2019, 179, 224–230. [Google Scholar] [CrossRef]
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An Estimate of Amyotrophic Lateral Sclerosis Heritability Using Twin Data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of Play in Amyotrophic Lateral Sclerosis Genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet. Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. β-Amyloid 42 Accumulation in the Lumbar Spinal Cord Motor Neurons of Amyotrophic Lateral Sclerosis Patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative Damage to Protein in Sporadic Motor Neuron Disease Spinal Cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef]
- Ihara, Y.; Nobukuni, K.; Takata, H.; Hayabara, T. Oxidative Stress and Metal Content in Blood and Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients with and without a Cu, Zn-Superoxide Dismutase Mutation. Neurol. Res. 2005, 27, 105–108. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Hüther, G.; et al. Reduced Oxidative Damage in ALS by High-Dose Enteral Melatonin Treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H.; Beal, M.F. Superoxide Dismutase Activity, Oxidative Damage, and Mitochondrial Energy Metabolism in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of Axonal Transport Defects in Motor Neuron Diseases: Opportunities for Translational Research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Simões, R.F.; Carvalho, M.; Ferreiro, E.; Silva, F.S.G. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells 2024, 13, 248. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Minczuk, M. In D-Loop: 40 Years of Mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef]
- Pereira, F.; Soares, P.; Carneiro, J.; Pereira, L.; Richards, M.B.; Samuels, D.C.; Amorim, A. Evidence for Variable Selective Pressures at a Large Secondary Structure of the Human Mitochondrial DNA Control Region. Mol. Biol. Evol. 2008, 25, 2759–2770. [Google Scholar] [CrossRef]
- Sbisà, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian Mitochondrial D-Loop Region Structural Analysis: Identification of New Conserved Sequences and Their Functional and Evolutionary Implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Basu, U.; Bostwick, A.M.; Das, K.; Dittenhafer-Reed, K.E.; Patel, S.S. Structure, Mechanism, and Regulation of Mitochondrial DNA Transcription Initiation. J. Biol. Chem. 2020, 295, 18406–18425. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular Mechanisms and Physiological Functions of Mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Di Nottia, M.; Verrigni, D.; Torraco, A.; Rizza, T.; Bertini, E.; Carrozzo, R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes 2021, 12, 247. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef]
- Seguin, A.; Jia, X.; Earl, A.M.; Li, L.; Wallace, J.; Qiu, A.; Bradley, T.; Shrestha, R.; Troadec, M.B.; Hockin, M.; et al. The Mitochondrial Metal Transporters Mitoferrin1 and Mitoferrin2 Are Required for Liver Regeneration and Cell Proliferation in Mice. J. Biol. Chem. 2020, 295, 11002–11020. [Google Scholar] [CrossRef]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC Regulation of Mitochondrial Calcium Flux: From Channel Biophysics to Disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An Updated Mitochondrial Proteome Now with Sub-Organelle Localization and Pathway Annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Sutovsky, P.; Moreno, R.D.; Ramalho-Santos, J.; Dominko, T.; Simerly, C.; Schatten, G. Ubiquitin Tag for Sperm Mitochondria. Nature 1999, 402, 371–372. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent Impacts of Aging on Mitochondrial DNA Quantity and Quality in Humans. BMC Genom. 2017, 18, 890. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Ashar, F.N.; Moes, A.; Moore, A.Z.; Grove, M.L.; Chaves, P.H.M.; Coresh, J.; Newman, A.B.; Matteini, A.M.; Bandeen-Roche, K.; Boerwinkle, E.; et al. Association of Mitochondrial DNA Levels with Frailty and All-Cause Mortality. J. Mol. Med. 2015, 93, 177–186. [Google Scholar] [CrossRef]
- Mehta, A.R.; Walters, R.; Waldron, F.M.; Pal, S.; Selvaraj, B.T.; Macleod, M.R.; Hardingham, G.E.; Chandran, S.; Gregory, J.M. Targeting Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Brain Commun. 2019, 1, fcz009. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Ladd, A.C.; Keeney, P.M.; Govind, M.M.; Bennett, J.P. Mitochondrial Oxidative Phosphorylation Transcriptome Alterations in Human Amyotrophic Lateral Sclerosis Spinal Cord and Blood. NeuroMolecular Med. 2014, 16, 714–726. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive Transcriptome Sequencing of Human Spinal Cord Tissues Provides New Insights into Motor Neuron Degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Kuhn, R.M.; Haussler, D.; James Kent, W. The UCSC Genome Browser and Associated Tools. Brief. Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and Revision of the Cambridge Reference Sequence for Human Mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Nacu, S. Fast and SNP-Tolerant Detection of Complex Variants and Splicing in Short Reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Simone, D.; Calabrese, F.M.; Lang, M.; Gasparre, G.; Attimonelli, M. The Reference Human Nuclear Mitochondrial Sequences Compilation Validated and Implemented on the UCSC Genome Browser. BMC Genom. 2011, 12, 517. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. Mitochondrial Genomes Gleaned from Human Whole-Exome Sequencing. Nat. Methods 2012, 9, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Gutta, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. MToolBox: A Highly Automated Pipeline for Heteroplasmy Annotation and Prioritization Analysis of Human Mitochondrial Variants in High-Throughput Sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef] [PubMed]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-Generation Sequencing Alignment Data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- Alkanaq, A.N.; Hamanaka, K.; Sekiguchi, F.; Taguri, M.; Takata, A.; Miyake, N.; Miyatake, S.; Mizuguchi, T.; Matsumoto, N. Comparison of Mitochondrial DNA Variants Detection Using Short- and Long-Read Sequencing. J. Hum. Genet. 2019, 64, 1107–1116. [Google Scholar] [CrossRef]
- Rebolledo-Jaramillo, B.; Su, M.S.W.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal Age Effect and Severe Germ-Line Bottleneck in the Inheritance of Human Mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified Representation of Genetic Variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila Melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Vihinen, M. PON-Mt-TRNA: A Multifactorial Probability-Based Method for Classification of Mitochondrial TRNA Variations. Nucleic Acids Res. 2016, 44, 2020–2027. [Google Scholar] [CrossRef]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the Pathogenicity of Novel Variants in Mitochondrial TRNA with MitoTIP. PLOS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef]
- Castellana, S.; Biagini, T.; Petrizzelli, F.; Parca, L.; Panzironi, N.; Caputo, V.; Vescovi, A.L.; Carella, M.; Mazza, T. MitImpact 3: Modeling the Residue Interaction Network of the Respiratory Chain Subunits. Nucleic Acids Res. 2021, 49, D1282–D1288. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.D.; Parca, L.; Petrizzelli, F.; Biagini, T.; Giovannetti, A.; Liorni, N.; Napoli, A.; Carella, M.; Procaccio, V.; Lott, M.T.; et al. APOGEE 2: Multi-Layer Machine-Learning Model for the Interpretable Prediction of Mitochondrial Missense Variants. Nat. Commun. 2023, 14, 5058. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Krishnan, V.G.; Mort, M.E.; Xin, F.; Kamati, K.K.; Cooper, D.N.; Mooney, S.D.; Radivojac, P. Automated Inference of Molecular Mechanisms of Disease from Amino Acid Substitutions. Bioinformatics 2009, 25, 2744–2750. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting Disease-Causing Genetic Variants Using Position-Specific Evolutionary Preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A Webserver and Lightweight Tool for Scoring Single Nucleotide Variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, M.; Savojardo, C.; Martelli, P.L.; Casadio, R. E-SNPs&GO: Embedding of Protein Sequence and Function Improves the Annotation of Human Pathogenic Variants. Bioinformatics 2022, 38, 5168–5174. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Zhou, H.; Arapoglou, T.; Li, X.; Li, Z.; Zheng, X.; Moore, J.; Asok, A.; Kumar, S.; Blue, E.E.; Buyske, S.; et al. FAVOR: Functional Annotation of Variants Online Resource and Annotator for Variation across the Human Genome. Nucleic Acids Res. 2023, 51, D1300–D1311. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate Prediction of Pathogenic Point Mutations via Extended Features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, Á.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A Comprehensive Platform Integrating Information on Human Disease-Associated Genes and Variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Gunning, A.C.; Fryer, V.; Fasham, J.; Crosby, A.H.; Ellard, S.; Baple, E.L.; Wright, C.F. Assessing Performance of Pathogenicity Predictors Using Clinically Relevant Variant Datasets. J. Med. Genet. 2021, 58, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in Silico Algorithms for Use with ACMG/AMP Clinical Variant Interpretation Guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING Database in 2017: Quality-Controlled Protein–Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Holze, H.; Kirsch, R.; Nastou, K.C.; Cuesta-Astroz, Y.; Rattei, T.; Szklarczyk, D.; von Mering, C.; Jensen, L.J. Cytoscape StringApp 2.0: Analysis and Visualization of Heterogeneous Biological Networks. J. Proteome Res. 2023, 22, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W.V. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Battle, S.L.; Puiu, D.; Verlouw, J.; Broer, L.; Boerwinkle, E.; Taylor, K.D.; Rotter, J.I.; Rich, S.S.; Grove, M.L.; Pankratz, N.; et al. A Bioinformatics Pipeline for Estimating Mitochondrial DNA Copy Number and Heteroplasmy Levels from Whole Genome Sequencing Data. NAR Genom. Bioinforma. 2022, 4, lqac034. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 Interaction Slows Progression of Amyotrophic Lateral Sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A User-Friendly Online Bioinformatics Tool for Amyotrophic Lateral Sclerosis Genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Lev, N.; Ickowicz, D.; Melamed, E.; Offen, D. Oxidative Insults Induce DJ-1 Upregulation and Redistribution: Implications for Neuroprotection. Neurotoxicology 2008, 29, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, C.; Sun, Q.; Pan, H.; Huang, P.; Ding, J.; Chen, S. MicroRNA-4639 Is a Regulator of DJ-1 Expression and a Potential Early Diagnostic Marker for Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 269182. [Google Scholar] [CrossRef] [PubMed]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA Target Prediction Easy, Fast and Flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Furuta, A.; Nishioka, K.I.; Suzuki, S.O.; Nakabeppu, Y.; Iwaki, T. Impairment of Mitochondrial DNA Repair Enzymes against Accumulation of 8-Oxo-Guanine in the Spinal Motor Neurons of Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2002, 103, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Ledahawsky, L.M.; Terzenidou, M.E.; Edwards, R.; Kline, R.A.; Graham, L.C.; Eaton, S.L.; van der Hoorn, D.; Chaytow, H.; Huang, Y.T.; Groen, E.J.N.; et al. The Mitochondrial Protein Sideroflexin 3 (SFXN3) Influences Neurodegeneration Pathways in Vivo. Febs J. 2022, 289, 3894. [Google Scholar] [CrossRef] [PubMed]
- van Oven, M.; Kayser, M. Updated Comprehensive Phylogenetic Tree of Global Human Mitochondrial DNA Variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef]
- Umbria, M.; Ramos, A.; Aluja, M.P.; Santos, C. The Role of Control Region Mitochondrial DNA Mutations in Cardiovascular Disease: Stroke and Myocardial Infarction. Sci. Rep. 2020, 10, 2766. [Google Scholar] [CrossRef]
- Belle, E.M.S.; Piganeau, G.; Gardner, M.; Eyre-Walker, A. An Investigation of the Variation in the Transition Bias among Various Animal Mitochondrial DNA. Gene 2005, 355, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Dolce, V.; Palmieri, L.; Ventura, M.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the Human Mitochondrial Oxodicarboxylate Carrier. J. Biol. Chem. 2001, 276, 8225–8230. [Google Scholar] [CrossRef] [PubMed]
- Ioakeimidis, F.; Ott, C.; Kozjak-Pavlovic, V.; Violitzi, F.; Rinotas, V.; Makrinou, E.; Eliopoulos, E.; Fasseas, C.; Kollias, G.; Douni, E. A Splicing Mutation in the Novel Mitochondrial Protein DNAJC11 Causes Motor Neuron Pathology Associated with Cristae Disorganization, and Lymphoid Abnormalities in Mice. PLoS ONE 2014, 9, 104237. [Google Scholar] [CrossRef] [PubMed]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Knippenberg, S.; Sipos, J.; Thau-Habermann, N.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Altered Expression of DJ-1 and PINK1 in Sporadic ALS and in the SOD1G93A ALS Mouse Model. J. Neuropathol. Exp. Neurol. 2013, 72, 1052–1061. [Google Scholar] [CrossRef]
- Guo, S.; Zhou, K.; Yuan, Q.; Su, L.; Liu, Y.; Ji, X.; Gu, X.; Guo, X.; Xing, J. An Innovative Data Analysis Strategy for Accurate Next-Generation Sequencing Detection of Tumor Mitochondrial DNA Mutations. Mol. Ther. Nucleic Acids 2021, 23, 232. [Google Scholar] [CrossRef]
- Laricchia, K.M.; Lake, N.J.; Watts, N.A.; Shand, M.; Haessly, A.; Gauthier, L.; Benjamin, D.; Banks, E.; Soto, J.; Garimella, K.; et al. Mitochondrial DNA Variation across 56,434 Individuals in GnomAD. Genome Res. 2022, 32, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Keeney, P.M.; Bennett, J.P. ALS Spinal Neurons Show Varied and Reduced MtDNA Gene Copy Numbers and Increased MtDNA Gene Deletions. Mol. Neurodegener. 2010, 5, 21. [Google Scholar] [CrossRef]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppedè, F. Mitochondrial DNA Copy Number and D-Loop Region Methylation in Carriers of Amyotrophic Lateral Sclerosis Gene Mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced Mitochondrial D-Loop Methylation Levels in Sporadic Amyotrophic Lateral Sclerosis. Clin. Epigenetics 2020, 12, 137. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Lin, Z.F.; Xu, H.B.; Wang, J.Y.; Lin, Q.; Ruan, Z.; Liu, F.B.; Jin, W.; Huang, H.H.; Chen, X. SIRT5 Desuccinylates and Activates SOD1 to Eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef]
- Kaufmann, P.; Thompson, J.L.P.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II Trial of CoQ10 for ALS Finds Insufficient Evidence to Justify Phase III. Ann. Neurol. 2009, 66, 235–244. [Google Scholar] [CrossRef]
- Yáñez, M.; Galán, L.; Matías-Guiu, J.; Vela, A.; Guerrero, A.; García, A.G. CSF from Amyotrophic Lateral Sclerosis Patients Produces Glutamate Independent Death of Rat Motor Brain Cortical Neurons: Protection by Resveratrol but Not Riluzole. Brain Res. 2011, 1423, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Tang, L.; Zhang, N.; Fan, D. Protective Effects of Resveratrol through the Up-Regulation of SIRT1 Expression in the Mutant HSOD1-G93A-Bearing Motor Neuron-like Cell Culture Model of Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2011, 503, 250–255. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-Specific MtDNA Abundance from Exome Data and Its Correlation with Mitochondrial Transcription, Mass and Respiratory Activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Mavraki, E.; Labrum, R.; Sergeant, K.; Alston, C.L.; Woodward, C.; Smith, C.; Knowles, C.V.Y.; Patel, Y.; Hodsdon, P.; Baines, J.P.; et al. Genetic Testing for Mitochondrial Disease: The United Kingdom Best Practice Guidelines. Eur. J. Hum. Genet. 2022, 31, 148–163. [Google Scholar] [CrossRef]
- Macken, W.L.; Falabella, M.; Pizzamiglio, C.; Woodward, C.E.; Scotchman, E.; Chitty, L.S.; Polke, J.M.; Bugiardini, E.; Hanna, M.G.; Vandrovcova, J.; et al. Enhanced Mitochondrial Genome Analysis: Bioinformatic and Long-Read Sequencing Advances and Their Diagnostic Implications. Expert. Rev. Mol. Diagn. 2023, 23, 797–814. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and Beyond: Multiple Functions of the Human Mitochondrial Transcription Machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Posse, V.; Milenkovic, D.; Mourier, A.; Siira, S.J.; Bonekamp, N.A.; Neumann, U.; Filipovska, A.; Polosa, P.L.; et al. POLRMT Regulates the Switch between Replication Primer Formation and Gene Expression of Mammalian MtDNA. Sci. Adv. 2016, 2, e1600963. [Google Scholar] [CrossRef]
- Doda, J.N.; Wright, C.T.; Clayton, D.A. Elongation of Displacement-Loop Strands in Human and Mouse Mitochondrial DNA Is Arrested near Specific Template Sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 6116–6120. [Google Scholar] [CrossRef] [PubMed]
- Jemt, E.; Persson, Ö.; Shi, Y.; Mehmedovic, M.; Uhler, J.P.; López, M.D.; Freyer, C.; Gustafsson, C.M.; Samuelsson, T.; Falkenberg, M. Regulation of DNA Replication at the End of the Mitochondrial D-Loop Involves the Helicase TWINKLE and a Conserved Sequence Element. Nucleic Acids Res. 2015, 43, 9262–9275. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, A.; Rice, E.J.; Kundaje, A.; Danko, C.G.; Mishmar, D. Initiation of MtDNA Transcription Is Followed by Pausing, and Diverges across Human Cell Types and during Evolution. Genome Res. 2017, 27, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; De Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 Inhibition Attenuates Neurotoxicity and Dopamine Release Deficits in Vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lloyd, R.S. Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA 2022, 2, 279–301. [Google Scholar] [CrossRef]
- Oka, S.; Leon, J.; Sakumi, K.; Abolhassani, N.; Sheng, Z.; Tsuchimoto, D.; LaFerla, F.M.; Nakabeppu, Y. MTH1 and OGG1 Maintain a Low Level of 8-Oxoguanine in Alzheimer’s Brain, and Prevent the Progression of Alzheimer’s Pathogenesis. Sci. Rep. 2021, 11, 5819. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Benítez-Buelga, C.; Calvo, P.A.; Hanna, B.M.F.; Mortusewicz, O.; Masuyer, G.; Davies, J.; Wallner, O.; Sanjiv, K.; Albers, J.J.; et al. Small-Molecule Activation of OGG1 Increases Oxidative DNA Damage Repair by Gaining a New Function. Science 2022, 376, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial Fusion and Fission Proteins Expression Dynamically Change in a Murine Model of Amyotrophic Lateral Sclerosis. Curr. Neurovascular Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-Beta Overproduction Causes Abnormal Mitochondrial Dynamics via Differential Modulation of Mitochondrial Fission/Fusion Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The Mitochondrial Permeability Transition Pore in Motor Neurons: Involvement in the Pathobiology of ALS Mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Méndez-lópez, I.; Sancho-bielsa, F.J.; Engel, T.; García, A.G.; Padín, J.F. Progressive Mitochondrial SOD1G93A Accumulation Causes Severe Structural, Metabolic and Functional Aberrations through OPA1 down-Regulation in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 8194. [Google Scholar] [CrossRef]
- Del Dotto, V.; Fogazza, M.; Lenaers, G.; Rugolo, M.; Carelli, V.; Zanna, C. OPA1: How Much Do We Know to Approach Therapy? Pharmacol. Res. 2018, 131, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Chanson, J.B.; Echaniz-Laguna, A.; Vogel, T.; Mohr, M.; Benoilid, A.; Kaltenbach, G.; Kiesmann, M. TDP43-Positive Intraneuronal Inclusions in a Patient with Motor Neuron Disease and Parkinson’s Disease. Neurodegener. Dis. 2010, 7, 260–264. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A Mitochondrial Origin for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis through CHCHD10 Involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate Up-Regulates the DJ-1 Protein and Protects Neurons in Cell Culture and in Animal Models of Parkinson Disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef]
- Lind-Holm Mogensen, F.; Scafidi, A.; Poli, A.; Michelucci, A. PARK7/DJ-1 in Microglia: Implications in Parkinson’s Disease and Relevance as a Therapeutic Target. J. Neuroinflam. 2023, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Olahova, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmüller, H.; et al. Mitochondrial Oxodicarboxylate Carrier Deficiency Is Associated with Mitochondrial DNA Depletion and Spinal Muscular Atrophy–like Disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.H.; Santosa, M.M.; Lim, V.J.W.; Ho, B.X.; Taylor, A.; Khong, Z.J.; Ravits, J.; Fan, Y.; Liou, Y.C.; Soh, B.S.; et al. ALS Motor Neurons Exhibit Hallmark Metabolic Defects That Are Rescued by SIRT3 Activation. Cell Death Differ. 2020, 28, 1379–1397. [Google Scholar] [CrossRef]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol Improves Motoneuron Function and Extends Survival in SOD1(G93A) ALS Mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef]
- Sun, Q.; Kang, R.R.; Chen, K.G.; Liu, K.; Ma, Z.; Liu, C.; Deng, Y.; Liu, W.; Xu, B. Sirtuin 3 Is Required for the Protective Effect of Resveratrol on Manganese-Induced Disruption of Mitochondrial Biogenesis in Primary Cultured Neurons. J. Neurochem. 2021, 156, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Dolinar, A.; Ravnik-Glavač, M.; Glavač, D. Epigenetic Mechanisms in Amyotrophic Lateral Sclerosis: A Short Review. Mech. Ageing Dev. 2018, 174, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Longchamps, R.J.; Castellani, C.A.; Yang, S.Y.; Newcomb, C.E.; Sumpter, J.A.; Lane, J.; Grove, M.L.; Guallar, E.; Pankratz, N.; Taylor, K.D.; et al. Evaluation of Mitochondrial DNA Copy Number Estimation Techniques. PLoS ONE 2020, 15, e0228166. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational Signatures of Ionizing Radiation in Second Malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef] [PubMed]
- Ehinger, J.K.; Morota, S.; Hansson, M.J.; Paul, G.; Elmér, E. Mitochondrial Dysfunction in Blood Cells from Amyotrophic Lateral Sclerosis Patients. J. Neurol. 2015, 262, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 Misplacing and Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis Pathogenesis. Front. Cell. Neurosci. 2015, 9, 156011. [Google Scholar] [CrossRef] [PubMed]
- Ballester, L.Y.; Luthra, R.; Kanagal-Shamanna, R.; Singh, R.R. Advances in Clinical Next-Generation Sequencing: Target Enrichment and Sequencing Technologies. Expert Rev. Mol. Diagn. 2016, 16, 357–372. [Google Scholar] [CrossRef]
- Kuźma-Kozakiewicz, M.; Kwieciński, H. The Genetics of Amyotrophic Lateral Sclerosis. Neurol. Neurochir. Pol. 2009, 43, 538–549. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | SAMPLE ID | Age | Sex | Tissue | Diagnosis | |
|---|---|---|---|---|---|---|
| WES dataset | NICHD | SAMN02768876 | 61 | Male | Spinal Cord Lumbar | ALS_1 |
| NICHD | SAMN02768878 | 54 | Male | Spinal Cord Lumbar | ALS_2 | |
| NICHD | SAMN02768879 | 59 | Male | Spinal Cord Lumbar | ALS_3 | |
| NICHD | SAMN02768883 | 56 | Male | Spinal Cord Lumbar | CNTR_1 | |
| NICHD | SAMN02768872 | / | Male | Spinal Cord Lumbar | CNTR_2 | |
| NICHD | SAMN02768875 | / | Male | Spinal Cord Lumbar | CNTR_3 | |
| WGS dataset | NYGS | CGND-HDA-00497 | 60 | Female | Spinal Cord Cervical | ALS1_497 |
| NYGS | CGND-HDA-00503 | 76 | Male | Spinal Cord Cervical | ALS2_503 | |
| NYGS | CGND-HDA-00536 | 58 | Female | Spinal Cord Cervical | ALS3_536 | |
| NYGS | CGND-HDA-00539 | 80 | Male | Spinal Cord Cervical | ALS4_539 | |
| NYGS | CGND-HDA-05608 | / | Male | Spinal Cord Lumbar | ALS5_5608 | |
| NYGS | CGND-HDA-05609 | / | Male | Spinal Cord Lumbar | ALS6_5609 | |
| NYGS | CGND-HDA-05610 | / | Female | Spinal Cord Lumbar | ALS7_5610 | |
| NYGS | CGND-HDA-05611 | 75 | Female | Spinal Cord Lumbar | ALS8_5611 | |
| NYGS | CGND-HDA-00596 | 65 | Female | Spinal Cord Cervical | CNTR1_596 | |
| NYGS | CGND-HDA-00597 | 50 | Female | Spinal Cord Cervical | CNTR2_597 | |
| NYGS | CGND-HDA-00598 | 16 | Female | Spinal Cord Cervical | CNTR3_598 | |
| NYGS | CGND-HDA-00599 | 61 | Male | Spinal Cord Cervical | CNTR4_599 |
| Official Gene Symbol | Gene Name | N° Variants WGS (WES) | WGS | WES | DisGeNET | ALSoD |
|---|---|---|---|---|---|---|
| FHIT | fragile histidine triad diadenosine triphosphatase | 26 (0) | X | |||
| MSRA | methionine sulfoxide reductase A | 20 (0) | X | |||
| SLC25A21 | solute carrier family 25 member 21 | 15 (3) | X | X | ||
| CLYBL | citramalyl-CoA lyase | 11 (0) | X | |||
| IMMP2L | inner mitochondrial membrane peptidase subunit 2 | 11 (0) | X | |||
| SND1 | staphylococcal nuclease and tudor domain containing 1 | 11 (0) | X | |||
| CHCHD3 | coiled-coil-helix-coiled-coil-helix domain containing 3 | 10 (0) | X | |||
| CHCHD6 | coiled-coil-helix-coiled-coil-helix domain containing 6 | 7 (0) | X | |||
| GPD2 | glycerol-3-phosphate dehydrogenase 2 | 7 (0) | X | |||
| OXR1 | oxidation resistance 1 | 7 (0) | X | X | ||
| SUGCT | succinyl-CoA:glutarate-CoA transferase | 7 (0) | X | |||
| AFG1L | AFG1 like ATPase | 6 (0) | X | |||
| BCKDHB | branched chain keto acid dehydrogenase E1 subunit beta | 6 (0) | X | |||
| DNAJC11 | DnaJ heat shock protein family | 4 (2) | X | X | ||
| MRRF | mitochondrial ribosome recycling factor | 5 (1) | X | X | ||
| PCCA | propionyl-CoA carboxylase subunit alpha | 6 (0) | X | |||
| PDSS2 | decaprenyl diphosphate synthase subunit 2 | 6 (0) | X | |||
| VPS13D | vacuolar protein sorting 13 homolog D | 1 (5) | X | X | ||
| ETFA | electron transfer flavoprotein subunit alpha | 5 (1) | X | X | ||
| IDE | insulin degrading enzyme | 5 (0) | X | |||
| METAP1D | methionyl aminopeptidase type 1D, mitochondrial | 5 (0) | X | |||
| MRPS6 | mitochondrial ribosomal protein S6 | 5 (0) | X | |||
| MTX2 | metaxin 2 | 5 (0) | X | |||
| PC | pyruvate carboxylase | 2 (3) | X | X | ||
| PRELID2 | PRELI domain containing 2 | 1 (4) | X | X | ||
| SFXN5 | sideroflexin 5 | 5 (0) | X | |||
| SLC25A26 | solute carrier family 25 member 26 | 5 (0) | X | |||
| STX17 | syntaxin 17 | 3 (2) | X | X | ||
| VWA8 | von Willebrand factor A domain containing 8 | 4 (1) | X | X | ||
| ACACA | acetyl-CoA carboxylase alpha | 4 (0) | X | |||
| ACSM3 | acyl-CoA synthetase medium chain family member 3 | 2 (2) | X | X | ||
| BCL2 | BCL2 apoptosis regulator | 4 (0) | X | |||
| GFER | growth factor, augmenter of liver regeneration | 2 (2) | X | X | ||
| HIBADH | 3-hydroxyisobutyrate dehydrogenase | 4 (0) | X | |||
| METTL8 | methyltransferase 8, tRNA N3-cytidine | 4 (0) | X | |||
| NARS2 | asparaginyl-tRNA synthetase 2, mitochondrial | 4 (0) | X | |||
| NUBPL | NUBP iron-sulfur cluster assembly factor, mitochondrial | 4 (0) | X | |||
| OGG1 | 8-oxoguanine DNA glycosylase | 2 (2) | X | X | X | |
| OPA1 | OPA1 mitochondrial dynamin like GTPase | 2 (2) | X | X | X | |
| PDE2A | phosphodiesterase 2A | 4 (0) | X | |||
| SFXN2 | sideroflexin 2 | 0 (4) | X | |||
| SPHKAP | SPHK1 interactor, AKAP domain containing | 4 (0) | X | |||
| NDUFV2 | NADH:ubiquinone oxidoreductase core subunit V2 | 4 (0) | X | |||
| SLC25A35 | solute carrier family 25 member 35 | 4 (0) | X | |||
| TSFM | Ts translation elongation factor, mitochondrial | 2 (1) | X | X | ||
| ALDH1L1 | aldehyde dehydrogenase 1 family member L1 | 3 (0) | X | |||
| ARG2 | arginase 2 | 3 (0) | X | |||
| ATP5F1D | ATP synthase F1 subunit delta | 3 (0) | X | |||
| BCL2L1 | BCL2 like 1 | 3 (0) | X | |||
| CCDC51 | coiled-coil domain containing 51 | 3 (0) | X | |||
| CLPB | ClpB family mitochondrial disaggregase | 1 (2) | X | X | ||
| COMT | catechol-O-methyltransferase | 3 (0) | X | |||
| CRLS1 | cardiolipin synthase 1 | 2 (1) | X | X | ||
| CYB5R3 | cytochrome b5 reductase 3 | 3 (0) | X | |||
| DBT | dihydrolipoamide branched chain transacylase E2 | 3 (0) | X | |||
| DELE1 | DAP3 binding cell death enhancer 1 | 2 (1) | X | X | ||
| DMGDH | dimethylglycine dehydrogenase | 3 (0) | X | |||
| DMPK | DM1 protein kinase | 1 (2) | X | X | ||
| GADD45GIP1 | GADD45G interacting protein 1 | 1 (2) | X | X | ||
| GATM | glycine amidinotransferase | 3 (0) | X | |||
| GLDC | glycine decarboxylase | 3 (0) | X | |||
| GLS | glutaminase | 3 (0) | X | X | ||
| LARS2 | leucyl-tRNA synthetase 2, mitochondrial | 2 (1) | X | X | ||
| LYRM2 | LYR motif containing 2 | 1 (2) | X | X | ||
| MCU | mitochondrial calcium uniporter | 3 (0) | X | X | ||
| MICU2 | mitochondrial calcium uptake 2 | 3 (0) | X | |||
| MRPL1 | mitochondrial ribosomal protein L1 | 3 (0) | X | |||
| MRPS27 | mitochondrial ribosomal protein S27 | 3 (0) | X | |||
| MTHFD1L | methylenetetrahydrofolate dehydrogenase | 3 (0) | X | |||
| NBR1 | NBR1 autophagy cargo receptor | 2 (1) | X | X | ||
| SHMT2 | serine hydroxymethyltransferase 2 | 4 (0) | X | |||
| NDUFS2 | NADH:ubiquinone oxidoreductase core subunit S2 | 3 (0) | X | |||
| NRDC | nardilysin convertase | 2 (1) | X | X | ||
| OCIAD1 | OCIA domain containing 1 | 2 (1) | X | X | ||
| OSBPL1A | oxysterol binding protein like 1A | 3 (0) | X | |||
| OXCT1 | 3-oxoacid CoA-transferase 1 | 3 (0) | X | |||
| PARK7 | Parkinsonism associated deglycase | 1 (2) | X | X | X | X |
| PDHX | pyruvate dehydrogenase complex component X | 2 (1) | X | X | ||
| PDSS1 | decaprenyl diphosphate synthase subunit 1 | 3 (0) | X | X | ||
| PNKD | PNKD metallo-beta-lactamase domain containing | 3 (0) | X | |||
| PNPLA8 | patatin like phospholipase domain containing 8 | 3 (0) | X | |||
| POLQ | DNA polymerase theta | 2 (1) | X | X | ||
| PREPL | prolyl endopeptidase like | 1 (2) | X | X | ||
| RARS2 | arginyl-tRNA synthetase 2, mitochondrial | 2 (1) | X | X | ||
| RTN4IP1 | reticulon 4 interacting protein 1 | 1 (2) | X | X | ||
| MTHFD2L | methylenetetrahydrofolate dehydrogenase | 5 (0) | X | X | ||
| SLC25A3 | solute carrier family 25 member 3 | 0 (3) | X | |||
| SPIRE1 | spire type actin nucleation factor 1 | 3 (0) | X | |||
| SUOX | sulfite oxidase | 1 (2) | X | X | ||
| TMEM65 | transmembrane protein 65 | 2 (1) | X | X |
| ALS | CNTR | p Value | |
|---|---|---|---|
| Type of variant | 0.0001 * | ||
| Het | 17/51 (33%) | 2/70 (3%) | |
| Hom | 34/51 (67%) | 68/70 (97%) | |
| Distribution of variants | 0.2250 * | ||
| Control Region | 18/51 (35%) | 17/70 (24%) | |
| Coding Region | 33/51 (65%) | 53/70 (76%) | |
| CR sites | 0.8061 | ||
| CR HV1 | 6/18 (52%) | 5/17 (29%) | |
| CR HV2 | 9/18 (48%) | 7/17 (41%) | |
| CR HV3 | 1/18 (6%) | 1/17 (6%) | |
| CR no-HV | 2/18 (11%) | 4/17 (24%) | |
| CR Functional Domains | |||
| CENTRAL DOMAIN | 3/7 (43%) | 6/6 (100%) | |
| CSB | 0/7 (0%) | 0/6 (0%) | |
| ETAS | 4/7 (57%) | 0/6 (0%) | |
| LSP | 0/7 (0%) | 0/6 (0%) | |
| Coding regions | 0.4854 | ||
| MT-ND1 | 2/20 (10%) | 2/36 (6%) | |
| MT-ND2 | 3/20 (15%) | 7/36 (19%) | |
| MT-CO1 | 4/20 (20%) | 3/36 (8%) | |
| MT-CO2 | 2/20 (10%) | 2/36 (6%) | |
| MT-ATP6 | 4/20 (20%) | 3/36 (8%) | |
| MT-CO3 | 0/20 (0%) | 2/36 (6%) | |
| MT-ND4 | 0/20 (0%) | 4/36 (11%) | |
| MT-ND5 | 1/20 (5%) | 5/36 (14%) | |
| MT-ND6 | 0/20 (0%) | 1/36 (3%) | |
| MT-CYB | 4/20 (20%) | 7/36 (19%) | |
| rRNA& tRNA genes | 0.1052 | ||
| 12s rRNA | 9/13 (69%) | 8/17 (47%) | |
| 16s rRNA | 1/13 (8%) | 7/17 (41%) | |
| tRNA | 3/13 (23%) | 1/17 (6%) | |
| Non-coding nt | 0/13 (0%) | 1/17 (6%) | |
| Type of substitution | 0.0527 | ||
| A>G | 21/51 (41%) | 34/69 (49%) | |
| C>T | 6/51 (12%) | 13/69 (19%) | |
| G>A | 4/51 (8%) | 10/69 (15%) | |
| T>C | 20/51 (39%) | 12/69 (17%) |
| ALS | CNTR | p Value | |
|---|---|---|---|
| Type of variant | 0.0833 * | ||
| Het | 154/321 (48%) | 178/324 (55%) | |
| Hom | 167/321 (52%) | 146/324 (45%) | |
| Distribution of variants | 0.0482 * | ||
| Control Region | 74/321 (23%) | 54/324 (17%) | |
| Coding Region | 247/321 (77%) | 270/324 (83%) | |
| CR sites | 0.7145 | ||
| CR HV1 | 18/74 (24%) | 17/54 (29%) | |
| CR HV2 | 32/74 (43%) | 23/54 (41%) | |
| CR HV3 | 8/74 (11%) | 6/54 (6%) | |
| CR no-HV | 16/74 (22%) | 8/54 (24%) | |
| CR Functional Domains | 0.3574 | ||
| CENTRAL DOMAIN | 16/29 (55%) | 10/26 (38%) | |
| CSB | 1/29 (3%) | 1/26 (4%) | |
| ETAS | 5/29 (17%) | 10/26 (38%) | |
| LSP | 7/29 (24%) | 5/26 (19%) | |
| Coding regions | 0.6290 | ||
| MT-ND1 | 15/190 (8%) | 14/211 (7%) | |
| MT-ND2 | 19/190 (10%) | 27/211 (13%) | |
| MT-CO1 | 12/190 (6%) | 21/211 (10%) | |
| MT-CO2 | 9/190 (5%) | 6/211 (3%) | |
| MT-ATP8 | 2/190 (1%) | 1/211 (0%) | |
| MT-ATP6 | 16/190 (8%) | 19/211 (9%) | |
| MT-CO3 | 8/190 (4%) | 11/211 (5%) | |
| MT-ND3 | 4/190 (2%) | 7/211 (3%) | |
| MT-ND4L | 6/190 (3%) | 8/211 (4%) | |
| MT-ND4 | 25/190 (13%) | 29/211 (14%) | |
| MT-ND5 | 28/190 (15%) | 33/211 (16%) | |
| MT-ND6 | 6/190 (3%) | 9/211 (4%) | |
| MT-CYB | 40/190 (21%) | 26/211 (12%) | |
| rRNA & tRNA genes | 0.3456 | ||
| 12s rRNA | 24/57 (42%) | 21/59 (36%) | |
| 16s rRNA | 22/57 (39%) | 28/59 (47%) | |
| tRNA | 9/57 (16%) | 5/59 (8%) | |
| Non-coding nt | 2/57 (3%) | 5/59 (8%) | |
| Type of substitution | 0.002 | ||
| A>G | 92/303 (30%) | 54/308 (17%) | |
| C>T | 46/303 (15%) | 64/308 (21%) | |
| G>A | 129/303 (43%) | 144/308 (46%) | |
| T>C | 36/303 (12%) | 46/308 (15%) | |
| Type of substitution (Het) | 0.675 | ||
| A>G | 8/139 (5%) | 8/166 (5%) | |
| C>T | 21/139 (15%) | 33/166 (20%) | |
| G>A | 105/139 (76%) | 121/166 (73%) | |
| T>C | 5/139 (4%) | 4/166 (2%) | |
| Type of substitution (Hom) | 0.007 | ||
| A>G | 84/164 (51%) | 46/142 (32%) | |
| C>T | 25/164 (15%) | 31/142 (22%) | |
| G>A | 24/164 (15%) | 23/142 (16%) | |
| T>C | 31/164 (19%) | 42/142 (30%) |
| Dataset | IPA Category | Pathway p-Value a | Ratio b | Gene Symbol |
|---|---|---|---|---|
| WES dataset | Mitochondrial Dysfunction | 1.01 × 10−7 | 15/345 | ATP5F1A, CYC1, MT-CO1, MT-CO2, DNM1L, GPX4, HTRA2, NDUFV1, NDUFB6, OPA1, PARK7, PDHX, PDHB, SIRT3, UQCR10 |
| Tryptophan Degradation III | 9.83 × 10−6 | 5/23 | GCDH, HADH, HSD17B4, KMO, PARK7 | |
| Glutaryl-CoA Degradation | 6.9 × 10−5 | 4/16 | GCDH, HADH, HSD17B4, PARK7 | |
| Oxidative Phosphorylation | 9.72 × 10−5 | 7/112 | ATP5F1A, CYC1, MT-CO1, MT-CO2, NDUFV1, NDUFB6, UQCR10, | |
| Sirtuin signaling Pathway | 5.83 × 10−3 | 8/292 | ATP5F1A, BCL2L11, CYC1, NDUFB6, NDUFV1, OGG1, SIRT3, SIRT4 | |
| WGS dataset | Mitochondrial Dysfunction | 2.85 × 10−32 | 58/345 | ACADL, ACO2, ADCY10, ARG2, ATP5F1A, ATP5F1D, ATP5PD, ATP5PO, ATPAF1, BBC3, BCL2, CACNA1G, CASP3, COX10, COX15, COX4I1, COX8C, CYB5B, CYB5R3, DLD, DNM1L, GPD2, GSR, ITPR3, MCU, MT-ATP6, MT-CO1, MT-CO2, MT-CYB, MT-ND1, MT-ND2, MT-ND4, MT-ND5, MT-ND6, MT-ND4L, NDUFA6, NDUFA7, NDUFA10, NDUFA4L2, NDUFB3, NDUFB5, NDUFS2, NDUFS5, NDUFV2, OPA1, PARK7, PDHX, PINK1, PRKACA, PRKN, RHOT1, SDHC, SOD1, SURF1, TFAM, UCP2, UQCRB, VDAC1 |
| Oxidative Phosphorylation | 1.98 × 10−22 | 30/112 | ATP5F1A, ATP5F1D, ATP5PD, ATP5PO, ATPAF1, COX10, COX15, COX4I1, COX8C, CYB5B, MT-ATP6, MT-CO1, MT-CO2, MT-CYB, MT-ND1, MT-ND2, MT-ND4, MT-ND5, MT-ND4L, NDUFA6, NDUFA7, NDUFA10, NDUFB3, NDUFB5, NDUFS2, NDUFS5, NDUFV2, SDHC, SURF1, UQCRB, | |
| Sirtuin signaling Pathway | 1.98 × 10−14 | 35/292 | ACADL, ARG2, ATP5F1A, ATP5F1D, CPS1, CPT1A, CPT1B, GLS, MT-ATP6, MT-CYB, MT-ND1, MT-ND2, MT-ND4, MT-ND5, MT-ND6, MT-ND4L, NDUFA6, NDUFA7, NDUFA10, NDUFA4L2, NDUFB3, NDUFB5, NDUFS2, NDUFS5, NDUFV2, OGG1, PCK2, SDHC, SIRT5, SLC25A5, SLC25A6, SOD1, TIMM23, UCP2, VDAC1 | |
| Granzyme A signalling | 4.22 × 10−10 | 7/112 | LMNB2, MT-ND1, MT-ND2, MT-ND4, MT-ND5, MT-ND6, MT-ND4L, NDUFA6, NDUFA7, NDUFA10, NDUFA4L2, NDUFB3, NDUFB5, NDUFS2, NDUFS5, NDUFV2 | |
| Valine Degradation I | 1.04 × 10−8 | 9/20 | ABAT, ACADSB, ALDH6A1, BCKDHA, BCKDHB, DBT, DLD, HADHA, HIBADH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cox, S.N.; Lo Giudice, C.; Lavecchia, A.; Poeta, M.L.; Chiara, M.; Picardi, E.; Pesole, G. Mitochondrial and Nuclear DNA Variants in Amyotrophic Lateral Sclerosis: Enrichment in the Mitochondrial Control Region and Sirtuin Pathway Genes in Spinal Cord Tissue. Biomolecules 2024, 14, 411. https://doi.org/10.3390/biom14040411
Cox SN, Lo Giudice C, Lavecchia A, Poeta ML, Chiara M, Picardi E, Pesole G. Mitochondrial and Nuclear DNA Variants in Amyotrophic Lateral Sclerosis: Enrichment in the Mitochondrial Control Region and Sirtuin Pathway Genes in Spinal Cord Tissue. Biomolecules. 2024; 14(4):411. https://doi.org/10.3390/biom14040411
Chicago/Turabian StyleCox, Sharon Natasha, Claudio Lo Giudice, Anna Lavecchia, Maria Luana Poeta, Matteo Chiara, Ernesto Picardi, and Graziano Pesole. 2024. "Mitochondrial and Nuclear DNA Variants in Amyotrophic Lateral Sclerosis: Enrichment in the Mitochondrial Control Region and Sirtuin Pathway Genes in Spinal Cord Tissue" Biomolecules 14, no. 4: 411. https://doi.org/10.3390/biom14040411
APA StyleCox, S. N., Lo Giudice, C., Lavecchia, A., Poeta, M. L., Chiara, M., Picardi, E., & Pesole, G. (2024). Mitochondrial and Nuclear DNA Variants in Amyotrophic Lateral Sclerosis: Enrichment in the Mitochondrial Control Region and Sirtuin Pathway Genes in Spinal Cord Tissue. Biomolecules, 14(4), 411. https://doi.org/10.3390/biom14040411