
Mapping Expression Quantitative Trait Loci Targeting Candidate Genes for Pregnancy in Beef Cows

 , , , and
, , , and
Abstract
1. Introduction
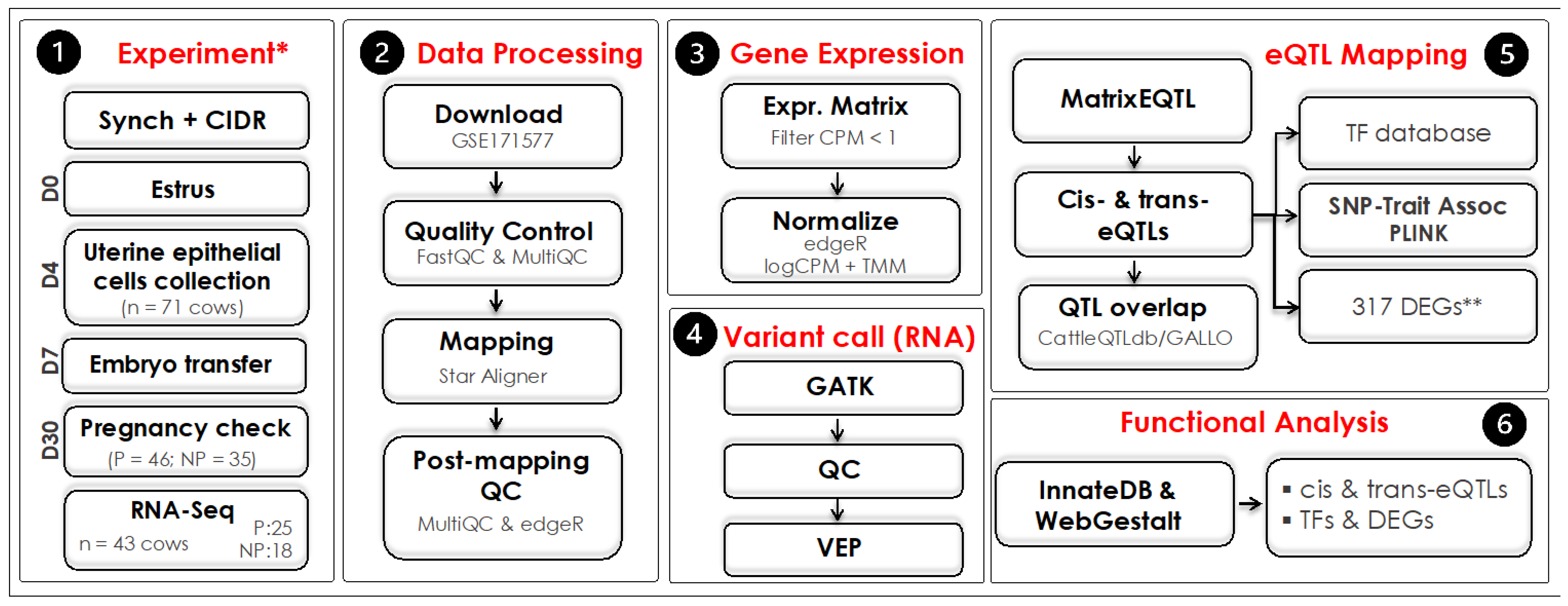
2. Materials and Methods
2.1. Data Collection, Quality Control, and RNA-Seq Mapping
2.2. RNA-Seq Variant Calling and Quality Control
2.3. Variant Functional Prediction
2.4. The eQTL Mapping and Annotation
2.5. SNP Trait Association and Cattle QTLdb Over-Representation Analyses
2.6. Functional Over-Representation Analysis
3. Results
- Identified the SNPs affecting the expression of genes from the uterine luminal epithelial cells;
- Investigated if the SNPs were associated with pregnancy outcomes, affected differentially expressed genes, and/or were harbored in known QTL regions for reproduction traits;
- Identified over-represented pathways and biological processes that underlie eQTL-modulated genes.
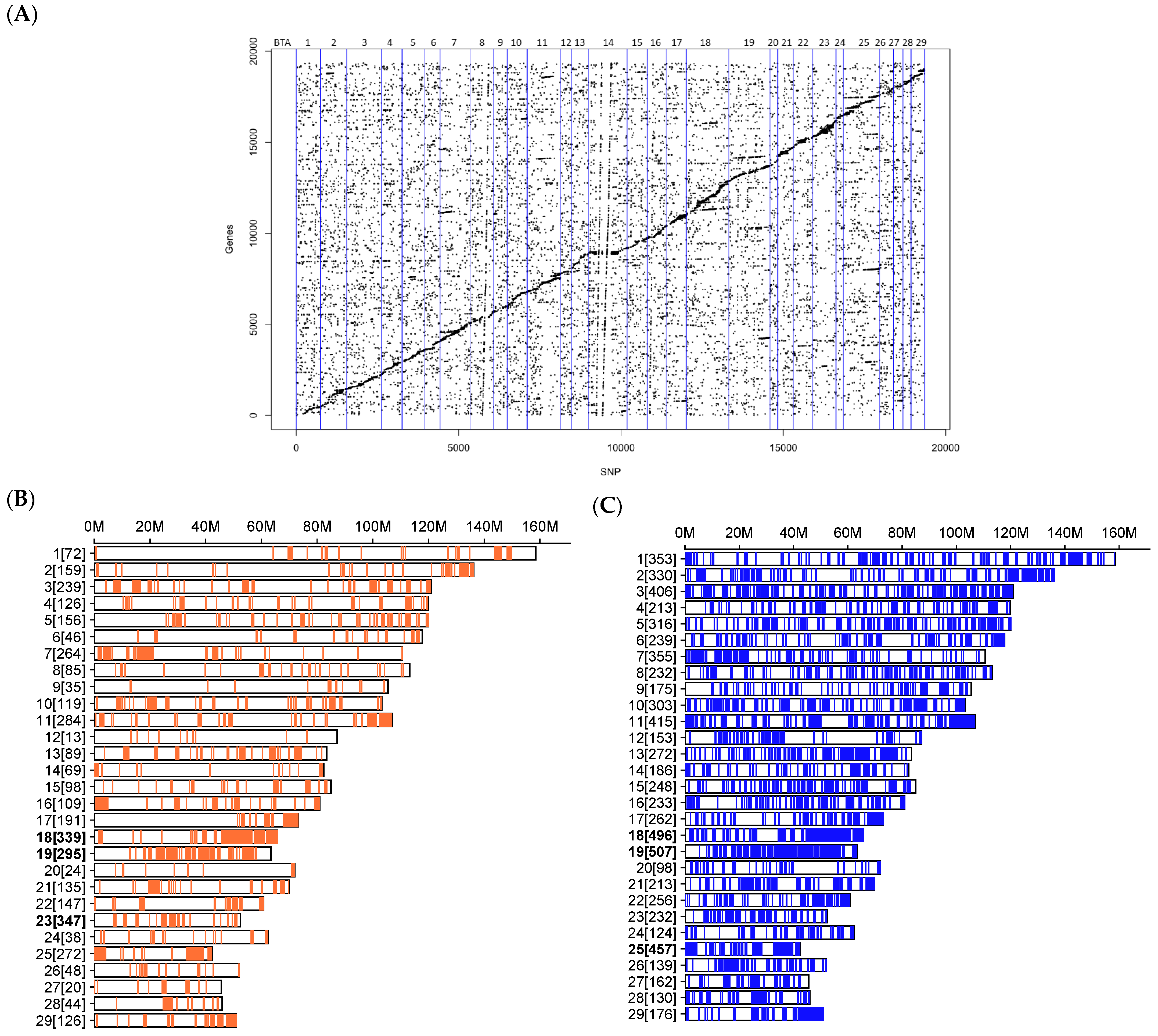
3.1. Variant Calling Analysis Retrieved 203,404 Unique SNPs
3.2. Cis and Trans eQTLs Affected the Expression of 3157 Genes
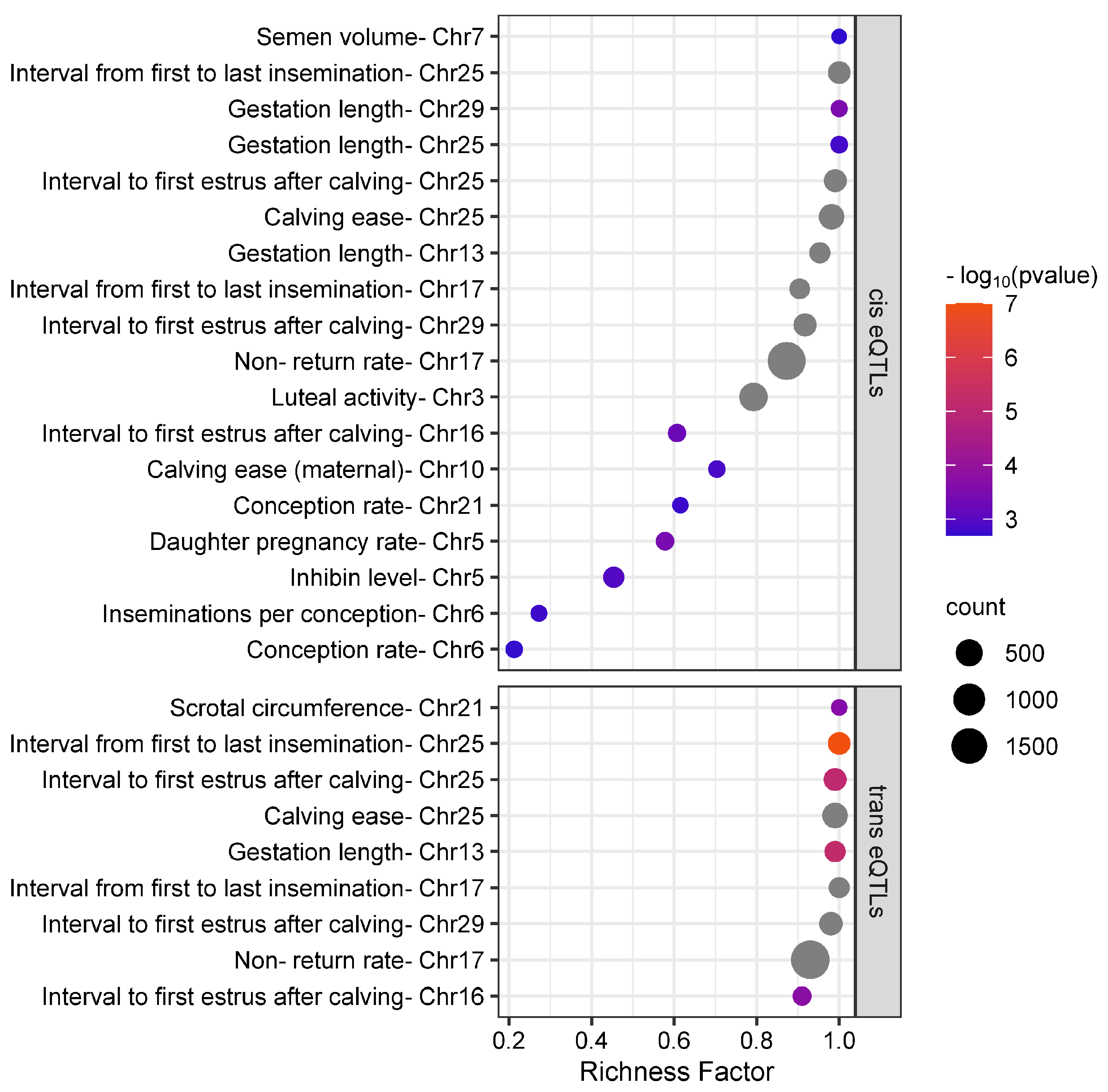
3.3. eQTLs Were Over-Represented in Reproduction-Related QTL Regions
3.4. Differentially Expressed Genes between Pregnant and Nonpregnant Cows Were Affected by eQTLs
3.5. eQTL-Modulated Genes Are Over-Represented in Pathways Related to the Metabolism and Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fleming, A.; Baes, C.F.; Martin, A.A.A.; Chud, T.C.S.; Malchiodi, F.; Brito, L.F.; Miglior, F. Symposium review: The choice and collection of new relevant phenotypes for fertility selection. J. Dairy Sci. 2019, 102, 3722–3734. [Google Scholar] [CrossRef]
- Veerkamp, R.F.; Beerda, B. Genetics and genomics to improve fertility in high producing dairy cows. Theriogenology 2007, 68, S266–S273. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E. Early pregnancy: Concepts, challenges, and potential solutions. Anim. Front. 2013, 3, 48–55. [Google Scholar] [CrossRef]
- Johnston, D.J. Genetic improvement of reproduction in beef cattle. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar]
- Alexandre, P.A.; Porto-Neto, L.R.; Hine, B.C.; Samaraweera, A.M.; Byrne, A.I.; Ingham, A.B.; Duff, C.J.; Reverter, A. Development of female fertility indicator traits for the Angus HeiferSELECT genomic tool. Proc. Assoc. Advmt. Anim. Breed. Genet. 2023, 25, 67–70. [Google Scholar]
- Van den Berg, I.V.; Hayes, B.J.; Chamberlain, A.J.; Goddard, M.E. Overlap between eQTL and QTL associated with production traits and fertility in dairy cattle. BMC Genom. 2019, 20, 291. [Google Scholar] [CrossRef]
- Stegemiller, M.R.; Murdoch, G.K.; Rowan, T.N.; Davenport, K.M.; Becker, G.M.; Hall, J.B.; Murdoch, B.M. Genome-wide association analyses of fertility traits in beef heifers. Genes 2021, 12, 217. [Google Scholar] [CrossRef]
- Minten, M.A.; Bilby, T.R.; Bruno, R.G.S.; Allen, C.C.; Madsen, C.A.; Wang, Z.; Sawyer, J.E.; Tibary, A.; Neibergs, H.L.; Geary, T.W.; et al. Effects of fertility on gene expression and function of the bovine endometrium. PLoS ONE 2013, 8, e69444. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Prioritizing candidate genes for fertility in dairy cows using gene-based analysis, functional annotation and differential gene expression. BMC Genom. 2019, 20, 255. [Google Scholar] [CrossRef] [PubMed]
- Forutan, M.; Engle, B.N.; Chamberlain, A.J.; Ross, E.M.; Nguyen, L.T.; D’occhio, M.; Snr, A.C.; Kho, E.A.; Fordyce, G.; Speight, S.; et al. Integrating genome-wide association and expression quantitative trait loci (eQTL) analyses identifies genes affecting fertility in cattle and suggests a common set of genes regulating fertility in mammals. Res. Sq. 2023. [Google Scholar] [CrossRef]
- McGettigan, P.A.; Browne, J.A.; Carrington, S.D.; Crowe, M.A.; Fair, T.; Forde, N.; Loftus, B.J.; Lohan, A.; Lonergan, P.; Pluta, K.; et al. Fertility and genomics: Comparison of gene expression in contrasting reproductive tissues of female cattle. Reprod. Fertil. Dev. 2016, 28, 11. [Google Scholar] [CrossRef]
- Ross, E.M.; Sanjana, H.; Nguyen, L.T.; Cheng, Y.Y.; Moore, S.S.; Hayes, B.J. Extensive variation in gene expression is revealed in 13 fertility-related genes using RNA-Seq, ISO-Seq, and CAGE-Seq from Brahman cattle. Front. Genet. 2022, 13, 784663. [Google Scholar] [CrossRef]
- Geary, T.W.; Burns, G.W.; Moraes, J.G.N.; Moss, J.I.; Denicol, A.C.; Dobbs, K.B.; Ortega, M.S.; Hansen, P.J.; Wehrman, M.E.; Neibergs, H.; et al. Identification of beef heifers with superior uterine capacity for pregnancy. Biol. Reprod. 2016, 95, 47. [Google Scholar] [CrossRef]
- Martins, T.; Sponchiado, M.; Silva, F.A.C.C.; Estrada-Cortés, E.; Hansen, P.J.; Peñagaricano, F.; Binelli, M. Progesterone-dependent and progesterone-independent modulation of luminal epithelial transcription to support pregnancy in cattle. Physiol. Genom. 2022, 54, 71–85. [Google Scholar] [CrossRef]
- Silva, F.A.C.C.; Martins, T.; Sponchiado, M.; Rocha, C.C.; Pohler, K.G.; Peñagaricano, F.; Binelli, M. Hormonal profile prior to luteolysis modulates the uterine luminal transcriptome in the subsequent cycle in beef cross-bred cows. Biol. Reprod. 2023, 108, 922–935. [Google Scholar] [CrossRef]
- Moraes, J.G.N.; Behura, S.K.; Geary, T.W.; Hansen, P.J.; Neibergs, H.L.; Spencer, T.E. Uterine influences on conceptus development in fertility-classified animals. Proc. Natl. Acad. Sci. USA 2018, 115, E1749–E1758. [Google Scholar] [CrossRef]
- Kertz, N.C.; Banerjee, P.; Dyce, P.W.; Diniz, W.J.S. Harnessing Genomics and Transcriptomics Approaches to Improve Female Fertility in Beef Cattle—A Review. Animals 2023, 13, 3284. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kadarmideen, H.N.; Mazzoni, G. Transcriptomics–genomics data integration and expression quantitative trait loci analyses in oocyte donors and embryo recipients for improving invitro production of dairy cattle embryos. Reprod. Fertil. Dev. 2019, 31, 55. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P. SRA-Explorer. Available online: https://sra-explorer.info/ (accessed on 13 May 2022).
- Diniz, W.J.S.; Banerjee, P.; Rodning, S.P.; Dyce, P.W. Machine learning-based co-expression network analysis unravels potential fertility-related genes in beef cows. Animals 2022, 12, 2715. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; FASTQC. A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 January 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinforma. 2013, 43, 1. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.R.; Crowgey, E.L. Accelerating next generation sequencing data analysis: An evaluation of optimized best practices for Genome Analysis Toolkit algorithms. Genom. Inform. 2020, 18, e10. [Google Scholar] [CrossRef] [PubMed]
- Brouard, J.-S.; Bissonnette, N. Variant Calling from RNA-seq Data Using the GATK Joint Genotyping Workflow. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2022; Volume 2493, pp. 205–233. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.W.; Brinkman, F.S.L.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Albert, F.W.; Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015, 16, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Griesemer, D.; Xue, J.R.; Reilly, S.K.; Ulirsch, J.C.; Kukreja, K.; Davis, J.R.; Kanai, M.; Yang, D.K.; Butts, J.C.; Guney, M.H.; et al. Genome-wide functional screen of 3′UTR variants uncovers causal variants for human disease and evolution. Cell 2021, 184, 5247–5260.e19. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; van den Berg, I.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. USA 2019, 116, 19398–19408. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Pastinen, T. The study of eQTL variations by RNA-seq: From SNPs to phenotypes. Trends Genet. 2011, 27, 72–79. [Google Scholar] [CrossRef] [PubMed]
- De Souza, M.M.; Zerlotini, A.; Geistlinger, L.; Tizioto, P.C.; Taylor, J.F.; Rocha, M.I.P.; Diniz, W.J.S.; Coutinho, L.L.; Regitano, L.C.A. A comprehensive manually-curated compendium of bovine transcription factors. Sci. Rep. 2018, 8, 13747. [Google Scholar] [CrossRef] [PubMed]
- Cheung, V.G.; Nayak, R.R.; Wang, I.X.; Elwyn, S.; Cousins, S.M.; Morley, M.; Spielman, R.S. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol. 2010, 8, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Imakawa, K.; Matsuno, Y.; Fujiwara, H. New roles for EVs, miRNA and lncRNA in bovine embryo implantation. Front. Vet. Sci. 2022, 9, 944370. [Google Scholar] [CrossRef]
- De Souza Fonseca, P.A.; Suárez-Vega, A.; Cánovas, A. Unrevealing functional candidate genes for bovine fertility through RNA sequencing meta-analysis and regulatory elements networks of co-expressed genes and lncRNAs. Funct. Integr. Genom. 2022, 22, 1361–1376. [Google Scholar] [CrossRef]
- Bazer, F.W. Uterine protein secretions: Relationship to development of the conceptus. J. Anim. Sci. 1975, 41, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- França, M.R.; da Silva, M.I.S.; Pugliesi, G.; Van Hoeck, V.; Binelli, M. Evidence of endometrial amino acid metabolism and transport modulation by peri-ovulatory endocrine profiles driving uterine receptivity. J. Anim. Sci. Biotechnol. 2017, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Forde, N.; Simintiras, C.A.; Sturmey, R.; Mamo, S.; Kelly, A.K.; Spencer, T.E.; Bazer, F.W.; Lonergan, P. Amino acids in the uterine luminal fluid reflects the temporal changes in transporter expression in the endometrium and conceptus during early pregnancy in cattle. PLoS ONE 2014, 9, e100010. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, A.A.; Vale-Cruz, D.S.; Simmen, F.A.; Simmen, R.C.M. Uterine androgen receptors: Roles in estrogen-mediated gene expression and DNA synthesis. Biol. Reprod. 2004, 70, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Ekman, J.; Almkvist, A.; Saji, S.; Wang, L.; Warner, M.; Gustafsson, J.-A. Involvement of androgen receptor in 17β-estradiol-induced cell proliferation in rat uterus. Biol. Reprod. 2002, 67, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, M.; Yang, F.; Zhang, Y.; Ma, S.; Zhang, D.; Wang, X.; Sferruzzi-Perri, A.N.; Wu, X.; Brännström, M.; et al. Increased uterine androgen receptor protein abundance results in implantation and mitochondrial defects in pregnant rats with hyperandrogenism and insulin resistance. J. Mol. Med. 2021, 99, 1427–1446. [Google Scholar] [CrossRef] [PubMed]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Bao, W.; Wang, J.; Yang, T.; He, X.; Liao, Y.; Wan, X. FOXA1 promotes tumor cell proliferation through AR involving the Notch pathway in endometrial cancer. BMC Cancer 2014, 14, 78. [Google Scholar] [CrossRef]
- Vasquez, Y.M.; Wang, X.; Wetendorf, M.; Franco, H.L.; Mo, Q.; Wang, T.; Lanz, R.B.; Young, S.L.; Lessey, B.A.; Spencer, T.E.; et al. FOXO1 regulates uterine epithelial integrity and progesterone receptor expression critical for embryo implantation. PLoS Genet. 2018, 14, e1007787. [Google Scholar] [CrossRef]
- Rubel, C.A.; Wu, S.-P.; Lin, L.; Wang, T.; Lanz, R.B.; Li, X.; Kommagani, R.; Franco, H.L.; Camper, S.A.; Tong, Q.; et al. A Gata2-Dependent Transcription Network Regulates Uterine Progesterone Responsiveness and Endometrial Function. Cell Rep. 2016, 17, 1414–1425. [Google Scholar] [CrossRef]
- Kelleher, A.M.; Peng, W.; Pru, J.K.; Pru, C.A.; DeMayo, F.J.; Spencer, T.E. Forkhead box a2 (FOXA2) is essential for uterine function and fertility. Proc. Natl. Acad. Sci. USA 2017, 114, E1018–E1026. [Google Scholar] [CrossRef] [PubMed]
- Forde, N.; Mehta, J.P.; McGettigan, P.A.; Mamo, S.; Bazer, F.W.; Spencer, T.E.; Lonergan, P. Alterations in expression of endometrial genes coding for proteins secreted into the uterine lumen during conceptus elongation in cattle. BMC Genom. 2013, 14, 321. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, G.; Pedersen, H.S.; Rabaglino, M.B.; Hyttel, P.; Callesen, H.; Kadarmideen, H.N. Characterization of the endometrial transcriptome in early diestrus influencing pregnancy status in dairy cattle after transfer of in vitro-produced embryos. Physiol. Genom. 2020, 52, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Wintermantel, T.M.; Campbell, R.E.; Porteous, R.; Bock, D.; Gröne, H.-J.; Todman, M.G.; Korach, K.S.; Greiner, E.; Pérez, C.A.; Schütz, G.; et al. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 2006, 52, 271–280. [Google Scholar] [CrossRef]
- Madoz, L.V.; Rabaglino, M.B.; Migliorisi, A.L.; Jaureguiberry, M.; Perez Wallace, S.; Lorenti, N.; Domínguez, G.; Giuliodori, M.J.; de la Sota, R.L. Association between progesterone concentration and endometrial gene expression in dairy cows. Domest. Anim. Endocrinol. 2021, 74, 106481. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Haller, M.E.; Chadchan, S.B.; Kommagani, R.; Ma, L. Signaling through retinoic acid receptors is essential for mammalian uterine receptivity and decidualization. JCI Insight 2021, 6, e150254. [Google Scholar] [CrossRef] [PubMed]
- Musavi, S.A.A.; Yamashita, S.; Fujihara, T.; Masaka, H.; Islam, M.R.; Kim, S.; Gotoh, T.; Kawahara, M.; Tashiro, K.; Yamauchi, N. Analysis of differentially expressed genes and the promoters in bovine endometrium throughout estrus cycle and early pregnancy. Anim. Sci. J. 2018, 89, 1609–1621. [Google Scholar] [CrossRef]
- Salilew-Wondim, D.; Hölker, M.; Rings, F.; Ghanem, N.; Ulas-Cinar, M.; Peippo, J.; Tholen, E.; Looft, C.; Schellander, K.; Tesfaye, D. Bovine pretransfer endometrium and embryo transcriptome fingerprints as predictors of pregnancy success after embryo transfer. Physiol. Genom. 2010, 42, 201–218. [Google Scholar] [CrossRef]
- Walker, C.G.; Meier, S.; Littlejohn, M.D.; Lehnert, K.; Roche, J.R.; Mitchell, M.D. Modulation of the maternal immune system by the pre-implantation embryo. BMC Genom. 2010, 11, 474. [Google Scholar] [CrossRef]
- Hansen, P. The immunology of early pregnancy in farm animals. Reprod. Domest. Anim. 2011, 46, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Fair, T. The contribution of the maternal immune system to the establishment of pregnancy in cattle. Front. Immunol. 2015, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Forde, N.; Lonergan, P. Interferon-tau and fertility in ruminants. Reproduction 2017, 154, F33–F43. [Google Scholar] [CrossRef] [PubMed]
- Coyral-castel, S.; Ramé, C.; Monniaux, D.; Fréret, S.; Fabre-nys, C.; Fritz, S. Ovarian parameters and fertility of dairy cows selected for one QTL located on BTA3. Theriogenology 2011, 75, 1239–1250. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl ID | Gene Name | Number of eQTLs | ||
|---|---|---|---|---|
| Cis | Trans | Total | ||
| ENSBTAG00000052527 | ENSBTAG00000052527 | 37 | 178 | 215 |
| ENSBTAG00000053827 | ENSBTAG00000053827 | 93 | 113 | 206 |
| ENSBTAG00000007816 | ENSBTAG00000007816 | 3 | 195 | 198 |
| ENSBTAG00000027397 | PPP1R3D | 1 | 192 | 193 |
| ENSBTAG00000048353 | ENSBTAG00000048353 | 30 | 151 | 181 |
| ENSBTAG00000048470 | IFITM1 | 33 | 143 | 176 |
| ENSBTAG00000027075 | ENSBTAG00000027075 | 2 | 171 | 173 |
| ENSBTAG00000038050 | ZNF420 | 35 | 128 | 163 |
| ENSBTAG00000025782 | TNFSF8 | 1 | 137 | 138 |
| ENSBTAG00000033449 | ENSBTAG00000033449 | 1 | 128 | 129 |
| Enriched Chromosomes a | Number of Genes | Genes b |
|---|---|---|
| 18 | 21 | ARHGEF1, CLASRP, ENSBTAG00000023367, BICRA, LMTK3, PRR12, NAPSA, ENSBATG00000049577, CD37, ENSBATG00000045880, ENSBTAG00000051367, ENSBTAG00000053237, VSTM1, ZNF581, KMT5C, PTPRH, PPP6R1, TMEM86B, EPS8L1, SMIM17, ZNF470 |
| 19 | 24 | SCARF1, P2RX5,PITPNM3, VMO1, CAMTA2, PLD2, TMEM95, DLG4, KCTD11, NLGN2, SOX15, TMEM88, KDM6B, PIK3R5, GAS7, RARA, ENSBTAG00000024839, STAT5A, CCR10, CNTNAP1, TMEM106A, HIGD1B, FMNL1, ENSBTAG00000055302 |
| 22 | 5 | SYN2, PPARG, CHST13, MGLL, SLC41A3 |
| 25 | 8 | LAT, CD19, GDPD3, DOC2A, ENSBTAG00000046752, TMEM265, FBXL19, SEPTIN14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diniz, W.J.S.; Afonso, J.; Kertz, N.C.; Dyce, P.W.; Banerjee, P. Mapping Expression Quantitative Trait Loci Targeting Candidate Genes for Pregnancy in Beef Cows. Biomolecules 2024, 14, 150. https://doi.org/10.3390/biom14020150
Diniz WJS, Afonso J, Kertz NC, Dyce PW, Banerjee P. Mapping Expression Quantitative Trait Loci Targeting Candidate Genes for Pregnancy in Beef Cows. Biomolecules. 2024; 14(2):150. https://doi.org/10.3390/biom14020150
Chicago/Turabian StyleDiniz, Wellison J. S., Juliana Afonso, Nicholas C. Kertz, Paul W. Dyce, and Priyanka Banerjee. 2024. "Mapping Expression Quantitative Trait Loci Targeting Candidate Genes for Pregnancy in Beef Cows" Biomolecules 14, no. 2: 150. https://doi.org/10.3390/biom14020150
APA StyleDiniz, W. J. S., Afonso, J., Kertz, N. C., Dyce, P. W., & Banerjee, P. (2024). Mapping Expression Quantitative Trait Loci Targeting Candidate Genes for Pregnancy in Beef Cows. Biomolecules, 14(2), 150. https://doi.org/10.3390/biom14020150