
Coupled Electrostatic and Hydrophobic Destabilisation of the Gelsolin-Actin Complex Enables Facile Detection of Ovarian Cancer Biomarker Lysophosphatidic Acid




Abstract
:1. Introduction
2. Materials and Methods
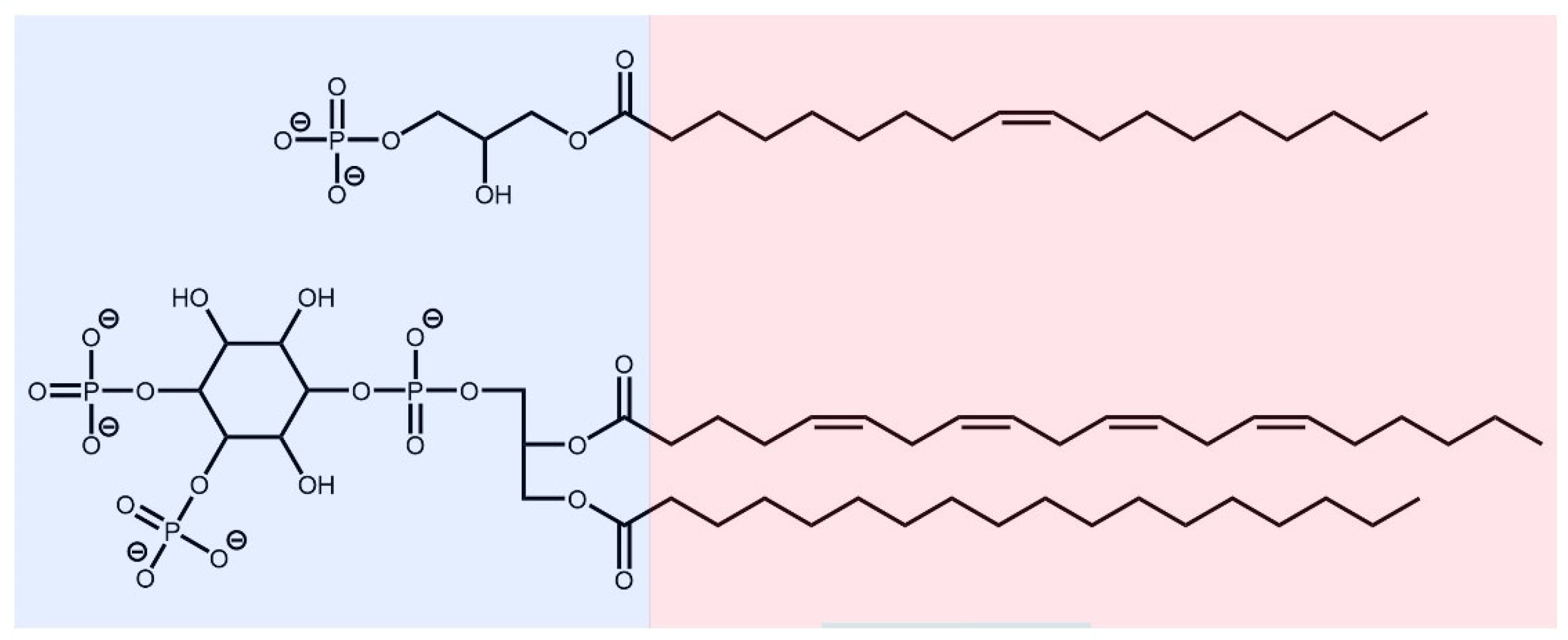
2.1. Source of Protein and LPA Structures
2.2. Macromolecular Docking
2.3. Molecular Dynamics Simulations
2.4. Model Analysis
3. Results and Discussion
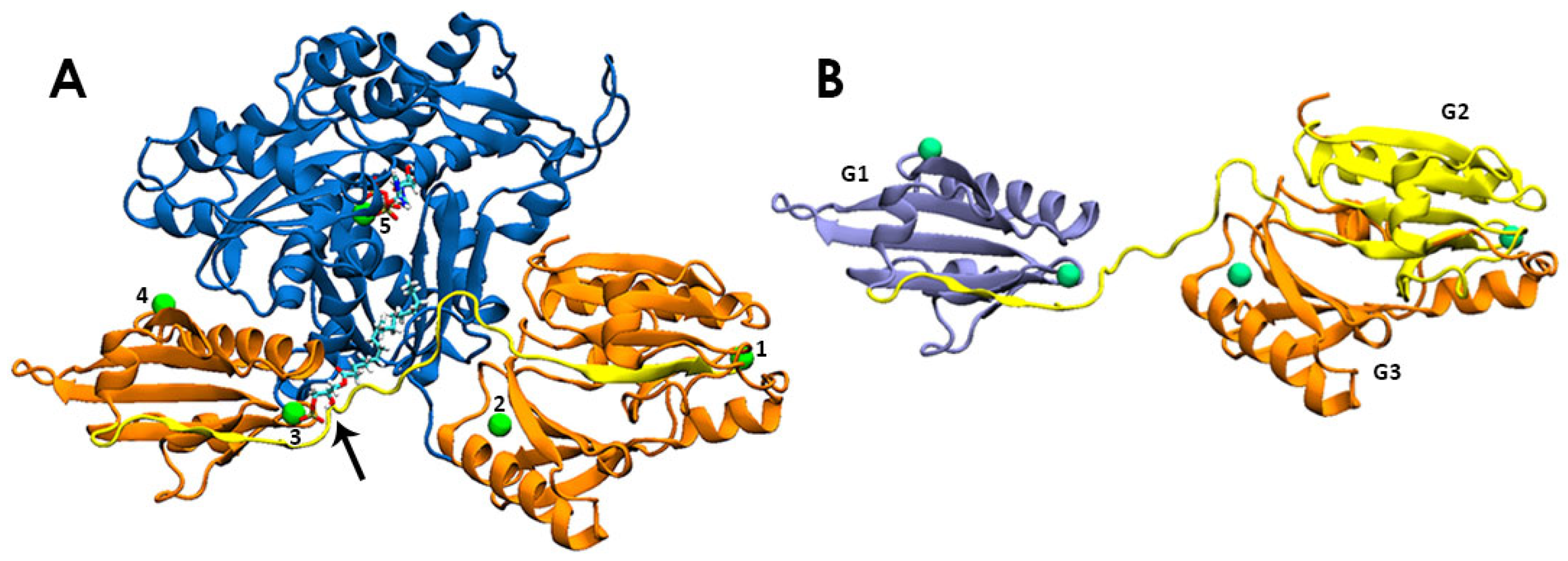
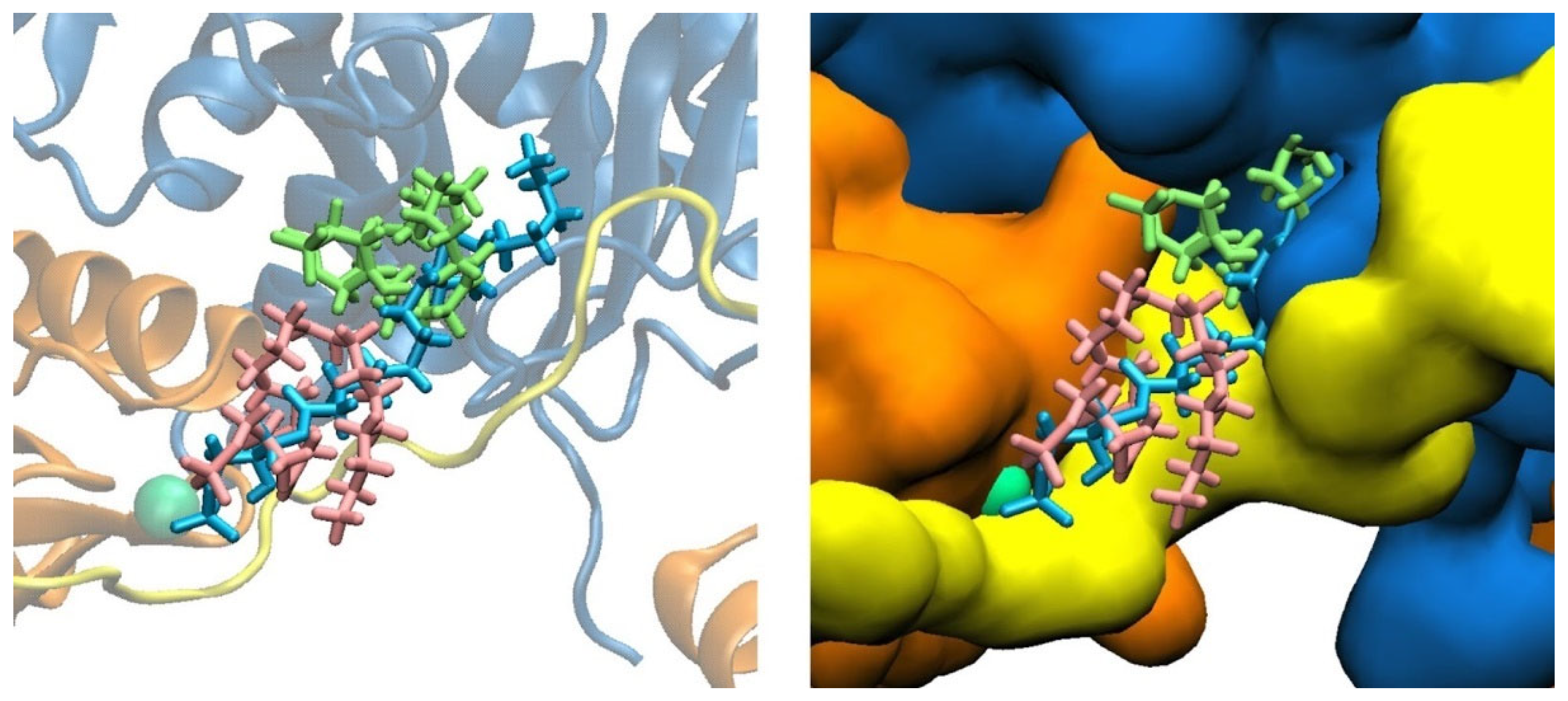
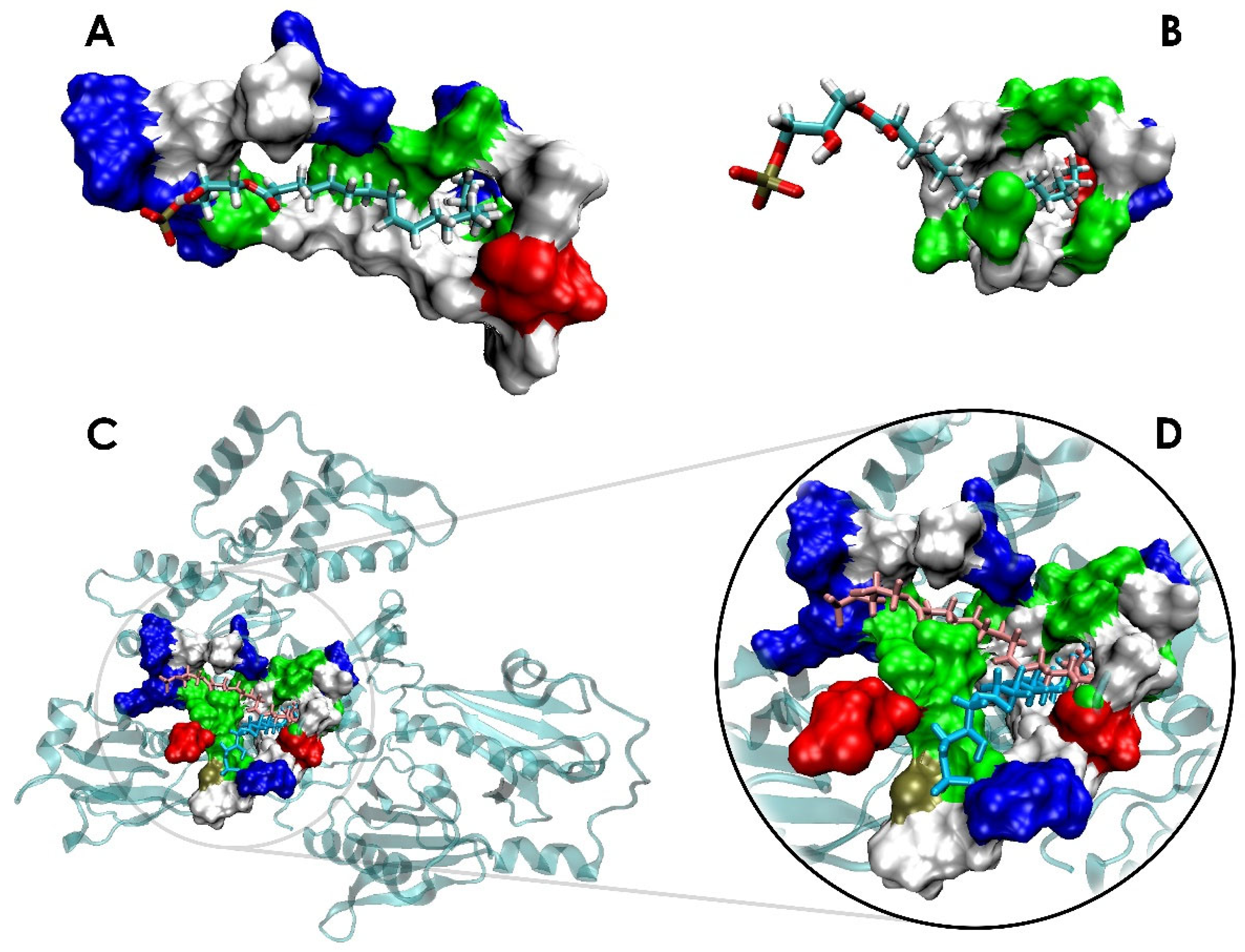
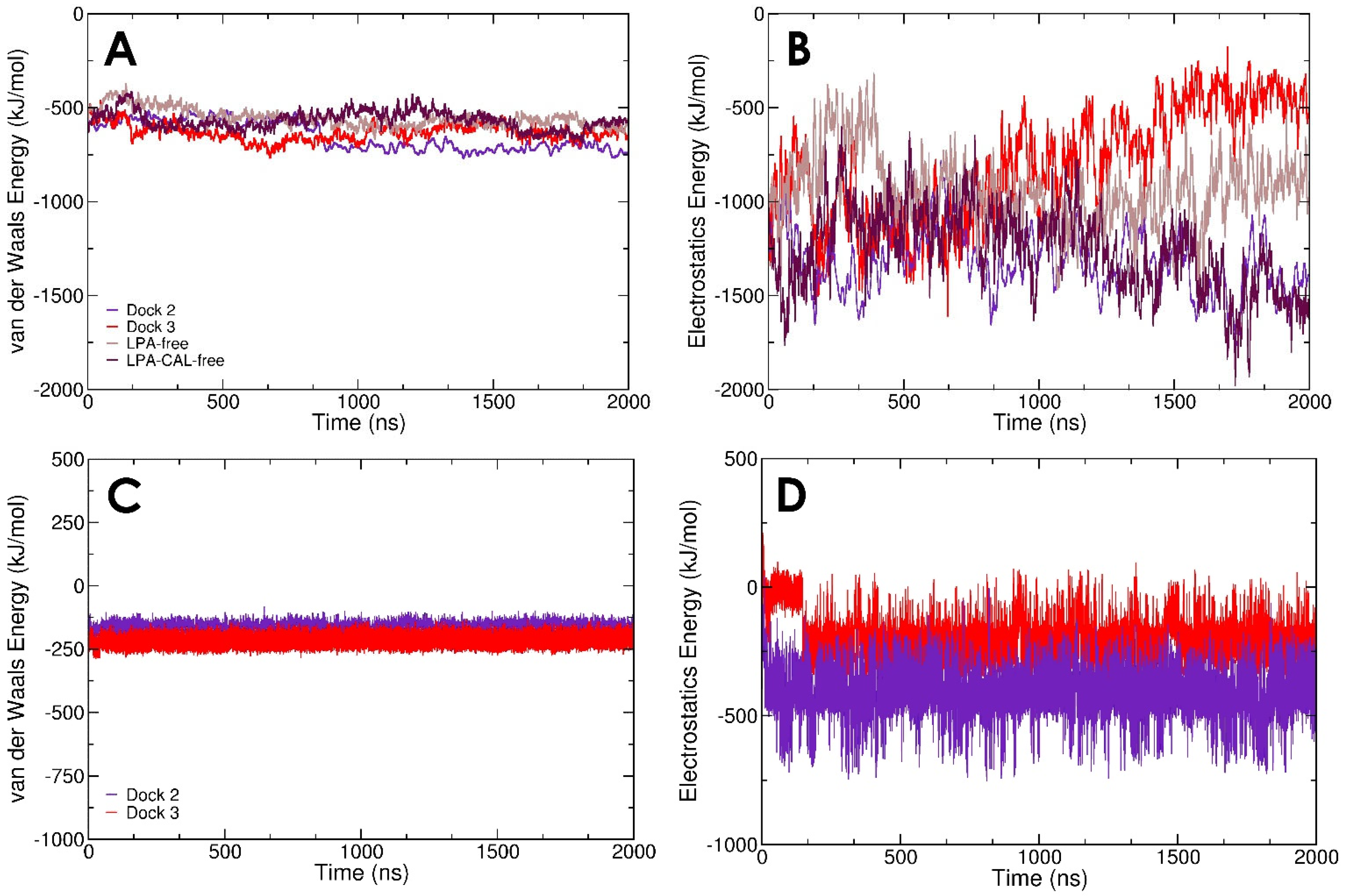
3.1. LPA Binding Is Not Restricted to the PIP2-Binding Domain of Gelsolin(1–3)
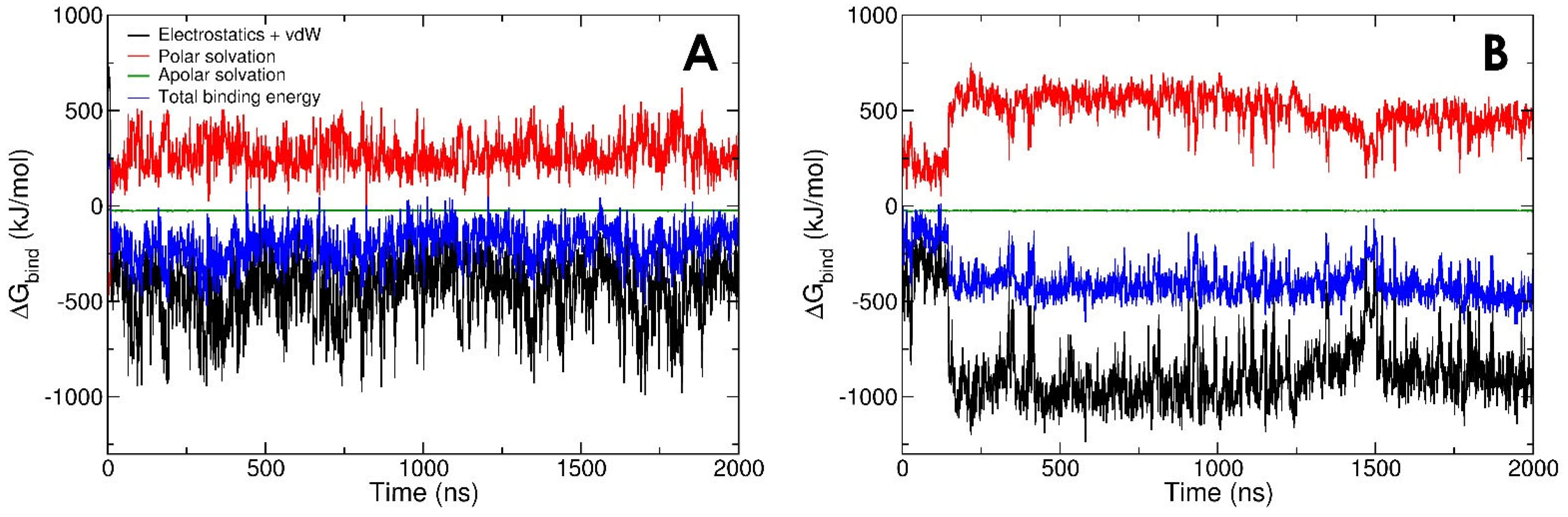
3.2. LPA-Binding Weakens the Gelsolin(1–3)-Actin Complex
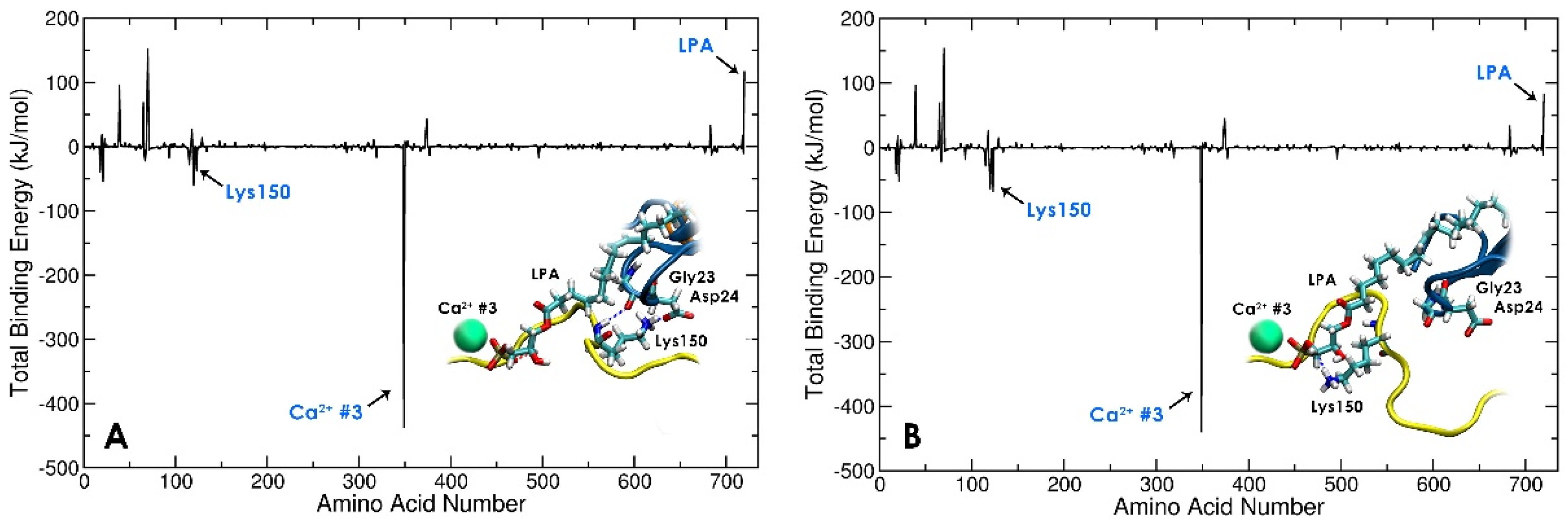
3.3. Local Conformational Rearrangements in Gelsolin(1–3)-Actin due to LPA Binding Predicts the Efficiency of LPA as an OC Biomarker
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandalaft, L.E.; Dangaj Laniti, D.; Coukos, G. Immunobiology of High-Grade Serous Ovarian Cancer: Lessons for Clinical Translation. Nat. Rev. Cancer 2022, 22, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Chan, J.K.; Tian, C.; Kesterson, J.P.; Monk, B.J.; Kapp, D.S.; Davidson, B.; Robertson, S.; Copeland, L.J.; Walker, J.L.; Wenham, R.M.; et al. Symptoms of Women with High-Risk Early-Stage Ovarian Cancer. Obstet. Gynecol. 2022, 139, 157–162. [Google Scholar] [CrossRef]
- Bankhead, C.R.; Collins, C.; Stokes-Lampard, H.; Rose, P.; Wilson, S.; Clements, A.; Mant, D.; Kehoe, S.T.; Austoker, J. Identifying Symptoms of Ovarian Cancer: A Qualitative and Quantitative Study. Int. J. Gynaecol. Obstet. 2008, 115, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Tate, A.R.; Martin, A.G.R.; Murray-Thomas, T.; Anderson, S.R.; Cassell, J.A. Determining the Date of Diagnosis—Is It a Simple Matter? The Impact of Different Approaches to Dating Diagnosis on Estimates of Delayed Care for Ovarian Cancer in UK Primary Care. BMC Med. Res. Methodol. 2009, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.D.; Skilling, J.S.; Udaltsova, N.V.; Smith, L.H. Early Symptoms of Ovarian Cancer: A Case–control Study Without Recall Bias. Fam. Pract. 2005, 22, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Jayde, V.; White, K.; Blomfield, P. Symptoms and Diagnostic Delay in Ovarian Cancer: A Summary of the Literature. Contemp. Nurse 2010, 34, 55–65. [Google Scholar] [CrossRef]
- Doubeni, C.A.; Doubeni, A.R.; Myers, A.E. Diagnosis and Management of Ovarian Cancer. Am. Fam. Physician 2016, 93, 937–944. [Google Scholar]
- SEER Cancer Statistics Review, 1975–2017. SEER. Available online: https://seer.cancer.gov/csr/1975_2017/index.html (accessed on 15 September 2022).
- Thompson, M.; Ahmadi, S.; Davoudian, K.; De La Franier, B.; Lotay, N.; Bernardini, M.Q. Sensor Detection in Gynaecological Medicine. Sens. Diagn. 2022, 1, 877–901. [Google Scholar] [CrossRef]
- Suri, A.; Perumal, V.; Ammalli, P.; Suryan, V.; Bansal, S.K. Diagnostic Measures Comparison for Ovarian Malignancy Risk in Epithelial Ovarian Cancer Patients: A Meta-Analysis. Sci. Rep. 2021, 11, 17308. [Google Scholar] [CrossRef]
- Mohaghegh, P.; Rockall, A.G. Imaging Strategy for Early Ovarian Cancer: Characterization of Adnexal Masses with Conventional and Advanced Imaging Techniques. Radiographics 2012, 32, 1751–1753. [Google Scholar] [CrossRef] [PubMed]
- Fischerova, D.; Burgetova, A. Imaging Techniques for the Evaluation of Ovarian Cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2014, 28, 697–720. [Google Scholar] [CrossRef] [PubMed]
- Essmat, A.A.-A.; Meleis, M.E.-S.; Rady, H.A.-S.; Zaki, I.I.A.; Khattab, A.S.A. Total Peritoneal Gutter Removal versus Random Peritoneal Biopsy in Cases of Ovarian Cancer. Open J. Obstet. Gynecol. 2021, 11, 1342–1350. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Y.; Fu, Z.; Dong, L.; Yang, S.; Meng, W.; Li, Y.; Zhang, W.; Zhang, J.; Zheng, C.; et al. Diagnostic Value of Plasma Lysophosphatidic Acid Levels in Ovarian Cancer Patients: A Case-control Study and Updated Meta-analysis. Open J. Obstet. Gynecol. 2015, 41, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Ueda, Y.; Matsuzaki, S.; Yokoyama, T.; Kimura, T.; Yoshino, K.; Fujita, M.; Kimura, T.; Enomoto, T. Biomarkers for Screening, Diagnosis, and Monitoring of Ovarian Cancer. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- De La Franier, B.; Thompson, M. Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. Biosensors 2020, 10, 13. [Google Scholar] [CrossRef]
- Noguchi, K.; Herr, D.; Mutoh, T.; Chun, J. Lysophosphatidic Acid (LPA) and Its Receptors. Curr. Opin. Pharmacol. 2009, 9, 15–23. [Google Scholar] [CrossRef]
- Kano, K.; Aoki, J.; Hla, T. Lysophospholipid Mediators in Health and Disease. Annu. Rev. Pathol. 2022, 17, 459–483. [Google Scholar] [CrossRef]
- Geraldo, L.H.M.; de Sampaio Spohr, T.C.L.; do Amaral, R.F.; da Fonseca, A.C.C.; Garcia, C.; de Almeida Mendes, F.; Freitas, C.; dos Santos, M.F.; Lima, F.R.S. Role of Lysophosphatidic Acid and Its Receptors in Health and Disease: Novel Therapeutic Strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The Autotaxin–LPA Axis Emerges as a Novel Regulator of Lymphocyte Homing and Inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef]
- Chen, X.; Gui, W.; Liu, H.; Ma, Q. A Novel CuZnInS Quantum Dot-Based ECL Sensing System for Lysophosphatidic Acid Detection. Analyst 2017, 142, 4142–4149. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic Acid as a Potential Biomarker for Ovarian and Other Gynecologic Cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Shoaib, M.; Kuschner, C.; Nishikimi, M.; Becker, L.B.; Lee, A.T.; Kim, J. Challenges and Inconsistencies in Using Lysophosphatidic Acid as a Biomarker for Ovarian Cancer. Cancers 2019, 11, 520. [Google Scholar] [CrossRef]
- Baker, D.L.; Morrison, P.; Miller, B.; Riely, C.A.; Tolley, B.; Westermann, A.M.; Bonfrer, J.M.G.; Bais, E.; Moolenaar, W.H.; Tigyi, G. Plasma Lysophosphatidic Acid Concentration and Ovarian Cancer. JAMA 2002, 287, 3081–3082. [Google Scholar] [CrossRef]
- Sedláková, I.; Vávrová, J.; Tošner, J.; Hanousek, L. Lysophosphatidic Acid (LPA)—A Perspective Marker in Ovarian Cancer. Tumor Biol. 2010, 32, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Meerschaert, K.; De Corte, V.; De Ville, Y.; Vandekerckhove, J.; Gettemans, J. Gelsolin and Functionally Similar Actin-Binding Proteins Are Regulated by Lysophosphatidic Acid. EMBO J. 1998, 17, 5923–5932. [Google Scholar] [CrossRef]
- Mintzer, E.; Sargsyan, H.; Bittman, R. Lysophosphatidic Acid and Lipopolysaccharide Bind to the PIP2-Binding Domain of Gelsolin. Biochim. Biophys. Acta. 2006, 1758, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Ma, Q.; Wang, H.; Chumnarnsilpa, S.; Lee, W.L.; Larsson, M.; Kannan, B.; Hernandez-Valladares, M.; Burtnick, L.D.; Robinson, R.C. Ca2+ Binding by Domain 2 Plays a Critical Role in the Activation and Stabilization of Gelsolin. Proc. Biol. Sci. 2009, 106, 13713–13718. [Google Scholar] [CrossRef]
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, a Multifunctional Actin Regulatory Protein. J. Biol. Chem. 1999, 274, 33179–33182. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Lee, H.; Azuma, T.; Stossel, T.P.; Turck, C.W.; Karliner, J.S. Gelsolin Binding and Cellular Presentation of Lysophosphatidic Acid. J. Biol. Chem. 2000, 275, 14573–14578. [Google Scholar] [CrossRef]
- Feng, L.; Mejillano, M.; Yin, H.L.; Chen, J.; Prestwich, G.D. Full-Contact Domain Labeling: Identification of a Novel Phosphoinositide Binding Site on Gelsolin That Requires the Complete Protein. Biochemistry 2001, 40, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Choe, H.; Burtnick, L.D.; Mejillano, M.; Yin, H.L.; Robinson, R.C.; Choe, S. The Calcium Activation of Gelsolin: Insights from the 3 Å Structure of the G4–G6/Actin Complex. J. Mol. Biol. 2002, 324, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kanzawa, N.; Ono, S. Calcium-Sensitive Activity and Conformation of Caenorhabditis elegans Gelsolin-like Protein 1 Are Altered by Mutations in the First Gelsolin-like Domain. J. Biol. Chem. 2011, 286, 34051–34059. [Google Scholar] [CrossRef]
- Liu, Z.; Klaavuniemi, T.; Ono, S. Distinct Roles of Four Gelsolin-like Domains of Caenorhabditis elegans Gelsolin-like Protein-1 in Actin Filament Severing, Barbed-end Capping, and Phosphoinositide Binding. Biochemistry 2010, 49, 4349–4360. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Larsson, M.; Robinson, R.C.; Burtnick, L.D. Gelsolin: The Tail of a Molecular Gymnast. Cytoskeleton 2013, 70, 360–384. [Google Scholar] [CrossRef]
- Keune, W.-J.; Hausmann, J.; Boiler, R.; Tolenaars, D.; Kremer, A.; Heidebrecht, T.; Joosten, R.P.; Sunkara, M.; Morris, A.J.; Matas-Rico, E.; et al. Steroid Binding to Autotaxin Links Bile Salts and Lysophosphatidic Acid Signalling. Nat. Commun. 2016, 7, 11248. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for pK a Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein–Protein and Protein–DNA/RNA docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hockney, R.W. The Potential Calculation and Some Applications. Methods Comput. Phys. 1970, 9, 135–211. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1998, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1998, 98, 10089. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Best, R.B.; Hummer, G.; Eaton, W.A. Native Contacts Determine Protein Folding Mechanisms in Atomistic Simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 17874–17879. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Mercadante, D.; Gräter, F.; Daday, C. CONAN: A Tool to Decode Dynamical Information from Molecular Interaction Maps. Biophys. J. 2018, 114, 1267–1273. [Google Scholar] [CrossRef]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA Producing Enzyme with Diverse Functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef]
- Salgado-Polo, F.; Fish, A.; Matsoukas, M.-T.; Heidebrecht, T.; Keune, W.J.; Perrakis, A. Lysophosphatidic Acid Produced by Autotaxin Acts as an Allosteric Modulator of Its Catalytic Efficiency. J. Biol. Chem. 2018, 293, 14312–14327. [Google Scholar] [CrossRef] [PubMed]
- Meloni, B.P.; Mastaglia, F.L.; Knuckey, N.W. Cationic Arginine-Rich Peptides (CARPs): A Novel Class of Neuroprotective Agents with a Multimodal Mechanism of Action. Front. Neurol. 2020, 11, 108. [Google Scholar] [CrossRef]
- Irobi, E.; Burtnick, L.D.; Urosev, D.; Narayan, K.; Robinson, R.C. From the First to the Second Domain of Gelsolin: A Common Path on the Surface of Actin? FEBS Lett. 2003, 552, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Irobi, E.; Aguda, A.H.; Larsson, M.; Guerin, C.; Yin, H.L.; Burtnick, L.D.; Blanchoin, L.; Robinson, R.C. Structural Basis of Actin Sequestration by Thymosin-β4: Implications for WH2 Proteins. EMBO J. 2004, 23, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Grossfield, A.; Zuckerman, D.M. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 2009, 5, 23–48. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Residues within 3 Å at 0 µs | Residues within 3 Å at 2 µs |
|---|---|---|
| Dock 1 | Gelsolin(1–3): Glu92, Ser94, Gln95, Ser147 *, Phe149 *, His151 * | N/A |
| Actin: Asp25 | N/A | |
| Dock 2 | Gelsolin(1–3): Gln95, Phe149 * | Gelsolin(1–3): Gln95, Arg120 |
| Actin: Gly23, Asp24, Asp25, Ala26, Glu334, Tyr337, Trp340, Ile341 | Actin: Lys18, Ala19, Gly20, Phe21, Ala22, Asp24, Asp25, Pro27, Ser145, Arg147, Ile330, Pro332, Glu334, Arg335, Tyr337, Ser338, Trp340, Ile341 | |
| Dock 3 | Gelsolin(1–3): Glu92, Ser94, Val145 *, Ala146 *, Ser147 *, Phe149 *, Lys150 *, His151 * | Gelsolin(1–3): Glu92, Ser94, Gln95, Val145 *, Ala146 *, Ser147 *, Phe149 *, Lys150 * |
| Actin: Val9, Asp11, Lys18, Asp24, Asp25, Ala26, Pro27, Trp340, Ile341 | Actin: Val9, Asp11, Lys18, Gly20, Ala22, Gly23, Pro27, Tyr337, Trp340, Ile341, Ser344 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davoudian, K.; Bhattacharya, S.; Thompson, D.; Thompson, M. Coupled Electrostatic and Hydrophobic Destabilisation of the Gelsolin-Actin Complex Enables Facile Detection of Ovarian Cancer Biomarker Lysophosphatidic Acid. Biomolecules 2023, 13, 1426. https://doi.org/10.3390/biom13091426
Davoudian K, Bhattacharya S, Thompson D, Thompson M. Coupled Electrostatic and Hydrophobic Destabilisation of the Gelsolin-Actin Complex Enables Facile Detection of Ovarian Cancer Biomarker Lysophosphatidic Acid. Biomolecules. 2023; 13(9):1426. https://doi.org/10.3390/biom13091426
Chicago/Turabian StyleDavoudian, Katharina, Shayon Bhattacharya, Damien Thompson, and Michael Thompson. 2023. "Coupled Electrostatic and Hydrophobic Destabilisation of the Gelsolin-Actin Complex Enables Facile Detection of Ovarian Cancer Biomarker Lysophosphatidic Acid" Biomolecules 13, no. 9: 1426. https://doi.org/10.3390/biom13091426
APA StyleDavoudian, K., Bhattacharya, S., Thompson, D., & Thompson, M. (2023). Coupled Electrostatic and Hydrophobic Destabilisation of the Gelsolin-Actin Complex Enables Facile Detection of Ovarian Cancer Biomarker Lysophosphatidic Acid. Biomolecules, 13(9), 1426. https://doi.org/10.3390/biom13091426