
Untangling the Context-Specificity of Essential Genes by Means of Machine Learning: A Constructive Experience






Abstract
1. Introduction
2. Common Properties of Essential Genes
3. Context-Specific Essentiality
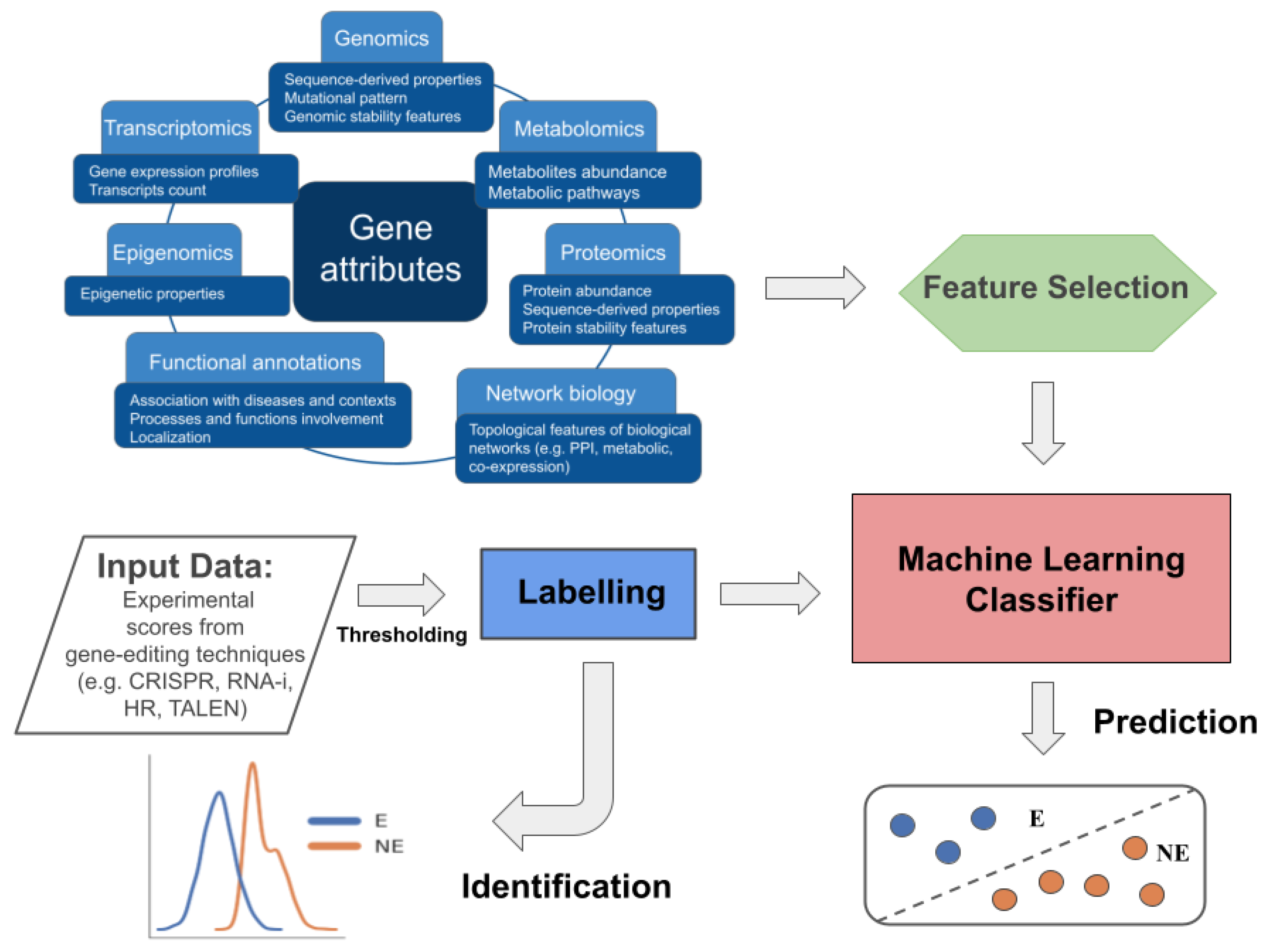
4. Computational Approaches to Define Gene Essentiality
4.1. Identification Methods
4.2. Predictive Models
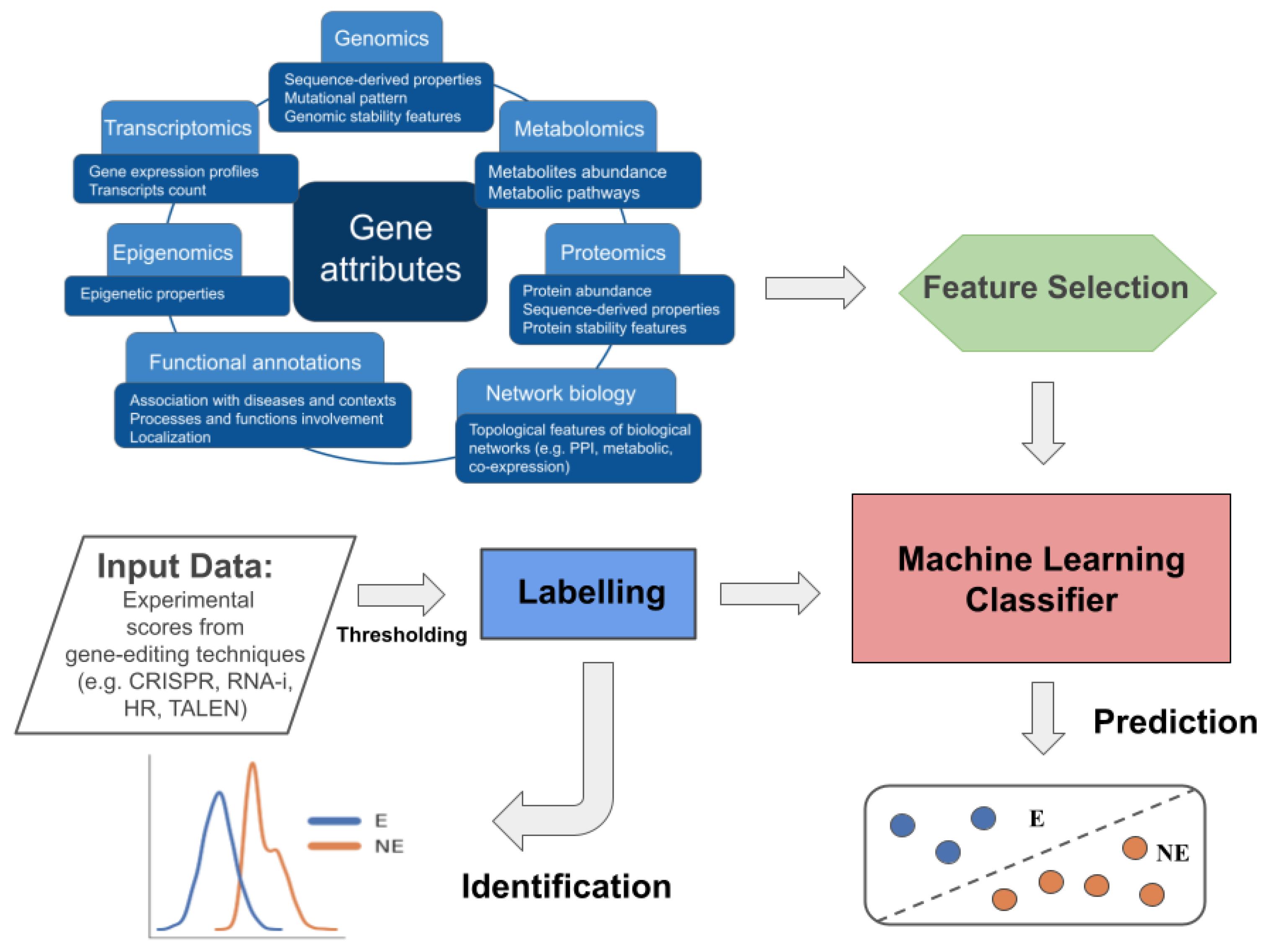
5. Experimental Study on Context-Specific EGs Identification and Prediction
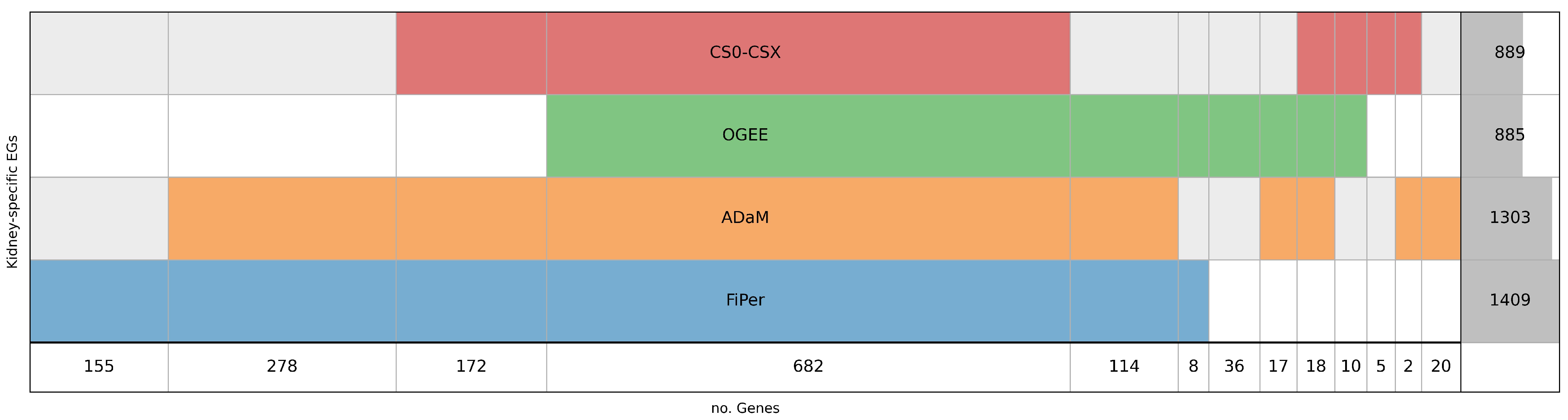
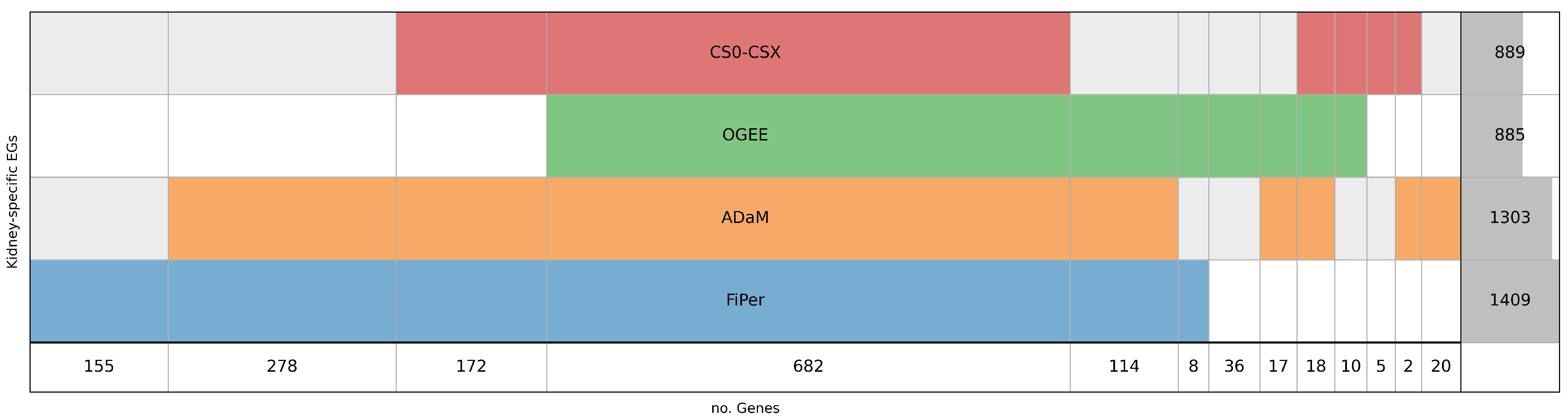
5.1. Identification of Human-Kidney-Specific CFGs
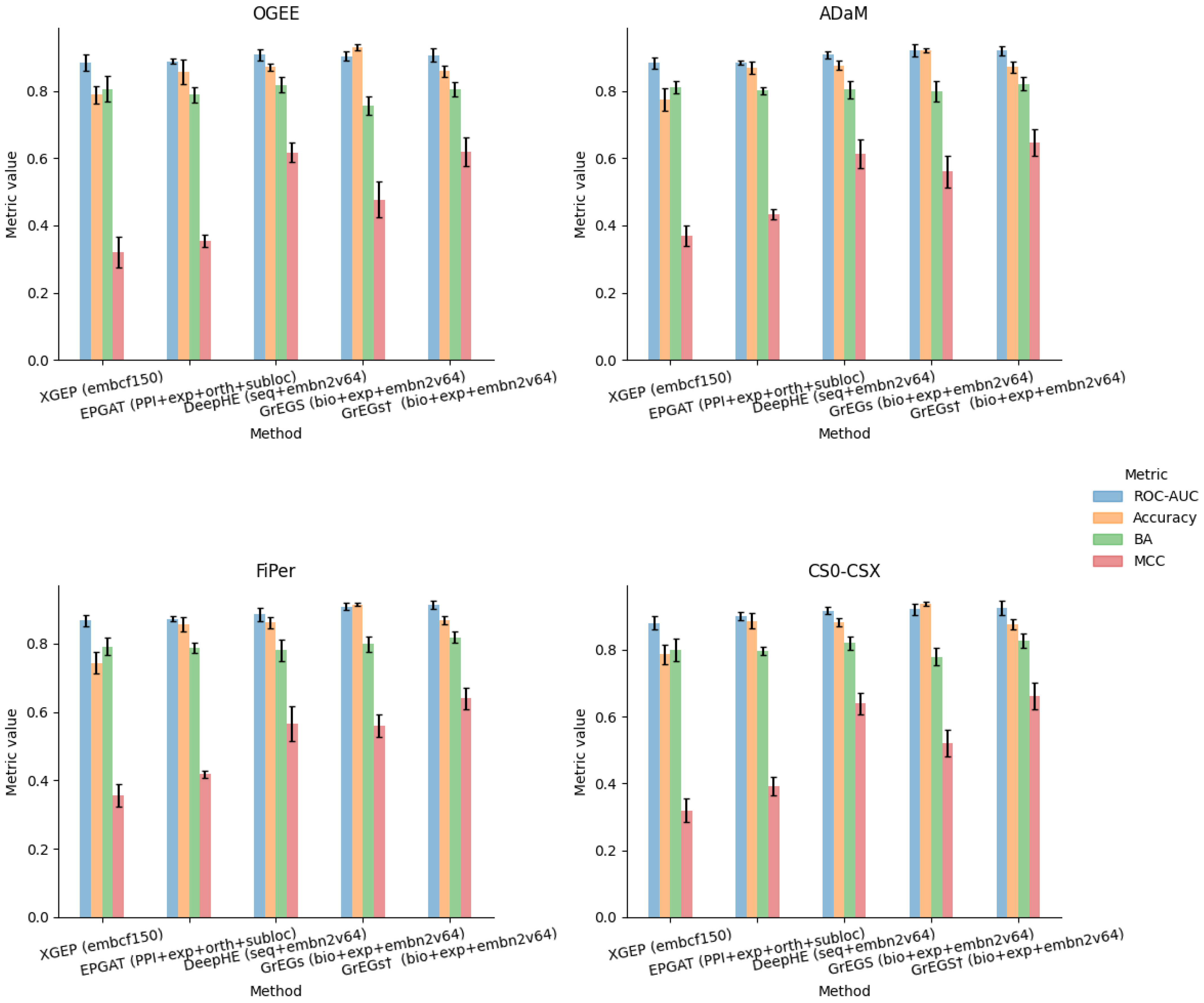
5.2. Prediction of Human-Kidney-Specific CFGs from Multiomics Data
- Features extracted from the PPI network by means of node2vec (named “embn2v<size>” in Table A4, Table A5, Table A6, Table A7, Table A8, Table A9, Table A10 and Table A11). The kidney-specific PPI was downloaded from the Integrated Interaction Database, which provides networks with comprehensive tissue, disease, cellular localisation and druggability annotations [62]. The tissue-specificity was obtained by filtering the edges by their tissue annotation;
- Features extracted from the correlation of TCGA transcriptomic data by means of collaborative embedding (named “embcf<size>” in Table A4, Table A5, Table A6, Table A7, Table A8, Table A9, Table A10 and Table A11). Expression data of the three subtypes of renal cancer (KIRP, KIRC and KICH) were downloaded from the GDC portal (https://portal.gdc.cancer.gov, accessed on 30 May 2023). Data were processed as described in [51] before being submitted to the collaborative embedding procedure;
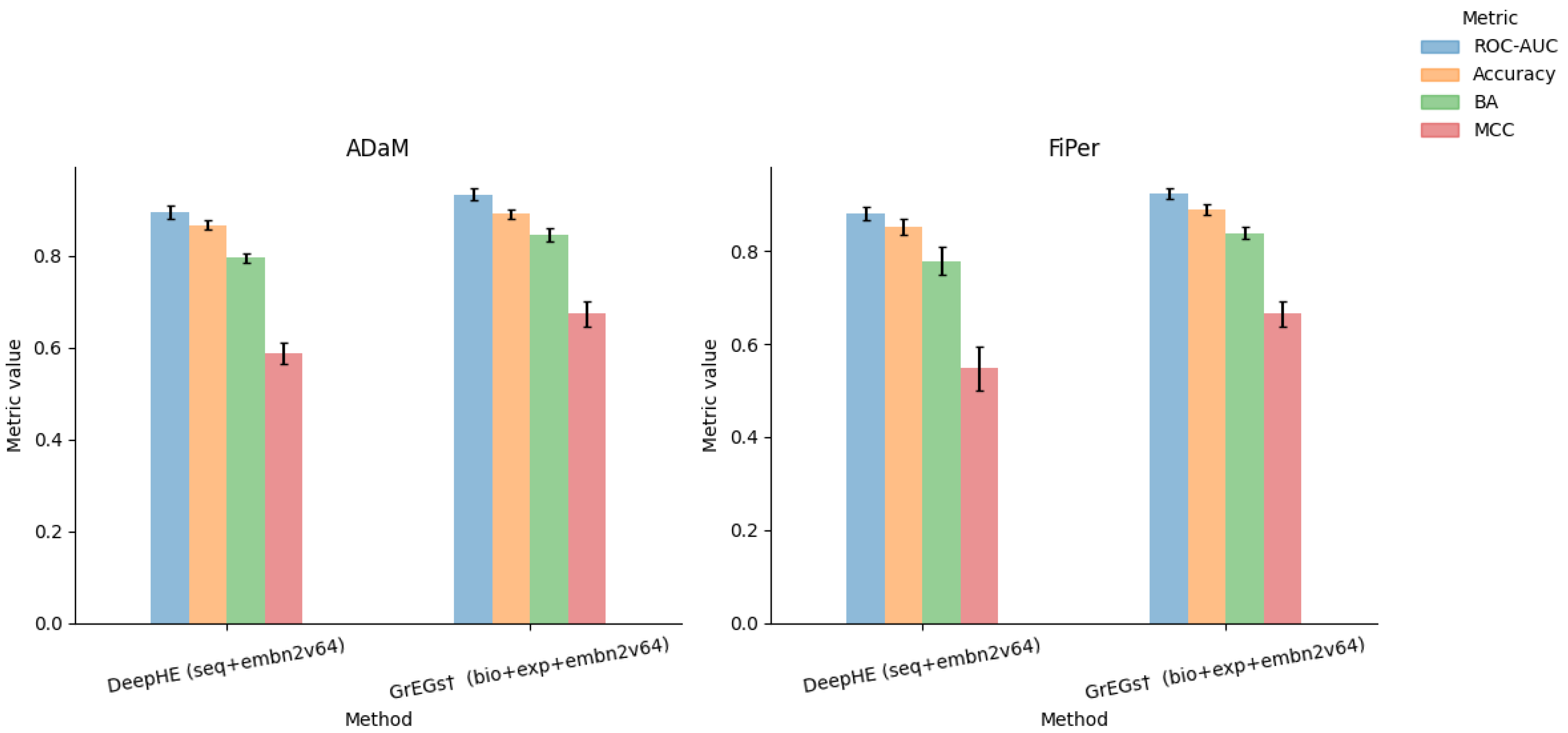
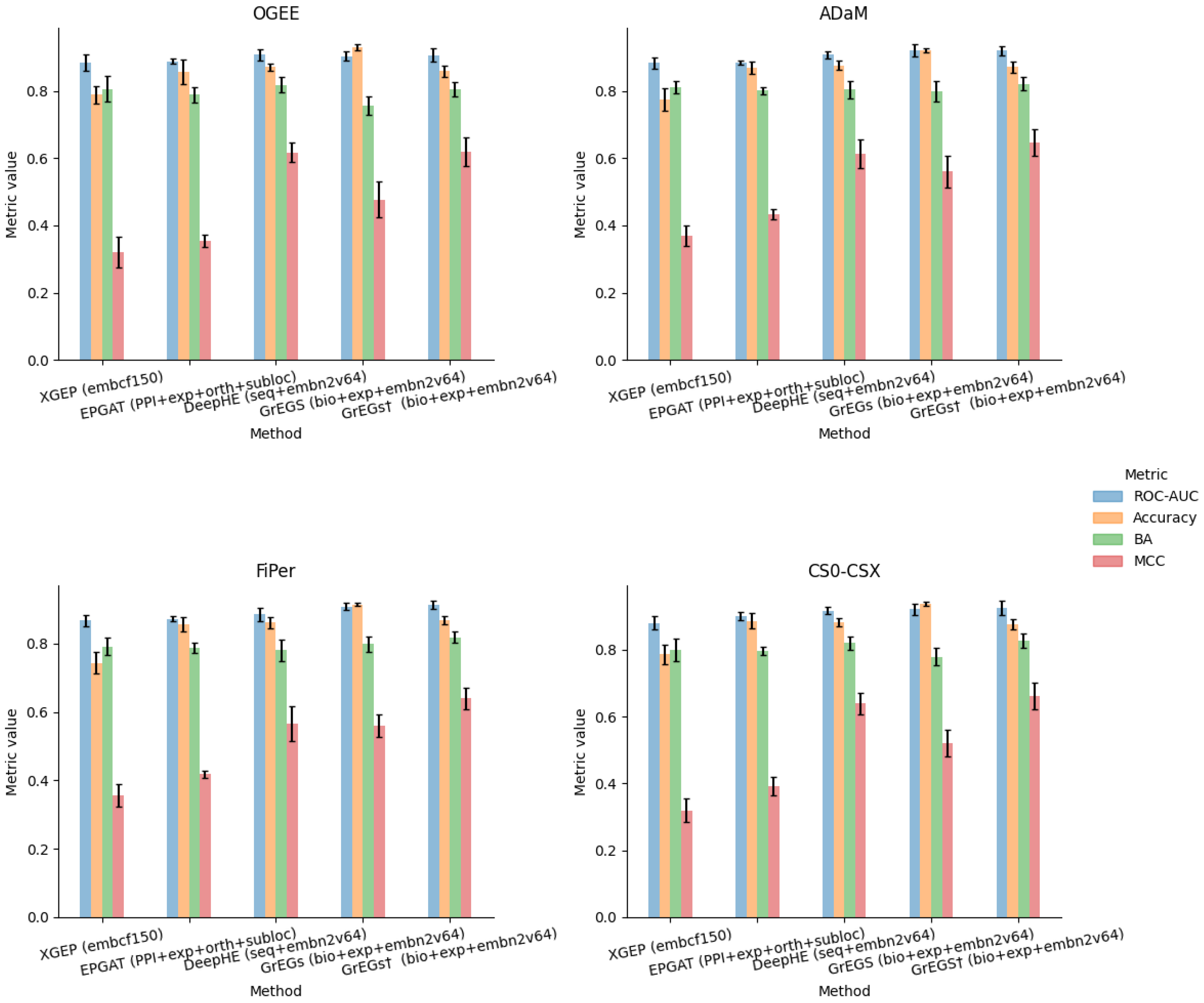
5.3. Analysis of Prediction Methods for Human-Kidney-Specific CFGs
5.4. Testing a Different Context: Identification and Prediction of Human Lung-Specific CFGs
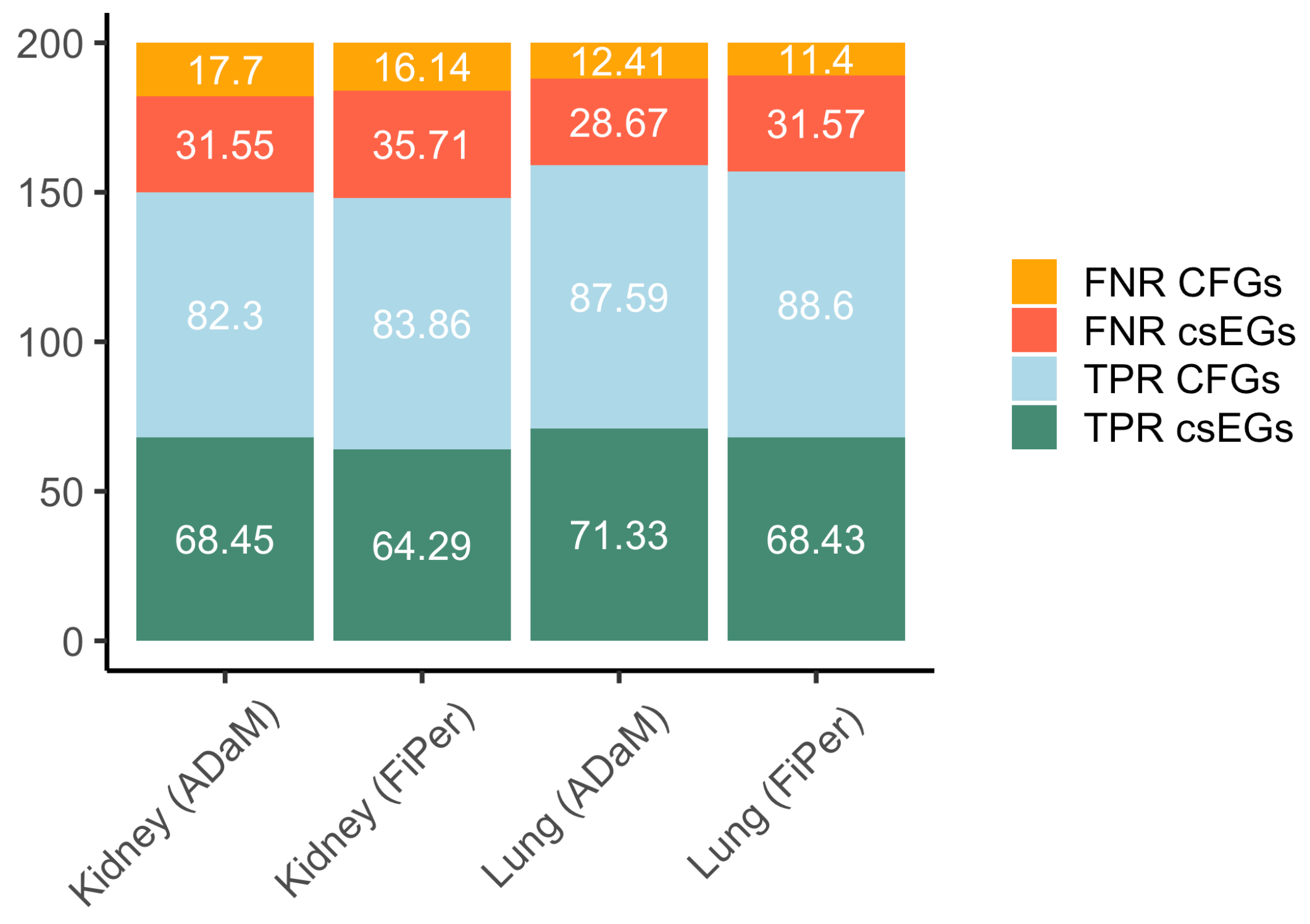
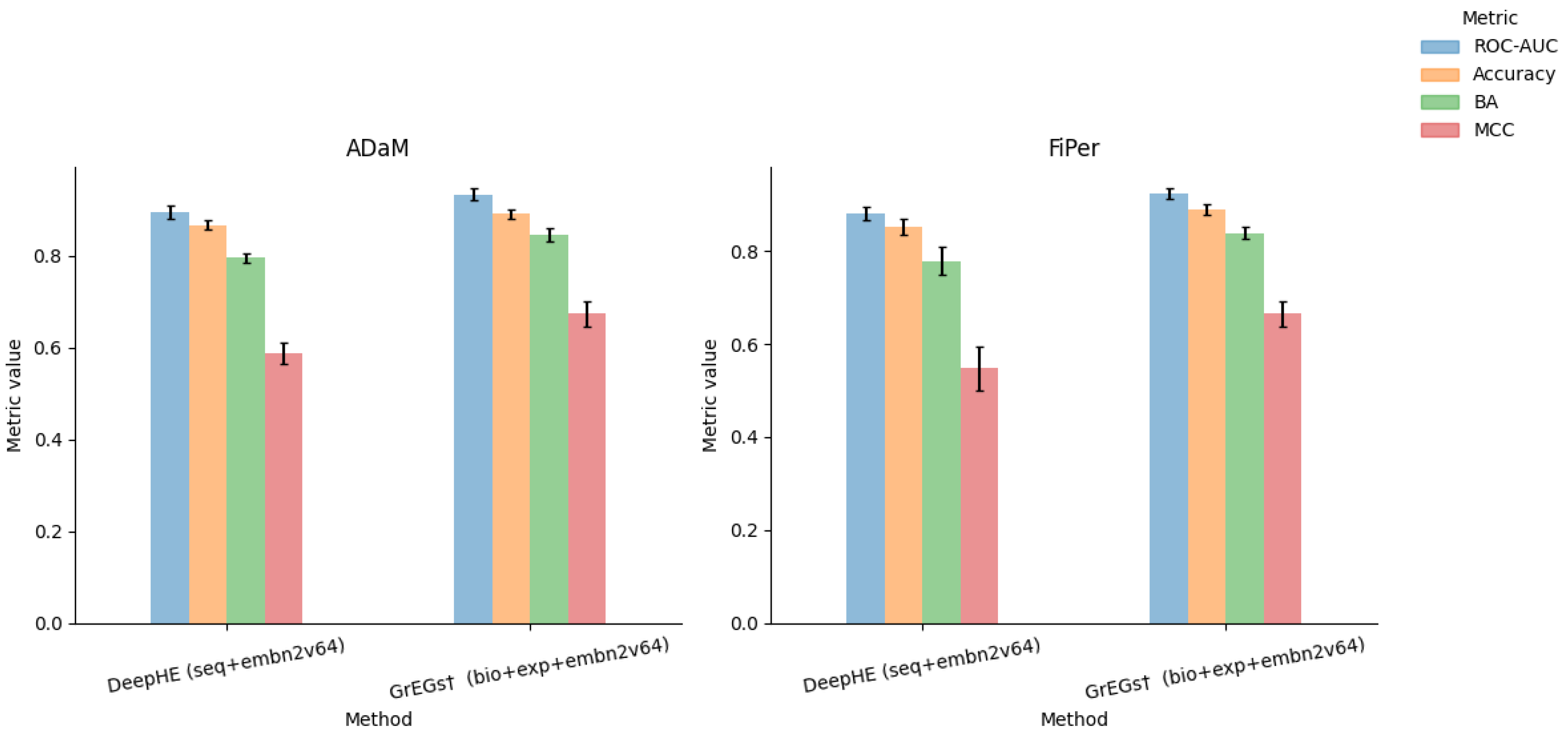
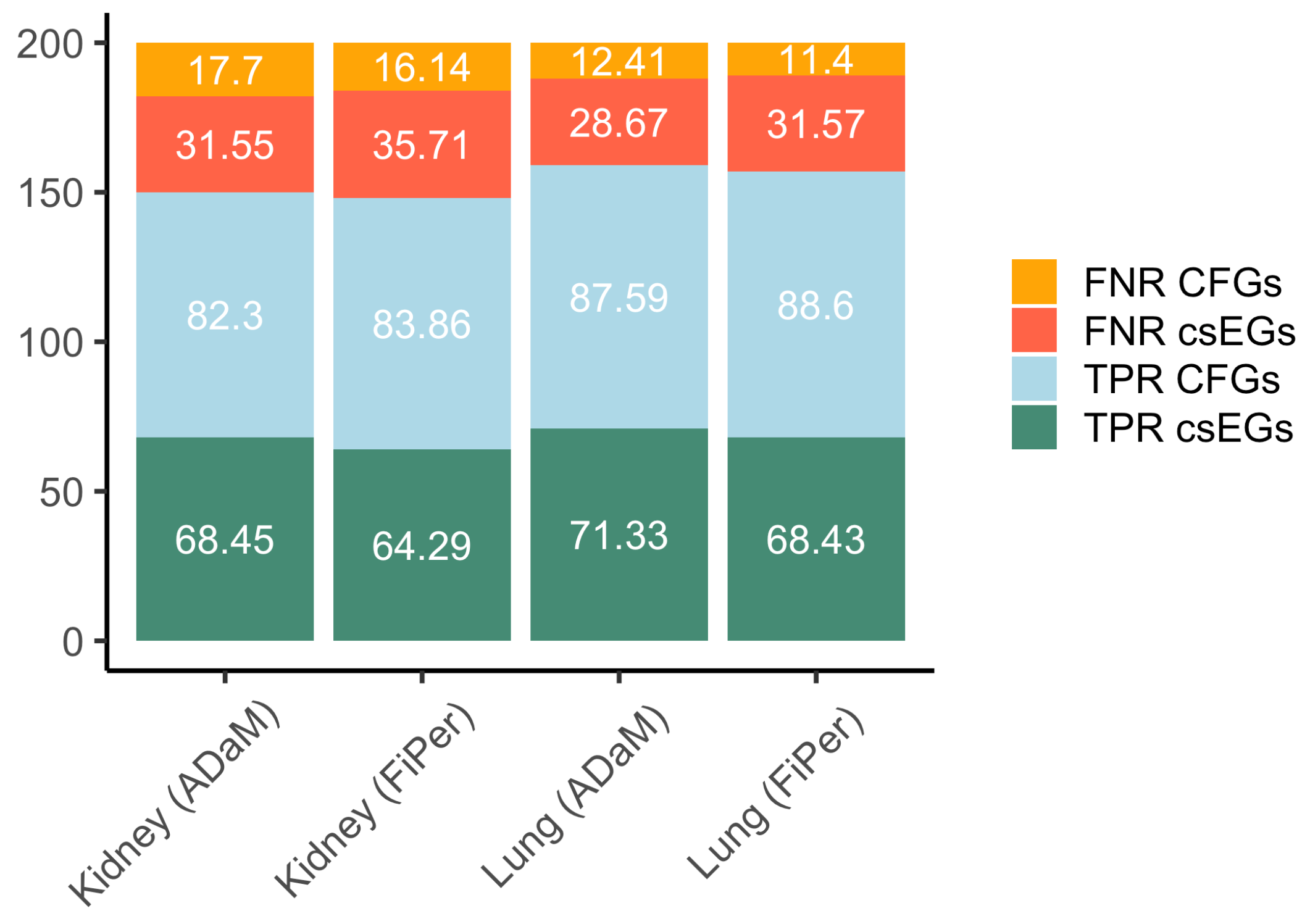
5.5. Performance Evaluation on csEGs and CFGs
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym (#) | Features |
|---|---|
| seq (90) | Gene.length GC.content, aaa, aac, aag, aat, aca, acc, acg, act, aga, agc, agg, agt, ata, atc, atg, att, caa, cac, cag, cat, cca, ccc, ccg, cct, cga, cgc, cgg, cgt, cta, ctc, ctg, ctt, gaa, gac, gag, gat, gca, gcc, gcg, gct, gga, ggc, ggg, ggt, gta, gtc, gtg, gtt, taa, tac, tag, tat, tca, tcc, tcg, tct, tga, tgc, tgg, tgt, tta, ttc, ttg, ttt, RSCUmax, CAI, A, R, N, D, C, E, Q, G, H, I, L, K, M, F, P, S, T, W, Y, V, symb, protein_length |
| exp (89 for kidney, 578 for lung) | GTEX-<sampleID>, … |
| subloc (11) | Mitochondrion, Nucleus, Endosome, Golgi apparatus, Cytosol, Plasma membrane, Endoplasmic reticulum, Cytoskeleton, Peroxisome, Lysosome, Extracellular space |
| orth (162) | H.sapiens-I.multifiliis, H.sapiens-P.tetraurelia, H.sapiens-I.scapularis, H.sapiens-P.trichocarpa, H.sapiens-K.africana, H.sapiens-P.tricornutum, H.sapiens-K.cryptofilum, H.sapiens-P.tritici-repentis, H.sapiens-K.lactis, H.sapiens-P.troglodytes, H.sapiens-K.pastoris, H.sapiens-P.ultimum, H.sapiens-L.africana, H.sapiens-P.vivax, H.sapiens-L.bicolor, H.sapiens-P.yoelii, H.sapiens-L.braziliensis, H.sapiens-R.baltica, H.sapiens-L.chalumnae, H.sapiens-R.communis, H.sapiens-L.elongisporus, H.sapiens-R.delemar, H.sapiens-L.infantum, H.sapiens-R.glutinis, H.sapiens-L.interrogans, H.sapiens-R.norvegicus, H.sapiens-L.loa, H.sapiens-Salpingoeca.sp., H.sapiens-L.maculans, H.sapiens-S.bicolor, H.sapiens-L.major, H.sapiens-S.cerevisiae, H.sapiens-L.thermotolerans, H.sapiens-S.coelicolor, H.sapiens-M.acetivorans, H.sapiens-S.commune, H.sapiens-M.acridum, H.sapiens-S.harrisii, H.sapiens-M.brevicollis, H.sapiens-S.invicta, H.sapiens-M.brunnea, H.sapiens-S.italica |
| bio (15) | GO-BP, GO-MF, GO-CC, UP_tissue, KEGG, UCSC_TFBS, GC_content, BIOGRID, REACTOME, Orth_count, OncoDB_DEG, Gene_Disease_ass_count, Gene_length, HPA_kidney|lung, GTEx_kidney|lung |
Appendix B
| Metric | Description | Formula |
|---|---|---|
| Accuracy (Acc) | Percentage of correctly classified samples | |
| Specificity (TNR) | Percentage of negative samples correctly classified | |
| Sensitivity (TPR) | Percentage of positive samples correctly classified | |
| False positive rate (FPR) | Percentage of positive samples incorrectly classified | |
| False negative rate (FNR) | Percentage of negative samples incorrectly classified | |
| BA | Balanced accuracy | |
| ROC-AUC | Area under the receiver operating characteristic curve | |
| MCC | Matthews correlation coefficient (MCC) |
Appendix C
| Method | Parameters | |||||
|---|---|---|---|---|---|---|
| DeepHE | no. hl = 3 | nodes per hl = 128, 256, 512 | epochs = 50 | lr = 0.001 | dropout = 20% | batch size = 32 |
| EPGAT | no. hl = 2 | nodes per hl = 8, 1 | epochs = 1000 | lr = 0.0005 | dropout = 40% | att. heads per layer = 8, 1 |
| XGEP | no. hl = 3 | nodes per hl = 150, 32, 11 | epochs = 20 | lr = 0.00127 | dropout = 10.6% | batch size = 128 |
| GrEGs | boosting = gbdt | no. leaves = 31 | max depth = None | lr = 0.1 | no. estimators = 100 | class_weight = balanced |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| XGEP (embcf150_KIRP) | 0.8233 ± 0.0454 | 0.7036 ± 0.0548 | 0.7572 ± 0.0459 | 0.2485 ± 0.0520 |
| XGEP (embcf150_KIRC) | 0.8749 ± 0.0427 | 0.7956 ± 0.0456 | 0.7922 ± 0.0468 | 0.3143 ± 0.0658 |
| XGEP (embcf150_KICH) | 0.8527 ± 0.0780 | 0.7945 ± 0.1072 | 0.7767 ± 0.0817 | 0.3171 ± 0.1154 |
| XGEP (embcf150_PAN_RenalCancer) | 0.8848 ± 0.0243 | 0.7889 ± 0.0260 | 0.8061 ± 0.0377 | 0.3203 ± 0.0458 |
| EPGAT (PPI+exp) | 0.8291 ± 0.0240 | 0.7457 ± 0.0260 | 0.7540 ± 0.0300 | 0.2556 ± 0.0330 |
| EPGAT (PPI+ortho) | 0.8825 ± 0.0120 | 0.8788 ± 0.0400 | 0.7920 ± 0.0190 | 0.3745 ± 0.0280 |
| EPGAT (PPI+subloc) | 0.8660 ± 0.0020 | 0.8330 ± 0.0530 | 0.7760 ± 0.0280 | 0.3250 ± 0.0120 |
| EPGAT (PPI+exp+orth+subloc) | 0.8884 ± 0.0080 | 0.8571 ± 0.0365 | 0.7896 ± 0.0233 | 0.3550 ± 0.0177 |
| DeepHE (seq) | 0.8283 ± 0.0243 | 0.8122 ± 0.0134 | 0.7238 ± 0.0352 | 0.4356 ± 0.0447 |
| DeepHE (embn2v64) | 0.8840 ± 0.0234 | 0.8588 ± 0.0135 | 0.7875 ± 0.0268 | 0.5665 ± 0.0451 |
| DeepHE (seq+embn2v64) | 0.9077 ± 0.0163 | 0.8718 ± 0.0113 | 0.8188 ± 0.0220 | 0.6174 ± 0.0291 |
| GrEGs (bio+exp) | 0.8724 ± 0.0103 | 0.9008 ± 0.0054 | 0.7158 ± 0.0190 | 0.3556 ± 0.0294 |
| GrEGs (embn2v64) | 0.8990 ± 0.0235 | 0.9311 ± 0.0058 | 0.7620 ± 0.0336 | 0.4406 ± 0.0491 |
| GrEGs (bio+exp+embn2v64) | 0.9040 ± 0.0137 | 0.9309 ± 0.0094 | 0.7556 ± 0.0276 | 0.4776 ± 0.0544 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| XGEP (embcf150_KIRP) | 0.8204 ± 0.0339 | 0.7274 ± 0.0401 | 0.7456 ± 0.0328 | 0.2819 ± 0.0392 |
| XGEP (embcf150_KIRC) | 0.8590 ± 0.0284 | 0.7529 ± 0.0474 | 0.7828 ± 0.0343 | 0.3314 ± 0.0463 |
| XGEP (embcf150_KICH) | 0.7902 ± 0.0615 | 0.7197 ± 0.0743 | 0.7299 ± 0.0587 | 0.2678 ± 0.0756 |
| XGEP (embcf150_PAN_RenalCancer) | 0.8830 ± 0.0177 | 0.7738 ± 0.0337 | 0.8108 ± 0.0187 | 0.3687 ± 0.0309 |
| EPGAT (PPI+exp) | 0.8035 ± 0.0250 | 0.7325 ± 0.0190 | 0.7414 ± 0.0160 | 0.2784 ± 0.0220 |
| EPGAT (PPI+ortho) | 0.8860 ± 0.0170 | 0.9000 ± 0.0080 | 0.7890 ± 0.0310 | 0.4680 ± 0.0230 |
| EPGAT (PPI+subloc) | 0.8880 ± 0.0040 | 0.8020 ± 0.0630 | 0.7990 ± 0.0000 | 0.3830 ± 0.0410 |
| EPGAT (PPI+exp+orth+subloc) | 0.8838 ± 0.0065 | 0.8685 ± 0.0195 | 0.8008 ± 0.0116 | 0.4330 ± 0.0146 |
| DeepHE (seq) | 0.8140 ± 0.0150 | 0.8012 ± 0.0094 | 0.7104 ± 0.0185 | 0.4063 ± 0.0236 |
| DeepHE (embn2v64) | 0.9068 ± 0.0125 | 0.8842 ± 0.0136 | 0.8184 ± 0.0257 | 0.6390 ± 0.0436 |
| DeepHE (seq+embn2v64) | 0.9071 ± 0.0097 | 0.8754 ± 0.0141 | 0.8034 ± 0.0250 | 0.6115 ± 0.0427 |
| GrEGs (bio+exp) | 0.8822 ± 0.0138 | 0.8762 ± 0.0127 | 0.7646 ± 0.0234 | 0.4340 ± 0.0424 |
| GrEGs (embn2v64) | 0.9063 ± 0.0145 | 0.9182 ± 0.0055 | 0.7975 ± 0.0164 | 0.5153 ± 0.0237 |
| GrEGs (bio+exp+embn2v64) | 0.9215 ± 0.0181 | 0.9207 ± 0.0073 | 0.7978 ± 0.0307 | 0.5604 ± 0.0470 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| XGEP (embcf150_KIRP) | 0.8052 ± 0.0149 | 0.6590 ± 0.0427 | 0.7349 ± 0.0167 | 0.2727 ± 0.0172 |
| XGEP (embcf150_KIRC) | 0.8371 ± 0.0250 | 0.7215 ± 0.0262 | 0.7700 ± 0.0317 | 0.3238 ± 0.0358 |
| XGEP (embcf150_KICH) | 0.7747 ± 0.0439 | 0.7632 ± 0.0443 | 0.6999 ± 0.0463 | 0.2617 ± 0.0627 |
| XGEP (embcf150_PAN_RenalCancer) | 0.8676 ± 0.0168 | 0.7443 ± 0.0308 | 0.7915 ± 0.0258 | 0.3557 ± 0.0320 |
| EPGAT (PPI+exp) | 0.7858 ± 0.0020 | 0.7028 ± 0.0130 | 0.7218 ± 0.0170 | 0.2580 ± 0.0140 |
| EPGAT (PPI+ortho) | 0.8700 ± 0.0130 | 0.8870 ± 0.0070 | 0.7710 ± 0.0120 | 0.4370 ± 0.0310 |
| EPGAT (PPI+subloc) | 0.8710 ± 0.0170 | 0.7790 ± 0.0080 | 0.7890 ± 0.0120 | 0.3580 ± 0.0410 |
| EPGAT (PPI+exp+orth+subloc) | 0.8730 ± 0.0086 | 0.8561 ± 0.0212 | 0.7890 ± 0.0150 | 0.4178 ± 0.0100 |
| DeepHE (seq) | 0.8020 ± 0.0206 | 0.7971 ± 0.0131 | 0.7097 ± 0.0372 | 0.4001 ± 0.0506 |
| DeepHE (embn2v64) | 0.8751 ± 0.0183 | 0.8628 ± 0.0097 | 0.7831 ± 0.0288 | 0.5690 ± 0.0401 |
| DeepHE (seq+embn2v64) | 0.8859 ± 0.0204 | 0.8614 ± 0.0171 | 0.7811 ± 0.0309 | 0.5663 ± 0.0521 |
| GrEGs (bio+exp) | 0.8709 ± 0.0121 | 0.8677 ± 0.0125 | 0.7532 ± 0.0201 | 0.4203 ± 0.0382 |
| GrEGs (embn2v64) | 0.8937 ± 0.0180 | 0.9126 ± 0.0059 | 0.7939 ± 0.0186 | 0.5122 ± 0.0318 |
| GrEGs (bio+exp+embn2v64) | 0.9086 ± 0.0104 | 0.9150 ± 0.0052 | 0.7992 ± 0.0222 | 0.5599 ± 0.0332 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| XGEP (embcf150_KIRP) | 0.8009 ± 0.0420 | 0.6659 ± 0.0392 | 0.7334 ± 0.0325 | 0.2223 ± 0.0367 |
| XGEP (embcf150_KIRC) | 0.8461 ± 0.0279 | 0.7277 ± 0.0701 | 0.7686 ± 0.0354 | 0.2713 ± 0.0453 |
| XGEP (embcf150_KICH) | 0.8076 ± 0.0520 | 0.7398 ± 0.0544 | 0.7223 ± 0.0477 | 0.2282 ± 0.0538 |
| XGEP (embcf150_PAN_RenalCancer) | 0.8806 ± 0.0204 | 0.7862 ± 0.0291 | 0.8007 ± 0.0334 | 0.3188 ± 0.0355 |
| EPGAT (PPI+exp) | 0.8230 ± 0.0030 | 0.7481 ± 0.0040 | 0.7710 ± 0.0100 | 0.2680 ± 0.0100 |
| EPGAT (PPI+ortho) | 0.9130 ± 0.0210 | 0.9300 ± 0.0080 | 0.7970 ± 0.0220 | 0.4760 ± 0.0310 |
| EPGAT (PPI+subloc) | 0.9020 ± 0.0150 | 0.8060 ± 0.0410 | 0.8360 ± 0.0120 | 0.3550 ± 0.0230 |
| EPGAT (PPI+exp+orth+subloc) | 0.9007 ± 0.0126 | 0.8861 ± 0.0225 | 0.7963 ± 0.0132 | 0.3917 ± 0.0273 |
| DeepHE (seq) | 0.8293 ± 0.0170 | 0.8068 ± 0.0184 | 0.7240 ± 0.0151 | 0.4286 ± 0.0320 |
| DeepHE (embn2v64) | 0.9034 ± 0.0137 | 0.8799 ± 0.0128 | 0.8076 ± 0.0188 | 0.6230 ± 0.0305 |
| DeepHE (seq+embn2v64) | 0.9169 ± 0.0104 | 0.8833 ± 0.0121 | 0.8204 ± 0.0204 | 0.6389 ± 0.0324 |
| GrEGs (bio+exp) | 0.8819 ± 0.0120 | 0.9057 ± 0.0084 | 0.7203 ± 0.0323 | 0.3677 ± 0.0524 |
| GrEGs (embn2v64) | 0.9094 ± 0.0255 | 0.9384 ± 0.0061 | 0.7847 ± 0.0275 | 0.4873 ± 0.0432 |
| GrEGs (bio+exp+embn2v64) | 0.9211 ± 0.0161 | 0.9376 ± 0.0048 | 0.7789 ± 0.0259 | 0.5199 ± 0.0403 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| DeepHE (seq) | 0.8283 ± 0.0243 | 0.8122 ± 0.0134 | 0.7238 ± 0.0352 | 0.4356 ± 0.0447 |
| DeepHE (bio+exp) | 0.8400 ± 0.0255 | 0.7843 ± 0.0126 | 0.7589 ± 0.0260 | 0.4517 ± 0.0377 |
| DeepHE (embn2v64) | 0.8840 ± 0.0234 | 0.8588 ± 0.0135 | 0.7875 ± 0.0268 | 0.5665 ± 0.0451 |
| DeepHE (seq+embn2v64) | 0.9077 ± 0.0163 | 0.8718 ± 0.0113 | 0.8188 ± 0.0220 | 0.6174 ± 0.0291 |
| DeepHE (bio+exp+embn2v64) | 0.8879 ± 0.0305 | 0.8579 ± 0.0224 | 0.7964 ± 0.0385 | 0.5742 ± 0.0684 |
| DeepHE (seq+bio+exp++embn2v64) | 0.8914 ± 0.0294 | 0.8607 ± 0.0225 | 0.7867 ± 0.0338 | 0.5714 ± 0.0640 |
| GrEGs† (seq) | 0.8291 ± 0.0185 | 0.8162 ± 0.0175 | 0.7086 ± 0.0277 | 0.4271 ± 0.0554 |
| GrEGs† (bio+exp) | 0.8658 ± 0.0164 | 0.8184 ± 0.0162 | 0.7620 ± 0.0294 | 0.5166 ± 0.0495 |
| GrEGs† (embn2v64) | 0.9042 ± 0.0136 | 0.8697 ± 0.0124 | 0.7996 ± 0.0218 | 0.6014 ± 0.0392 |
| GrEGs† (seq+embn2v64) | 0.9157 ± 0.0095 | 0.8809 ± 0.0072 | 0.8121 ± 0.0158 | 0.6327 ± 0.0227 |
| GrEGs† (bio+exp+embn2v64) | 0.9070 ± 0.0199 | 0.8598 ± 0.0166 | 0.8053 ± 0.0218 | 0.6190 ± 0.0429 |
| GrEGs† (seq+bio+exp+embn2v64) | 0.9104 ± 0.0158 | 0.8585 ± 0.0182 | 0.8006 ± 0.0276 | 0.6145 ± 0.0476 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| DeepHE (seq) | 0.8140 ± 0.0150 | 0.8012 ± 0.0094 | 0.7104 ± 0.0185 | 0.4063 ± 0.0236 |
| DeepHE (bio+exp) | 0.8429 ± 0.0205 | 0.8153 ± 0.0237 | 0.7349 ± 0.0236 | 0.4552 ± 0.0364 |
| DeepHE (embn2v64) | 0.9068 ± 0.0125 | 0.8842 ± 0.0136 | 0.8184 ± 0.0257 | 0.6390 ± 0.0436 |
| DeepHE (seq+embn2v64) | 0.9071 ± 0.0097 | 0.8754 ± 0.0141 | 0.8034 ± 0.0250 | 0.6115 ± 0.0427 |
| DeepHE (bio+exp+embn2v64) | 0.8979 ± 0.0094 | 0.8736 ± 0.0132 | 0.8025 ± 0.0180 | 0.6057 ± 0.0377 |
| DeepHE (seq+bio+exp+embn2v64) | 0.9009 ± 0.0120 | 0.8679 ± 0.0197 | 0.7985 ± 0.0193 | 0.5943 ± 0.0455 |
| GrEGs† (seq) | 0.8396 ± 0.0167 | 0.8201 ± 0.0169 | 0.7357 ± 0.0220 | 0.4608 ± 0.0452 |
| GrEGs† (bio+exp) | 0.8840 ± 0.0149 | 0.8267 ± 0.0195 | 0.7857 ± 0.0244 | 0.5507 ± 0.0479 |
| GrEGs† (embn2v64) | 0.9064 ± 0.0141 | 0.8712 ± 0.0140 | 0.8107 ± 0.0195 | 0.6115 ± 0.0387 |
| GrEGs† (seq+embn2v64) | 0.9180 ± 0.0145 | 0.8783 ± 0.0099 | 0.8129 ± 0.0174 | 0.6259 ± 0.0300 |
| GrEGs† (bio+exp+embn2v64) | 0.9197 ± 0.0132 | 0.8704 ± 0.0156 | 0.8215 ± 0.0188 | 0.6473 ± 0.0397 |
| GrEGs† (seq+bio+exp+embn2v64) | 0.9239 ± 0.0128 | 0.8784 ± 0.0161 | 0.8302 ± 0.0232 | 0.6673 ± 0.0439 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| DeepHE (seq) | 0.8020 ± 0.0206 | 0.7971 ± 0.0131 | 0.7097 ± 0.0372 | 0.4001 ± 0.0506 |
| DeepHE (bio+exp) | 0.8295 ± 0.0258 | 0.7950 ± 0.0148 | 0.7334 ± 0.0305 | 0.4278 ± 0.0451 |
| DeepHE (embn2v64) | 0.8751 ± 0.0183 | 0.8628 ± 0.0097 | 0.7831 ± 0.0288 | 0.5690 ± 0.0401 |
| DeepHE (seq+embn2v64) | 0.8859 ± 0.0204 | 0.8614 ± 0.0171 | 0.7811 ± 0.0309 | 0.5663 ± 0.0521 |
| DeepHE (bio+exp+embn2v64) | 0.8852 ± 0.0202 | 0.8753 ± 0.0144 | 0.7971 ± 0.0341 | 0.6044 ± 0.0515 |
| DeepHE (seq+bio+exp++embn2v64) | 0.8869 ± 0.0148 | 0.8668 ± 0.0147 | 0.7940 ± 0.0218 | 0.5873 ± 0.0373 |
| GrEGs† (seq) | 0.8343 ± 0.0141 | 0.8146 ± 0.0150 | 0.7326 ± 0.0188 | 0.4504 ± 0.0376 |
| GrEGs† (bio+exp) | 0.8678 ± 0.0190 | 0.8275 ± 0.0198 | 0.7830 ± 0.0286 | 0.5458 ± 0.0529 |
| GrEGs† (embn2v64) | 0.8973 ± 0.0142 | 0.8702 ± 0.0114 | 0.8055 ± 0.0182 | 0.6048 ± 0.0340 |
| GrEGs† (seq+embn2v64) | 0.9097 ± 0.0094 | 0.8752 ± 0.0099 | 0.8065 ± 0.0102 | 0.6150 ± 0.0260 |
| GrEGs† (bio+exp+embn2v64) | 0.9129 ± 0.0120 | 0.8693 ± 0.0110 | 0.8181 ± 0.0167 | 0.6398 ± 0.0305 |
| GrEGs† (seq+bio+exp+embn2v64) | 0.9155 ± 0.0090 | 0.8727 ± 0.0093 | 0.8226 ± 0.0187 | 0.6494 ± 0.0271 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| DeepHE (seq) | 0.8293 ± 0.0170 | 0.8068 ± 0.0184 | 0.7240 ± 0.0151 | 0.4286 ± 0.0320 |
| DeepHE (bio+exp) | 0.8501 ± 0.0212 | 0.8199 ± 0.0229 | 0.7568 ± 0.0251 | 0.4828 ± 0.0427 |
| DeepHE (embn2v64) | 0.9034 ± 0.0137 | 0.8799 ± 0.0128 | 0.8076 ± 0.0188 | 0.6230 ± 0.0305 |
| DeepHE (seq+embn2v64) | 0.9169 ± 0.0104 | 0.8833 ± 0.0121 | 0.8204 ± 0.0204 | 0.6389 ± 0.0324 |
| DeepHE (bio+exp+embn2v64) | 0.9038 ± 0.0229 | 0.8726 ± 0.0220 | 0.8121 ± 0.0288 | 0.6120 ± 0.0587 |
| DeepHE (seq+bio+exp+embn2v64) | 0.9137 ± 0.0173 | 0.8783 ± 0.0131 | 0.8079 ± 0.0232 | 0.6174 ± 0.0425 |
| GrEGs† (seq) | 0.8377 ± 0.0210 | 0.8242 ± 0.0215 | 0.7222 ± 0.0336 | 0.4533 ± 0.0660 |
| GrEGs† (bio+exp) | 0.8749 ± 0.0214 | 0.8127 ± 0.0256 | 0.7543 ± 0.0330 | 0.5005 ± 0.0660 |
| GrEGs† (embn2v64) | 0.9206 ± 0.0169 | 0.8853 ± 0.0150 | 0.8235 ± 0.0208 | 0.6496 ± 0.0401 |
| GrEGs† (seq+embn2v64) | 0.9295 ± 0.0144 | 0.8897 ± 0.0152 | 0.8234 ± 0.0225 | 0.6585 ± 0.0443 |
| GrEGs† (bio+exp+embn2v64) | 0.9253 ± 0.0205 | 0.8763 ± 0.0139 | 0.8271 ± 0.0212 | 0.6616 ± 0.0386 |
| GrEGs† (seq+bio+exp+embn2v64) | 0.9254 ± 0.0177 | 0.8819 ± 0.0184 | 0.8308 ± 0.0252 | 0.6751 ± 0.0500 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| DeepHE (seq) | 0.7972 ± 0.0227 | 0.7926 ± 0.0191 | 0.7097 ± 0.0320 | 0.3984 ± 0.0384 |
| DeepHE (embn2v64) | 0.8818 ± 0.0107 | 0.8624 ± 0.0090 | 0.7834 ± 0.0156 | 0.5693 ± 0.0276 |
| DeepHE (seq+embn2v64) | 0.8812 ± 0.0145 | 0.8508 ± 0.0169 | 0.7782 ± 0.0298 | 0.5476 ± 0.0472 |
| GrEGs† (bio+exp) | 0.9009 ± 0.0128 | 0.8592 ± 0.0092 | 0.8141 ± 0.0120 | 0.5933 ± 0.0226 |
| GrEGs† (embn2v64) | 0.8917 ± 0.0122 | 0.8631 ± 0.0103 | 0.8024 ± 0.0197 | 0.5896 ± 0.0334 |
| GrEGs† (bio+exp+embn2v64) | 0.9238 ± 0.0106 | 0.8894 ± 0.0105 | 0.8389 ± 0.0122 | 0.6648 ± 0.0270 |
| Method | ROC-AUC | Accuracy | BA | MCC |
|---|---|---|---|---|
| DeepHE (seq) | 0.8038 ± 0.0200 | 0.7838 ± 0.0205 | 0.7052 ± 0.0311 | 0.3833 ± 0.0425 |
| DeepHE (embn2v64) | 0.8898 ± 0.0130 | 0.8664 ± 0.0059 | 0.7957 ± 0.0181 | 0.5864 ± 0.0228 |
| DeepHE (seq+embn2v64) | 0.8952 ± 0.0135 | 0.8672 ± 0.0095 | 0.7959 ± 0.0103 | 0.5881 ± 0.0238 |
| GrEGs† (bio+exp) | 0.9163 ± 0.0157 | 0.8624 ± 0.0160 | 0.8246 ± 0.0158 | 0.6093 ± 0.0372 |
| GrEGs† (embn2v64) | 0.9016 ± 0.0142 | 0.8648 ± 0.0106 | 0.8083 ± 0.0175 | 0.5994 ± 0.0264 |
| GrEGs† (bio+exp+embn2v64) | 0.9338 ± 0.0126 | 0.8911 ± 0.0096 | 0.8459 ± 0.0145 | 0.6739 ± 0.0275 |
References
- Juhas, M.; Eberl, L.; Glass, J.I. Essence of life: Essential genes of minimal genomes. Trends Cell Biol. 2011, 21, 562–568. [Google Scholar] [CrossRef]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. BioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- Gurumayum, S.; Jiang, P.; Hao, X.; Campos, T.L.; Young, N.D.; Korhonen, P.K.; Gasser, R.B.; Bork, P.; Zhao, X.M.; He, L.j.; et al. OGEE v3: Online GEne Essentiality database with increased coverage of organisms and human cell lines. Nucleic Acids Res. 2021, 49, D998–D1003. [Google Scholar] [CrossRef]
- Ferreira, P.; Choupina, A.B. CRISPR/Cas9 a simple, inexpensive and effective technique for gene editing. Mol. Biol. Rep. 2022, 49, 7079–7086. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.; Yang, Y.; Dai, W.; Song, J. A novel method for identifying essential genes by fusing dynamic protein–protein interactive networks. Genes 2019, 10, 31. [Google Scholar] [CrossRef]
- Funk, L.; Su, K.C.; Ly, J.; Feldman, D.; Singh, A.; Moodie, B.; Blainey, P.C.; Cheeseman, I.M. The phenotypic landscape of essential human genes. Cell 2022, 185, 4634–4653. [Google Scholar] [CrossRef]
- Bartha, I.; Di Iulio, J.; Venter, J.C.; Telenti, A. Human gene essentiality. Nat. Rev. Genet. 2018, 19, 51–62. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef]
- McDonald, E.R.; De Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef]
- Larrimore, K.E.; Rancati, G. The conditional nature of gene essentiality. Curr. Opin. Genet. Dev. 2019, 58, 55–61. [Google Scholar] [CrossRef]
- Luo, H.; Lin, Y.; Liu, T.; Lai, F.L.; Zhang, C.T.; Gao, F.; Zhang, R. DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res. 2021, 49, D677–D686. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar]
- Chen, H.; Zhang, Z.; Jiang, S.; Li, R.; Li, W.; Zhao, C.; Hong, H.; Huang, X.; Li, H.; Bo, X. New insights on human essential genes based on integrated analysis and the construction of the HEGIAP web-based platform. Brief. Bioinform. 2020, 21, 1397–1410. [Google Scholar] [CrossRef]
- Fan, Y.; Tang, X.; Hu, X.; Wu, W.; Ping, Q. Prediction of essential proteins based on subcellular localization and gene expression correlation. BMC Bioinform. 2017, 18, 13–21. [Google Scholar] [CrossRef]
- Beder, T.; Aromolaran, O.; Dönitz, J.; Tapanelli, S.; Adedeji, E.O.; Adebiyi, E.; Bucher, G.; Koenig, R. Identifying essential genes across eukaryotes by machine learning. NAR Genom. Bioinform. 2021, 3, lqab110. [Google Scholar] [CrossRef]
- Cacheiro, P.; Muñoz-Fuentes, V.; Murray, S.A.; Dickinson, M.E.; Bucan, M.; Nutter, L.M.; Peterson, K.A.; Haselimashhadi, H.; Flenniken, A.M.; Morgan, H.; et al. Human and mouse essentiality screens as a resource for disease gene discovery. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Fogarty, N.M.; McCarthy, A.; Snijders, K.E.; Powell, B.E.; Kubikova, N.; Blakeley, P.; Lea, R.; Elder, K.; Wamaitha, S.E.; Kim, D.; et al. Genome editing reveals a role for OCT4 in human embryogenesis. Nature 2017, 550, 67–73. [Google Scholar] [CrossRef]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A systematic survey of centrality measures for protein–protein interaction networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef]
- Lachance, J.C.; Matteau, D.; Brodeur, J.; Lloyd, C.J.; Mih, N.; King, Z.A.; Knight, T.F.; Feist, A.M.; Monk, J.M.; Palsson, B.O.; et al. Genome-scale metabolic modeling reveals key features of a minimal gene set. Mol. Syst. Biol. 2021, 17, e10099. [Google Scholar] [CrossRef]
- Shimada, K.; Bachman, J.A.; Muhlich, J.L.; Mitchison, T.J. shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. eLife 2021, 10, e57116. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- López-Cortés, A.; Paz-y Miño, C.; Guerrero, S.; Cabrera-Andrade, A.; Barigye, S.J.; Munteanu, C.R.; González-Díaz, H.; Pazos, A.; Pérez-Castillo, Y.; Tejera, E. OncoOmics approaches to reveal essential genes in breast cancer: A panoramic view from pathogenesis to precision medicine. Sci. Rep. 2020, 10, 5285. [Google Scholar] [CrossRef]
- Mair, B.; Tomic, J.; Masud, S.N.; Tonge, P.; Weiss, A.; Usaj, M.; Tong, A.H.Y.; Kwan, J.J.; Brown, K.R.; Titus, E.; et al. Essential gene profiles for human pluripotent stem cells identify uncharacterized genes and substrate dependencies. Cell Rep. 2019, 27, 599–615. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Zhan, T.; Boutros, M. Towards a compendium of essential genes–from model organisms to synthetic lethality in cancer cells. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 74–85. [Google Scholar] [CrossRef]
- Byars, S.G.; Voskarides, K. Antagonistic pleiotropy in human disease. J. Mol. Evol. 2020, 88, 12–25. [Google Scholar] [CrossRef]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef]
- Nandi, S.; Ganguli, P.; Sarkar, R.R. Essential gene prediction using limited gene essentiality information—An integrative semi-supervised machine learning strategy. PLoS ONE 2020, 15, e0242943. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Colic, M.; Hart, T. Common computational tools for analyzing CRISPR screens. Emerg. Top. Life Sci. 2021, 5, 779–788. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, M.; Yang, D. Bioinformatics approaches to analyzing CRISPR screen data: From dropout screens to single-cell CRISPR screens. Quant. Biol. 2022, 10, 307. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef]
- Hart, T.; Moffat, J. BAGEL: A computational framework for identifying essential genes from pooled library screens. BMC Bioinform. 2016, 17, 164. [Google Scholar] [CrossRef]
- Hart, T.; Brown, K.R.; Sircoulomb, F.; Rottapel, R.; Moffat, J. Measuring error rates in genomic perturbation screens: Gold standards for human functional genomics. Mol. Syst. Biol. 2014, 10, 733. [Google Scholar] [CrossRef]
- Vinceti, A.; Karakoc, E.; Pacini, C.; Perron, U.; De Lucia, R.R.; Garnett, M.J.; Iorio, F. CoRe: A robustly benchmarked R package for identifying core-fitness genes in genome-wide pooled CRISPR-Cas9 screens. BMC Genom. 2021, 22, 828. [Google Scholar] [CrossRef]
- Sharma, S.; Dincer, C.; Weidemüller, P.; Wright, G.J.; Petsalaki, E. CEN-tools: An integrative platform to identify the contexts of essential genes. Mol. Syst. Biol. 2020, 16, e9698. [Google Scholar] [CrossRef]
- Manzo, M.; Giordano, M.; Maddalena, L.; Guarracino, M.R.; Granata, I. Novel Data Science Methodologies for Essential Genes Identification Based on Network Analysis. In Data Science in Applications; Springer: Berlin/Heidelberg, Germany, 2023; pp. 117–145. [Google Scholar] [CrossRef]
- Granata, I.; Giordano, M.; Maddalena, L.; Manzo, M.; Guarracino, M.R. Network-Based Computational Modeling to Unravel Gene Essentiality. In Trends in Biomathematics: Modeling Epidemiological, Neuronal, and Social Dynamics: Selected Works from the BIOMAT Consortium Lectures, Rio de Janeiro, Brazil, 2022; Mondaini, R.P., Ed.; Springer: Cham, Switzerland, 2023; pp. 29–56. [Google Scholar] [CrossRef]
- Rasti, S.; Vogiatzis, C. A survey of computational methods in protein–protein interaction networks. Ann. Oper. Res. 2019, 276, 35–87. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Zeng, M.; Zheng, R.; Li, M. Network-based methods for predicting essential genes or proteins: A survey. Brief. Bioinform. 2020, 21, 566–583. [Google Scholar] [CrossRef]
- Dong, C.; Jin, Y.T.; Hua, H.L.; Wen, Q.F.; Luo, S.; Zheng, W.X.; Guo, F.B. Comprehensive review of the identification of essential genes using computational methods: Focusing on feature implementation and assessment. Brief. Bioinform. 2020, 21, 171–181. [Google Scholar] [CrossRef]
- Aromolaran, O.; Aromolaran, D.; Isewon, I.; Oyelade, J. Machine learning approach to gene essentiality prediction: A review. Brief. Bioinform. 2021, 22, bbab128. [Google Scholar] [CrossRef]
- Aromolaran, O.; Beder, T.; Oswald, M.; Oyelade, J.; Adebiyi, E.; Koenig, R. Essential gene prediction in Drosophila melanogaster using machine learning approaches based on sequence and functional features. Comput. Struct. Biotechnol. J. 2020, 18, 612–621. [Google Scholar] [CrossRef]
- Dai, W.; Chang, Q.; Peng, W.; Zhong, J.; Li, Y. Network embedding the protein–protein interaction network for human essential genes identification. Genes 2020, 11, 153. [Google Scholar] [CrossRef]
- Zeng, M.; Li, M.; Wu, F.; Li, Y.; Pan, Y. DeepEP: A deep learning framework for identifying essential proteins. BMC Bioinform. 2019, 20-S, 506:1–506:10. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, W.; Xiao, W. DeepHE: Accurately predicting human essential genes based on deep learning. PLoS Comput. Biol. 2020, 16, e1008229. [Google Scholar] [CrossRef]
- Kuang, S.; Wei, Y.; Wang, L. Expression-based prediction of human essential genes and candidate lncRNAs in cancer cells. Bioinformatics 2021, 37, 396–403. [Google Scholar] [CrossRef]
- Zeng, M.; Li, M.; Fei, Z.; Wu, F.X.; Li, Y.; Pan, Y.; Wang, J. A Deep Learning Framework for Identifying Essential Proteins by Integrating Multiple Types of Biological Information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 296–305. [Google Scholar] [CrossRef]
- Schapke, J.; Tavares, A.; Recamonde-Mendoza, M. EPGAT: Gene Essentiality Prediction With Graph Attention Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1615–1626. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar] [CrossRef]
- Grover, A.; Leskovec, J. Node2vec: Scalable Feature Learning for Networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD ’16), San Francisco, CA, USA, 13–17 August 2016; pp. 855–864. [Google Scholar] [CrossRef]
- TCGA Research Network. The Cancer Genome Atlas Program (TCGA). 2023. Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga (accessed on 30 May 2023).
- Choy, C.T.; Wong, C.H.; Chan, S.L. Embedding of Genes Using Cancer Gene Expression Data: Biological Relevance and Potential Application on Biomarker Discovery. Front. Genet. 2019, 9, 682. [Google Scholar] [CrossRef]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2vec: Distributed representation of genes based on co-expression. BMC Genom. 2019, 20, 82. [Google Scholar] [CrossRef]
- Vincent, P.; Larochelle, H.; Lajoie, I.; Bengio, Y.; Manzagol, P.A. Stacked Denoising Autoencoders: Learning Useful Representations in a Deep Network with a Local Denoising Criterion. J. Mach. Learn. Res. 2010, 11, 3371–3408. [Google Scholar]
- Charif, D.; Lobry, J. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Biological and Medical Physics, Biomedical Engineering; Bastolla, U., Porto, M., Roman, H., Vendruscolo, M., Eds.; Springer: New York, NY, USA, 2007; pp. 207–232. ISBN 978-3-540-35305-8. [Google Scholar] [CrossRef]
- Xiao, N.; Cao, D.S.; Zhu, M.F.; Xu, Q.S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef]
- Kotlyar, M.; Pastrello, C.; Ahmed, Z.; Chee, J.; Varyova, Z.; Jurisica, I. IID 2021: Towards context-specific protein interaction analyses by increased coverage, enhanced annotation and enrichment analysis. Nucleic Acids Res. 2021, 50, D640–D647. [Google Scholar] [CrossRef]
- Theodoris, C.; Xiao, L.; Chopra, A.; Chaffin, M.D.; Al Sayed, Z.R.; Hill, M.C.; Mantineo, H.; Brydon, E.M.; Zeng, Z.; Liu, X.S.; et al. Transfer learning enables predictions in network biology. Nature 2023, 618, 616–624. [Google Scholar] [CrossRef]

| Feature Context | Attributes |
|---|---|
| Structural stability | Gene/protein length |
| GC content | |
| Transcripts count | |
| Gene/protein sequence-derived properties | |
| Gene/protein expression | Transcripts abundance |
| Protein abundance | |
| Function and localisation | Functional annotation |
| Pathway involvement | |
| Subcellular localisation | |
| Epigenetics | Transcription factor binding |
| Chromatin accessibility | |
| DNA methylation | |
| Histone modification | |
| Conservation/evolution | Orthologs count |
| Protein stability | |
| Evolutionary age | |
| Association with disease | Gene–disease association |
| Cancer driver mutation | |
| Differential expression | |
| Embryonic development | Gene expression pattern |
| Network biology | Topological attributes |
| First Author | Years | Taxonomy |
|---|---|---|
| & Refs. | Covered | |
| Rasti [43] | 2001–2017 | (1) Network topology-based |
(2) Integrating PPINs and biological information
| ||
| Li [44] | 1987–2018 | (1) Network topology-based (exploiting neighbourhood, path, eigenvector information, or their combination) |
| (2) Integrating PPINs and biological information | ||
| (3) Dynamic network-based | ||
| (4) Machine learning-based | ||
| Dong [45] | 1996–2018 | Modeling methods implementing/combining five types of features: |
| (1) Evolutionary conservation | ||
| (2) Domain information | ||
| (3) Network topology | ||
| (4) Sequence component | ||
| (5) Expression level | ||
| Aromolaran [46] | 2004–2021 | (1) Homology mapping |
| (2) Constraint-based | ||
| (3) Machine learning-based (intrinsic/extrinsic features) | ||
| Granata [42] | 2019–2021 | (1) Network topology-based |
| (2) Classical machine learning-based | ||
| (3) Deep learning-based |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, M.; Falbo, E.; Maddalena, L.; Piccirillo, M.; Granata, I. Untangling the Context-Specificity of Essential Genes by Means of Machine Learning: A Constructive Experience. Biomolecules 2024, 14, 18. https://doi.org/10.3390/biom14010018
Giordano M, Falbo E, Maddalena L, Piccirillo M, Granata I. Untangling the Context-Specificity of Essential Genes by Means of Machine Learning: A Constructive Experience. Biomolecules. 2024; 14(1):18. https://doi.org/10.3390/biom14010018
Chicago/Turabian StyleGiordano, Maurizio, Emanuele Falbo, Lucia Maddalena, Marina Piccirillo, and Ilaria Granata. 2024. "Untangling the Context-Specificity of Essential Genes by Means of Machine Learning: A Constructive Experience" Biomolecules 14, no. 1: 18. https://doi.org/10.3390/biom14010018
APA StyleGiordano, M., Falbo, E., Maddalena, L., Piccirillo, M., & Granata, I. (2024). Untangling the Context-Specificity of Essential Genes by Means of Machine Learning: A Constructive Experience. Biomolecules, 14(1), 18. https://doi.org/10.3390/biom14010018