
Identification of Molecular Markers Associated with Prostate Cancer Subtypes: An Integrative Bioinformatics Approach



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Differential Expression Analysis
- Metastatic versus primary PCa groups for GSE3325, GSE3933 and GSE68882 datasets;
- mCRPC versus PCa groups for GSE32269, GSE6811, GSE70770, GSE6752 and GSE35988 datasets;
- AR-driven versus non AR-driven groups for GSE101607;
- CHGA negative versus CHGA positive/SYP positive/SR negative groups were compared for GSE77930.
2.3. Over-Representation Analysis
2.4. Statistical Analysis
3. Results
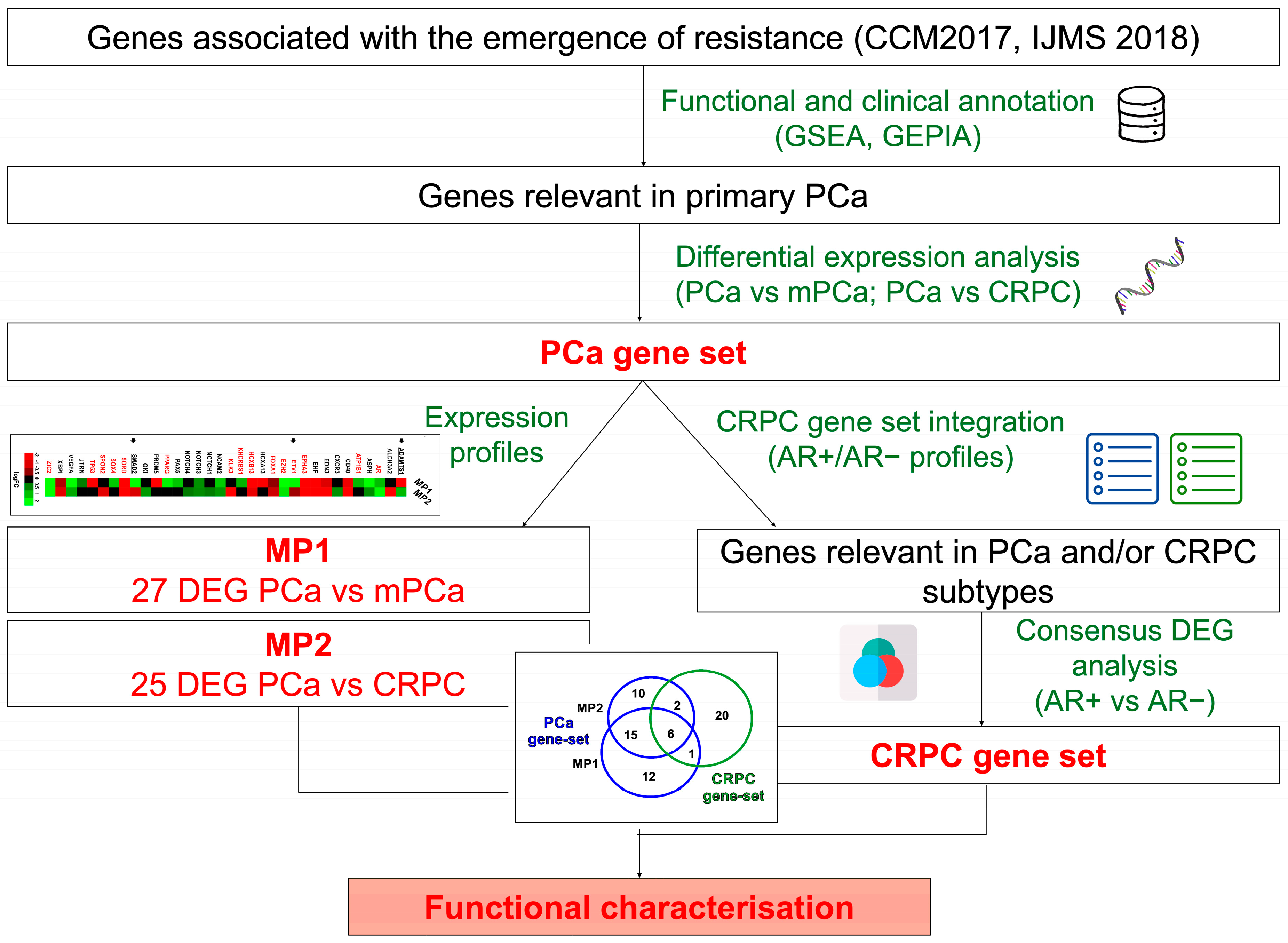
3.1. Identification and Characterization of Clinically Relevant Gene Sets in PCa Progression to Metastatic and Castration-Resistant Phenotypes
3.2. Evaluation of 50 Gene Set Alterations in PCa Tissue Samples at Different Evolutive Stages
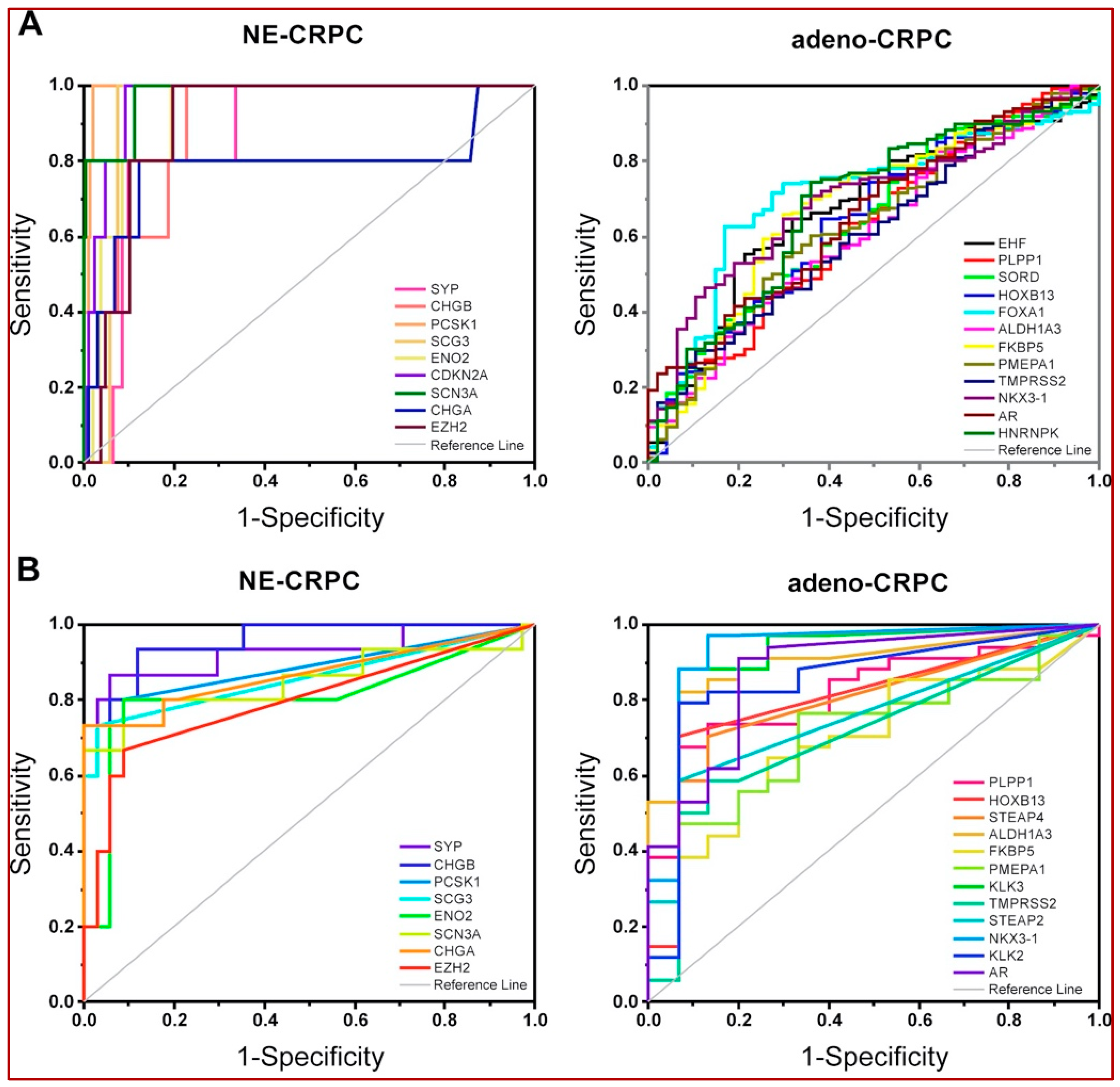
3.3. Identification of Genes Associated with Different CRPC Phenotypes
3.4. Validation of CRPC-Gene Set on Independent Datasets
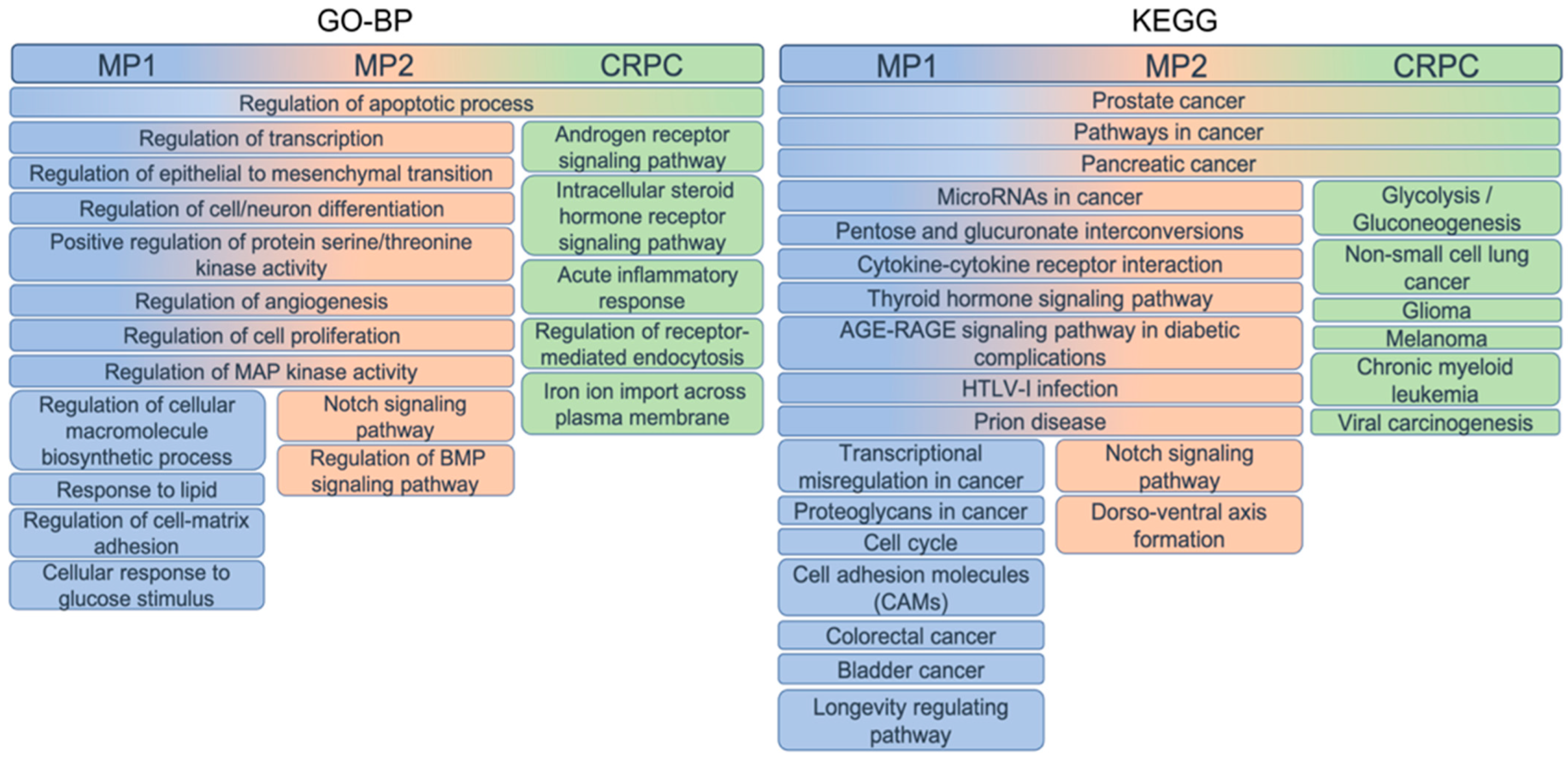
3.5. Functional Enrichment of Gene Sets
3.6. Investigating the Context-Specific Essentiality
3.7. Computational Drug Identification Based on the PCa- and CRPC-Gene Sets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pilleron, S.; Sarfati, D.; Janssen-Heijnen, M.; Vignat, J.; Ferlay, J.; Bray, F.; Soerjomataram, I. Global cancer incidence in older adults, 2012 and 2035: A population-based study: Global Cancer Incidence in Older Adults. Int. J. Cancer 2019, 144, 49–58. [Google Scholar] [CrossRef]
- Bungaro, M.; Buttigliero, C.; Tucci, M. Overcoming the mechanisms of primary and acquired resistance to new generation hormonal therapies in advanced prostate cancer: Focus on androgen receptor independent pathways. Cancer Drug Resist. 2020, 3, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; Izumi, K.; Mizokami, A. Undesirable Status of Prostate Cancer Cells after Intensive Inhibition of AR Signaling: Post-AR Era of CRPC Treatment. Biomedicines 2021, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Cattrini, C.; Zanardi, E.; Vallome, G.; Cavo, A.; Cerbone, L.; Di Meglio, A.; Fabbroni, C.; Latocca, M.; Rizzo, F.; Messina, C.; et al. Targeting androgen-independent pathways: New chances for patients with prostate cancer? Crit. Rev. Oncol. 2017, 118, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; Granata, I.; Capaia, M.; Piccirillo, M.; Guarracino, M.R.; Venè, R.; Brizzolara, A.; Petretto, A.; Inglese, E.; Morini, M.; et al. Adaptive phenotype drives resistance to androgen deprivation therapy in prostate cancer. Cell Commun. Signal. 2017, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, A.V.; Ellis, L. Epigenetic reprogramming: A key mechanism driving therapeutic resistance. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Roubaud, G.; Liaw, B.C.; Oh, W.K.; Mulholland, D.J. Strategies to avoid treatment-induced lineage crisis in advanced prostate cancer. Nat. Rev. Clin. Oncol. 2017, 14, 269–283. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.; Bedard, P.; Tortora, G.; Douillard, J.-Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin. Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Vellano, C.P.; White, M.G.; Andrews, M.C.; Chelvanambi, M.; Witt, R.G.; Daniele, J.R.; Titus, M.; McQuade, J.L.; Conforti, F.; Burton, E.M.; et al. Androgen receptor blockade promotes response to BRAF/MEK-targeted therapy. Nature 2022, 606, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sue, A.C.-H.; Bin Goh, W.W. Examining the practical limits of batch effect-correction algorithms: When should you care about batch effects? J. Genet. Genom. 2019, 46, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Zięba, A.; Stępnicki, P.; Matosiuk, D.; Kaczor, A.A. What are the challenges with multi-targeted drug design for complex diseases? Expert Opin. Drug Discov. 2022, 17, 673–683. [Google Scholar] [CrossRef]
- Catara, G.; Colanzi, A.; Spano, D. Combinatorial Strategies to Target Molecular and Signaling Pathways to Disarm Cancer Stem Cells. Front. Oncol. 2021, 11, 689131. [Google Scholar] [CrossRef] [PubMed]
- Bhal, S.; Kundu, C.N. Targeting crosstalk of signaling pathways in cancer stem cells: A promising approach for development of novel anti-cancer therapeutics. Med. Oncol. 2023, 40, 82. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene Expression Profiling Identifies Clinically Relevant Subtypes of Prostate Cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef]
- LaTulippe, E.; Satagopan, J.; Smith, A.; Scher, H.; Scardino, P.; Reuter, V.; Gerald, W.L. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002, 62, 4499–4506. [Google Scholar]
- Cai, C.; Wang, H.; He, H.H.; Chen, S.; He, L.; Ma, F.; Mucci, L.; Wang, Q.; Fiore, C.; Sowalsky, A.G.; et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Investig. 2013, 123, 1109–1122. [Google Scholar] [CrossRef]
- Tamura, K.; Furihata, M.; Tsunoda, T.; Ashida, S.; Takata, R.; Obara, W.; Yoshioka, H.; Daigo, Y.; Nasu, Y.; Kumon, H.; et al. Molecular Features of Hormone-Refractory Prostate Cancer Cells by Genome-Wide Gene Expression Profiles. Cancer Res. 2007, 67, 5117–5125. [Google Scholar] [CrossRef] [PubMed]
- Ross-Adams, H.; Lamb, A.D.; Dunning, M.J.; Halim, S.; Lindberg, J.; Massie, C.M.; Egevad, L.A.; Russell, R.; Ramos-Montoya, A.; Vowler, S.L.; et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine 2015, 2, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Chandran, U.R.; Ma, C.; Dhir, R.; Bisceglia, M.; Lyons-Weiler, M.; Liang, W.; Michalopoulos, G.; Becich, M.; Monzon, F.A. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 2007, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Ylitalo, E.B.; Thysell, E.; Jernberg, E.; Lundholm, M.; Crnalic, S.; Egevad, L.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Subgroups of Castration-resistant Prostate Cancer Bone Metastases Defined through an Inverse Relationship between Androgen Receptor Activity and Immune Response. Eur. Urol. 2017, 71, 776–787. [Google Scholar] [CrossRef]
- Tsai, H.K.; Lehrer, J.; Alshalalfa, M.; Erho, N.; Davicioni, E.; Lotan, T.L. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer 2017, 17, 759. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Capaia, M.; Granata, I.; Guarracino, M.; Petretto, A.; Inglese, E.; Cattrini, C.; Ferrari, N.; Boccardo, F.; Barboro, P. A hnRNP K–AR-Related Signature Reflects Progression toward Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1920. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Lewis, M. PCAtools: Everything Principal Components Analysis. 2019. Available online: https://github.com/kevinblighe/PCAtools (accessed on 10 November 2023).
- Jackson, S.; Cimentada, J.; Ruiz, E. Corrr: Correlations in R. Available online: https://github.com/drsimonj/corrr (accessed on 15 October 2023).
- Győrffy, B. Discovery and ranking of the most robust prognostic biomarkers in serous ovarian cancer. GeroScience 2023, 45, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kumari, S.; Hu, Q.; Senapati, D.; Venkadakrishnan, V.B.; Wang, D.; DePriest, A.D.; Schlanger, S.E.; Ben-Salem, S.; Valenzuela, M.M.; et al. A Comprehensive Analysis of Coregulator Recruitment, Androgen Receptor Function and Gene Expression in Prostate Cancer. eLife 2017, 6, e28482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, J.; Keller, J.M.; Yeung, K.; Keller, E.T.; Fu, Z. Transcriptional Regulation of RKIP Expression by Androgen in Prostate Cells. Cell. Physiol. Biochem. 2012, 30, 1340–1350. [Google Scholar] [CrossRef]
- Han, Z.-D.; Zhang, Y.-Q.; He, H.-C.; Dai, Q.-S.; Qin, G.-Q.; Chen, J.-H.; Cai, C.; Fu, X.; Bi, X.-C.; Zhu, J.-G.; et al. Identification of novel serological tumor markers for human prostate cancer using integrative transcriptome and proteome analysis. Med. Oncol. 2012, 29, 2877–2888. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef]
- Mendiratta, P.; Mostaghel, E.; Guinney, J.; Tewari, A.K.; Porrello, A.; Barry, W.T.; Nelson, P.S.; Febbo, P.G. Genomic Strategy for Targeting Therapy in Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2009, 27, 2022–2029. [Google Scholar] [CrossRef]
- Tien, A.H.; Sadar, M.D. Androgen-Responsive Gene Expression in Prostate Cancer Progression. In Androgen-Responsive Genes in Prostate Cancer; Wang, Z., Ed.; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Larkin, S.E.; Zeidan, B.; Taylor, M.G.; Bickers, B.; Al-Ruwaili, J.; Aukim-Hastie, C.; Townsend, P.A. Proteomics in prostate cancer biomarker discovery. Expert Rev. Proteom. 2010, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hübschmann, D.; et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018, 34, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- van Bokhoven, A.; Varella-Garcia, M.; Korch, C.; Johannes, W.U.; Smith, E.E.; Miller, H.L.; Nordeen, S.K.; Miller, G.J.; Lucia, M.S. Molecular characterization of human prostate carcinoma cell lines. Prostate 2003, 57, 205–225. [Google Scholar] [CrossRef]
- Gravina, G.L.; Marampon, F.; Giusti, I.; Carosa, E.; Di Sante, S.; Ricevuto, E.; Dolo, V.; Tombolini, V.; Jannini, E.A.; Festuccia, C. Differential effects of PXD101 (belinostat) on androgen-dependent and androgen-independent prostate cancer models. Int. J. Oncol. 2012, 40, 711–720. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Alumkal, J.J.; Stein, M.N.; Taplin, M.-E.; Babb, J.S.; Barnett, E.S.; Gomez-Pinillos, A.; Liu, X.; Moore, D.F.; DiPaola, R.S.; et al. Epigenetic Therapy with Panobinostat Combined with Bicalutamide Rechallenge in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone Deacetylases Are Required for Androgen Receptor Function in Hormone-Sensitive and Castrate-Resistant Prostate Cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef]
- He, Y.; Xu, W.; Xiao, Y.-T.; Huang, H.; Gu, D.; Ren, S. Targeting signaling pathways in prostate cancer: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 198. [Google Scholar] [CrossRef]
- Majera, D.; Skrott, Z.; Bouchal, J.; Bartkova, J.; Simkova, D.; Gachechiladze, M.; Steigerova, J.; Kurfurstova, D.; Gursky, J.; Korinkova, G.; et al. Targeting genotoxic and proteotoxic stress-response pathways in human prostate cancer by clinically available PARP inhibitors, vorinostat and disulfiram. Prostate 2019, 79, 352–362. [Google Scholar] [CrossRef]
- Ghosh, T.M.; White, J.; Davis, J.; Mazumder, S.; Kansom, T.; Skarupa, E.; Barnett, G.S.; Piazza, G.A.; Bird, R.C.; Mitra, A.K.; et al. Identification and Characterization of Key Differentially Expressed Genes Associated with Metronomic Dosing of Topotecan in Human Prostate Cancer. Front. Pharmacol. 2021, 12, 736951. [Google Scholar] [CrossRef] [PubMed]
- Cattrini, C.; Capaia, M.; Boccardo, F.; Barboro, P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: Data from a translational study. Cancer Treat. Res. Commun. 2020, 25, 100221. [Google Scholar] [CrossRef] [PubMed]
- Alabi, B.R.; Liu, S.; Stoyanova, T. Current and emerging therapies for neuroendocrine prostate cancer. Pharmacol. Ther. 2022, 238, 108255. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Li, C.; Xu, K.; Su, W.; Mao, X.; Zou, Y.; Li, B. The Effect of Prostate Cancer-Targeting Doxorubicin Nanomicelles Combined with Photothermal Therapy on Castration-Resistant Prostate Cancer. J. Biomed. Nanotechnol. 2022, 18, 1276–1288. [Google Scholar] [CrossRef]
- Li, K.; Zhan, W.; Chen, Y.; Jha, R.K.; Chen, X. Docetaxel and Doxorubicin Codelivery by Nanocarriers for Synergistic Treatment of Prostate Cancer. Front. Pharmacol. 2019, 10, 1436. [Google Scholar] [CrossRef]
- Dhani, N.C.; Emmenegger, U.; Adams, L.; Jongstra, J.; Tannock, I.F.; Sridhar, S.S.; Knox, J.J.; Day, J.R.; Groskopf, J.; Joshua, A.M. Phase II study of cytarabine in men with docetaxel-refractory, castration-resistant prostate cancer with evaluation of TMPRSS2-ERG and SPINK1 as serum biomarkers: Phase II study of cytarabine in CRPC. BJU Int. 2012, 110, 840–845. [Google Scholar] [CrossRef]
- Ueki, T.; Uemura, H.; Nagashima, Y.; Ohta, S.; Ishiguro, H.; Kubota, Y. Antitumour effect of electrochemotherapy with bleomycin on human prostate cancer xenograft. BJU Int. 2008, 102, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Iannantuono, G.M.; Torino, F.; Rosenfeld, R.; Guerriero, S.; Carlucci, M.; Sganga, S.; Capotondi, B.; Riondino, S.; Roselli, M. The Role of Histology-Agnostic Drugs in the Treatment of Metastatic Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 8535. [Google Scholar] [CrossRef]
- Li, J.; Wu, H.; Lv, S.; Quan, D.; Yang, D.; Xu, J.; Chen, B.; Ou, B.; Wu, S.; Wei, Q. Enhanced antitumor efficacy by combining afatinib with MDV3100 in castration-resistant prostate cancer. Pharmazie 2022, 77, 59–66. [Google Scholar] [CrossRef]
- Matheux, A.; Gassiot, M.; Fromont, G.; Leenhardt, F.; Boulahtouf, A.; Fabbrizio, E.; Marchive, C.; Garcin, A.; Agherbi, H.; Combès, E.; et al. PXR Modulates the Prostate Cancer Cell Response to Afatinib by Regulating the Expression of the Monocarboxylate Transporter SLC16A1. Cancers 2021, 13, 3635. [Google Scholar] [CrossRef]
- Vignani, F.; Bertaglia, V.; Buttigliero, C.; Tucci, M.; Scagliotti, G.V.; Di Maio, M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat. Rev. 2016, 44, 61–73. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Ocio, E.M.; Crusoe, E.; Santamaria, C.; Hernández-Campo, P.; Blanco, J.F.; Sanchez-Guijo, F.M.; Hernández-Iglesias, T.; Briñón, J.G.; Fisac-Herrero, R.M.; et al. Dasatinib as a Bone-Modifying Agent: Anabolic and Anti-Resorptive Effects. PLoS ONE 2012, 7, e34914. [Google Scholar] [CrossRef] [PubMed]
- Tong, D. Unravelling the molecular mechanisms of prostate cancer evolution from genotype to phenotype. Crit. Rev. Oncol. 2021, 163, 103370. [Google Scholar] [CrossRef]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Inamura, S.; Ito, H.; Taga, M.; Tsuchiyama, K.; Hoshino, H.; Kobayashi, M.; Yokoyama, O. Low-dose Docetaxel Enhanced the Anticancer Effect of Temsirolimus by Overcoming Autophagy in Prostate Cancer Cells. Anticancer Res. 2019, 39, 5417–5425. [Google Scholar] [CrossRef]
- Shariatifar, H.; Ranjbarian, F.; Hajiahmadi, F.; Farasat, A. A comprehensive review on methotrexate containing nanoparticles; an appropriate tool for cancer treatment. Mol. Biol. Rep. 2022, 49, 11049–11060. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-L.; Leu, W.-J.; Hsu, L.-C.; Ho, C.-H.; Liu, S.-P.; Guh, J.-H. Phosphodiesterase Type 5 Inhibitors Synergize Vincristine in Killing Castration-Resistant Prostate Cancer through Amplifying Mitotic Arrest Signaling. Front. Oncol. 2020, 10, 1274. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, S. Reposition of the Fungicide Ciclopirox for Cancer Treatment. Recent Patents Anti-Cancer Drug Discov. 2021, 16, 122–135. [Google Scholar] [CrossRef]
- Wilson, J.L.; Steinberg, E.; Racz, R.; Altman, R.B.; Shah, N.; Grimes, K. A network paradigm predicts drug synergistic effects using downstream protein–protein interactions. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 1527–1538. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Gil da Costa, R.M.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef]
- Menssouri, N.; Poiraudeau, L.; Helissey, C.; Bigot, L.; Sabio, J.; Ibrahim, T.; Pobel, C.; Nicotra, C.; Ngo-Camus, M.; Lacroix, L.; et al. Genomic Profiling of Metastatic Castration-Resistant Prostate Cancer Samples Resistant to Androgen Receptor Pathway Inhibitors. Clin. Cancer Res. 2023, 29, 4504–4517. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-D.; Pang, K.; Wu, Z.-X.; Dong, Y.; Hao, L.; Qin, J.-X.; Wang, W.; Chen, Z.-S.; Han, C.-H. Tumor cell plasticity in targeted therapy-induced resistance: Mechanisms and new strategies. Signal Transduct. Target. Ther. 2023, 8, 113. [Google Scholar] [CrossRef]
- Sailer, V.; von Amsberg, G.; Duensing, S.; Kirfel, J.; Lieb, V.; Metzger, E.; Offermann, A.; Pantel, K.; Schuele, R.; Taubert, H.; et al. Experimental in vitro, ex vivo and in vivo models in prostate cancer research. Nat. Rev. Urol. 2023, 20, 158–178. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.S.; Hostetter, G.; Tang, L.; Frank, S.B.; Saboda, K.; Mehra, R.; Wang, L.; Li, X.; Keller, E.T.; Miranti, C.K. Notch3 promotes prostate cancer-induced bone lesion development via MMP-3. Oncogene 2020, 39, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Deng, L.; Li, C.; He, Q.; He, Y.; Hu, X.; Cai, Y.; Gan, Y. Loss of EHF facilitates the development of treatment-induced neuroendocrine prostate cancer. Cell Death Dis. 2021, 12, 46. [Google Scholar] [CrossRef]
- Püschel, J.; Dubrovska, A.; Gorodetska, I. The Multifaceted Role of Aldehyde Dehydrogenases in Prostate Cancer Stem Cells. Cancers 2021, 13, 4703. [Google Scholar] [CrossRef] [PubMed]
- Farah, E.; Li, C.; Cheng, L.; Kong, Y.; Lanman, N.A.; Pascuzzi, P.; Lorenz, G.R.; Zhang, Y.; Ahmad, N.; Li, L.; et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J. Biol. Chem. 2019, 294, 8543–8554. [Google Scholar] [CrossRef]
- Han, H.; Park, C.K.; Choi, Y.-D.; Cho, N.H.; Lee, J.; Cho, K.S. Androgen-Independent Prostate Cancer Is Sensitive to CDC42-PAK7 Kinase Inhibition. Biomedicines 2022, 11, 101. [Google Scholar] [CrossRef]
- Tan, I.d.A.; Ricciardelli, C.; Russell, D.L. The metalloproteinase ADAMTS1: A comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer 2013, 133, 2263–2276. [Google Scholar] [CrossRef]
- Serrano-Garrido, O.; Peris-Torres, C.; Redondo-García, S.; Asenjo, H.G.; Plaza-Calonge, M.D.C.; Fernandez-Luna, J.L.; Rodríguez-Manzaneque, J.C. ADAMTS1 Supports Endothelial Plasticity of Glioblastoma Cells with Relevance for Glioma Progression. Biomolecules 2020, 11, 44. [Google Scholar] [CrossRef]
- Camacho, L.; Zabala-Letona, A.; Cortazar, A.R.; Astobiza, I.; Dominguez-Herrera, A.; Ercilla, A.; Crespo, J.; Viera, C.; Fernández-Ruiz, S.; Martinez-Gonzalez, A.; et al. Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens. Cancers 2021, 13, 4307. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.-P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef]
- Ramalingam, S.; Eisenberg, A.; Foo, W.C.; Freedman, J.; Armstrong, A.J.; Moss, L.G.; Harrison, M.R. Treatment-related neuroendocrine prostate cancer resulting in Cushing′s syndrome. Int. J. Urol. 2016, 23, 1038–1041. [Google Scholar] [CrossRef]
- Ramos-Montoya, A.; Lamb, A.D.; Russell, R.; Carroll, T.; Jurmeister, S.; Galeano-Dalmau, N.; Massie, C.E.; Boren, J.; Bon, H.; Theodorou, V.; et al. HES6 drives a critical AR transcriptional programme to induce castration-resistant prostate cancer through activation of an E2F1-mediated cell cycle network. EMBO Mol. Med. 2014, 6, 651–661. [Google Scholar] [CrossRef]
- Sreekumar, A.; Saini, S. Role of transcription factors and chromatin modifiers in driving lineage reprogramming in treatment-induced neuroendocrine prostate cancer. Front. Cell Dev. Biol. 2023, 11, 1075707. [Google Scholar] [CrossRef]
- Liu, S.; Alabi, B.R.; Yin, Q.; Stoyanova, T. Molecular mechanisms underlying the development of neuroendocrine prostate cancer. Semin. Cancer Biol. 2022, 86, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Blee, A. Lineage plasticity-mediated therapy resistance in prostate cancer. Asian J. Androl. 2019, 21, 241–248. [Google Scholar] [CrossRef]
- Hankey, W.; Zhong, C.; Qianben, W. Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors. Cancer Res. 2020, 80, 2427–2436. [Google Scholar] [CrossRef] [PubMed]
- Baena, E.; Shao, Z.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fujiwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef]
- Oh, S.; Shin, S.; Song, H.; Grande, J.P.; Janknecht, R. Relationship between ETS Transcription Factor ETV1 and TGF-β-regulated SMAD Proteins in Prostate Cancer. Sci. Rep. 2019, 9, 8186. [Google Scholar] [CrossRef]
- Thompson-Elliott, B.; Johnson, R.; Khan, S.A. Alterations in TGFβ Signaling during Prostate Cancer Progression. Am. J. Clin. Exp. Urol. 2021, 9, 318–328. [Google Scholar] [PubMed]
- Calvo, M.B.; Fernández, V.B.; Villaamil, V.M.; Gallego, G.A.; Prado, S.D.; Pulido, E.G. Biology of BMP signalling and cancer. Clin. Transl. Oncol. 2009, 11, 126–137. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granata, I.; Barboro, P. Identification of Molecular Markers Associated with Prostate Cancer Subtypes: An Integrative Bioinformatics Approach. Biomolecules 2024, 14, 87. https://doi.org/10.3390/biom14010087
Granata I, Barboro P. Identification of Molecular Markers Associated with Prostate Cancer Subtypes: An Integrative Bioinformatics Approach. Biomolecules. 2024; 14(1):87. https://doi.org/10.3390/biom14010087
Chicago/Turabian StyleGranata, Ilaria, and Paola Barboro. 2024. "Identification of Molecular Markers Associated with Prostate Cancer Subtypes: An Integrative Bioinformatics Approach" Biomolecules 14, no. 1: 87. https://doi.org/10.3390/biom14010087
APA StyleGranata, I., & Barboro, P. (2024). Identification of Molecular Markers Associated with Prostate Cancer Subtypes: An Integrative Bioinformatics Approach. Biomolecules, 14(1), 87. https://doi.org/10.3390/biom14010087