
Integrated Metabolomic and Transcriptomic Analysis of the Quinoa Seedling Response to High Relative Humidity Stress

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Materials and High RH Treatment
2.2. Analysis of Morphological and Physiological Indicators
2.3. Metabolite and Transcript Determination and Analysis
2.3.1. Metabolite Extraction and Liquid Chromatography-Mass Spectrometry Analysis
2.3.2. Qualitative and Quantitative Analysis of Metabolites
2.4. Transcript Profiling and Data Analysis
2.4.1. RNA Extraction and Sequencing
2.4.2. Identification of Differentially Expressed Genes and Enrichment Analysis
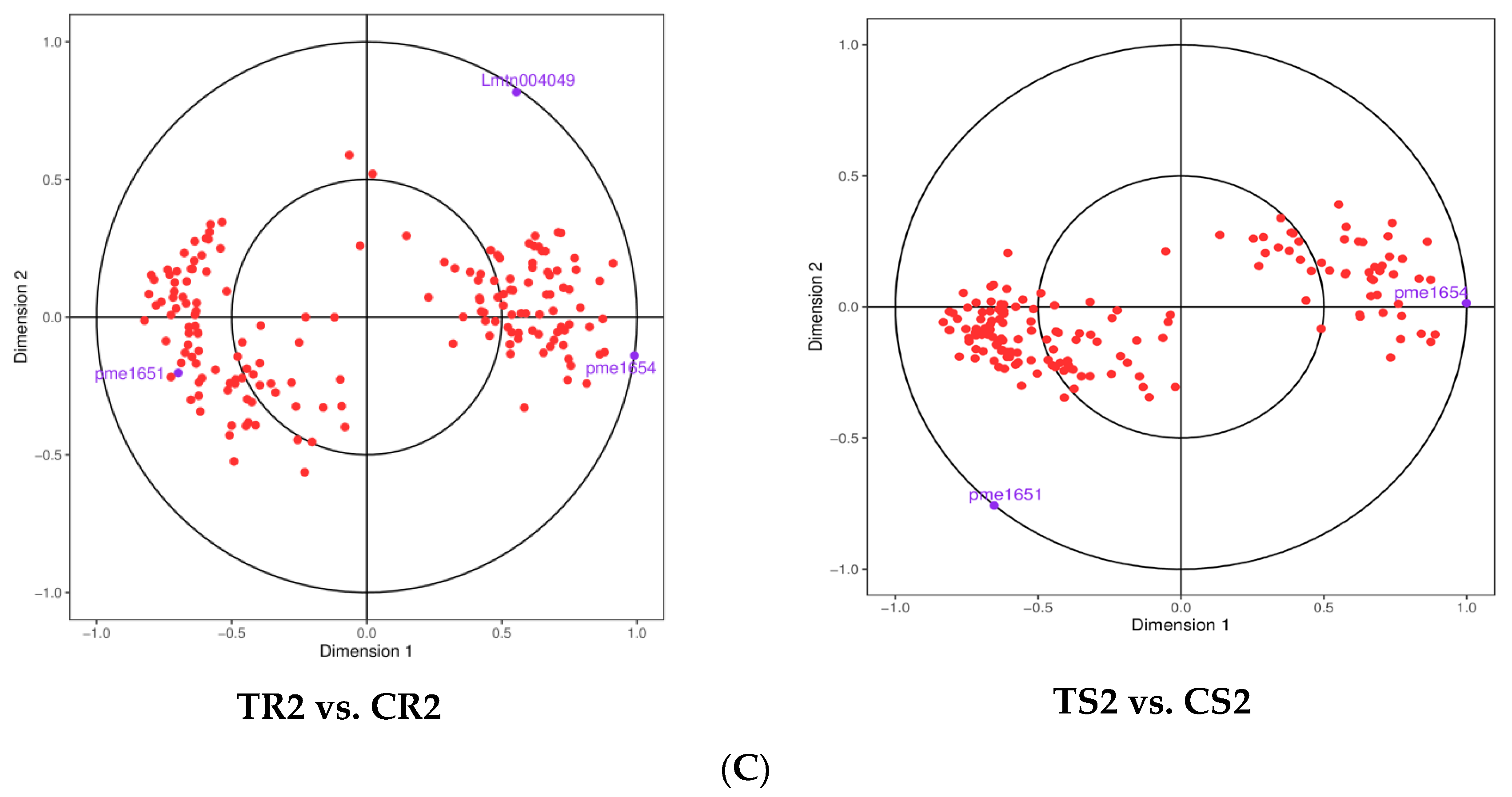
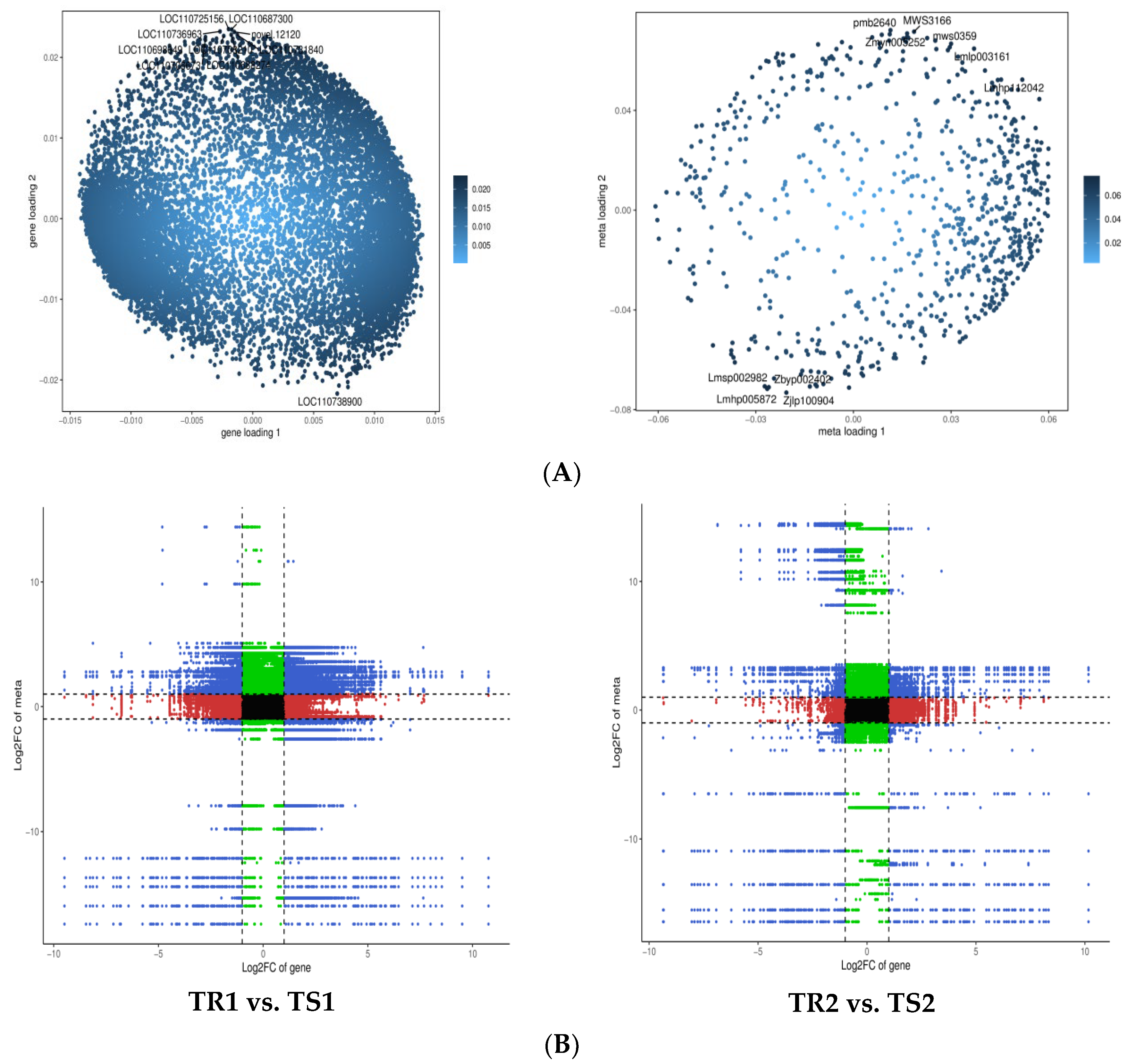
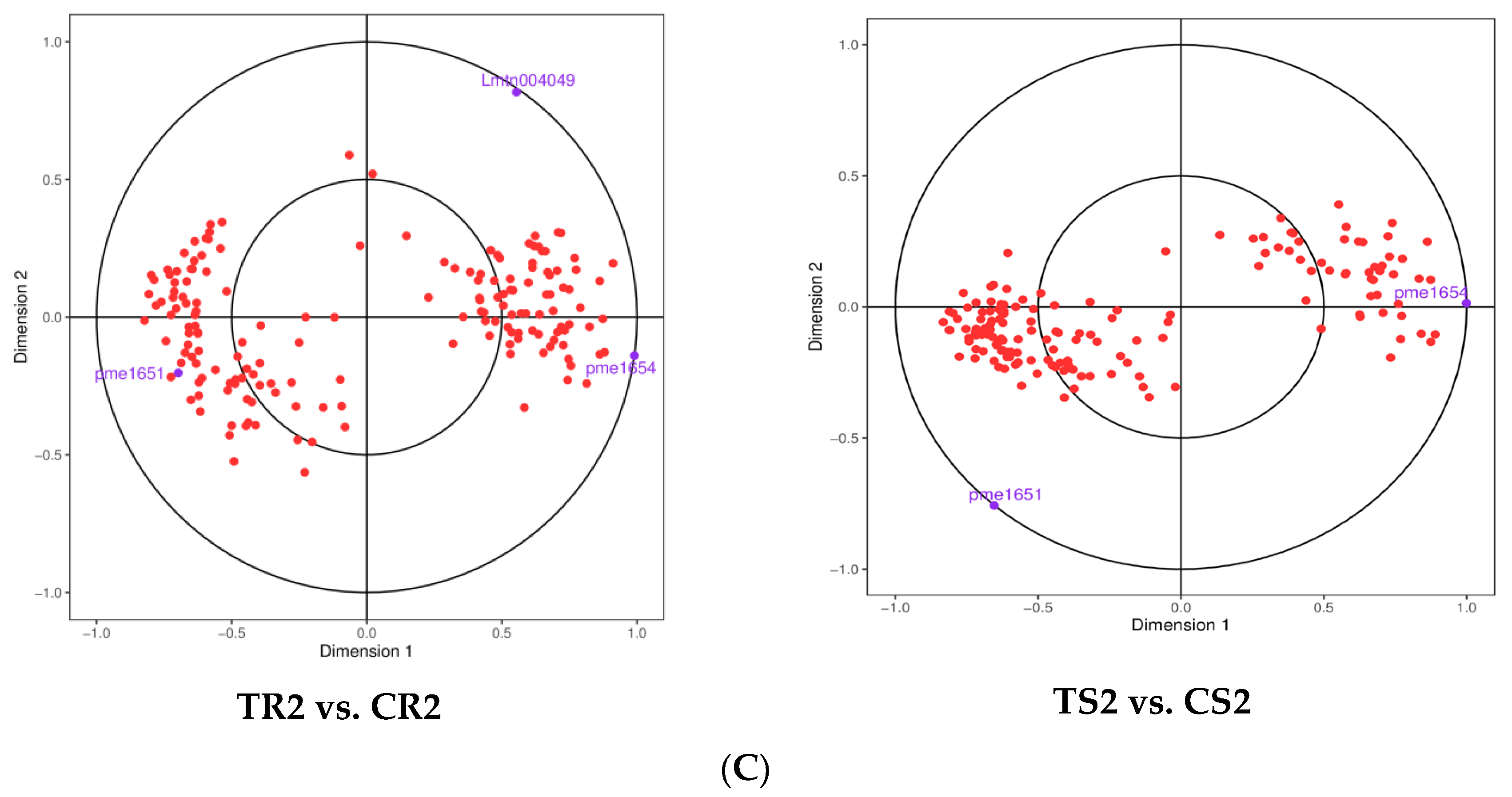
2.5. Joint Transcriptome and Metabolome Analysis
2.6. Transcription Factor Identification, Weighted Gene Co-Expression Network Analysis, and Gene Network Visualization
2.7. Quantitative Reverse Transcription-PCR
2.8. Statistical Analysis
3. Results
3.1. Changes in Morphological Indicators under High RH Stress
3.2. Changes in Physiological Indicators under High RH Stress
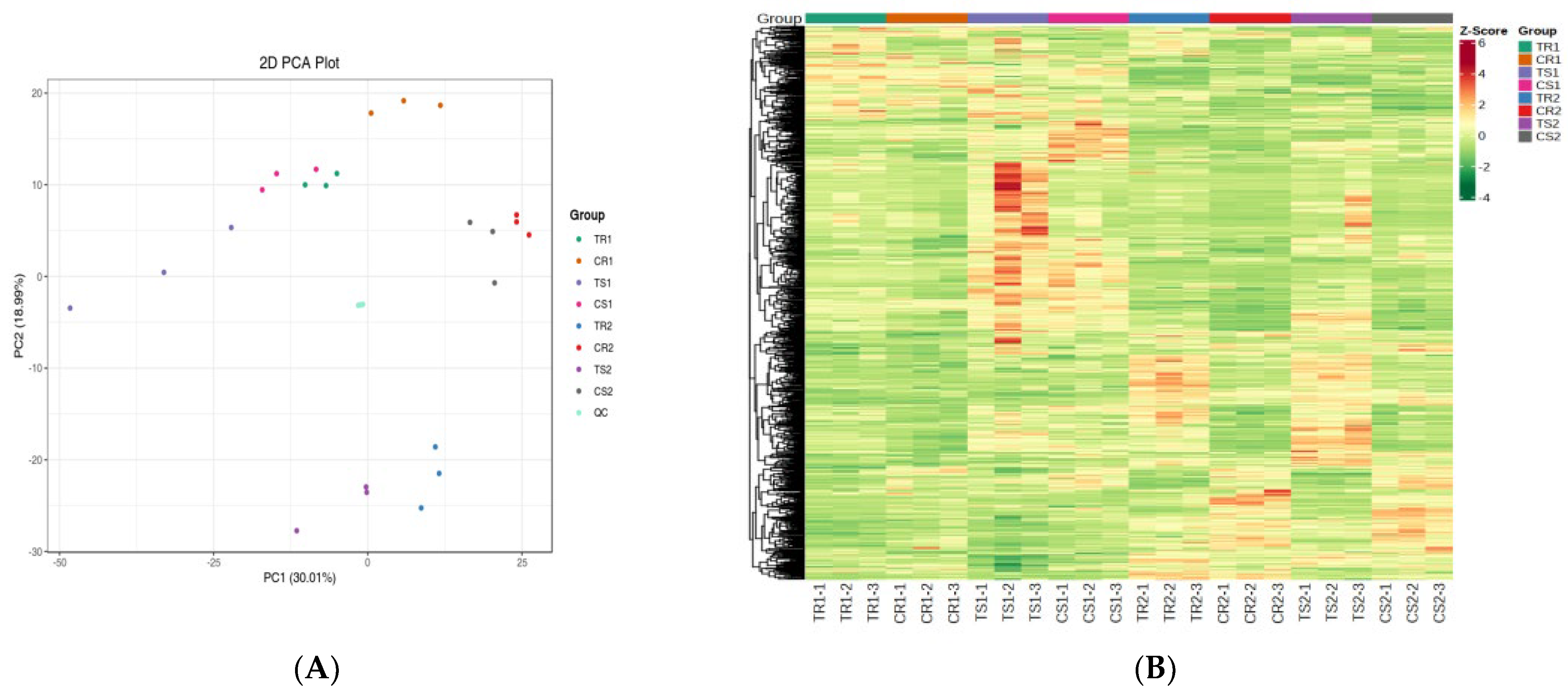
3.3. Metabolic Profiling of Quinoa Seedlings under High RH Stress
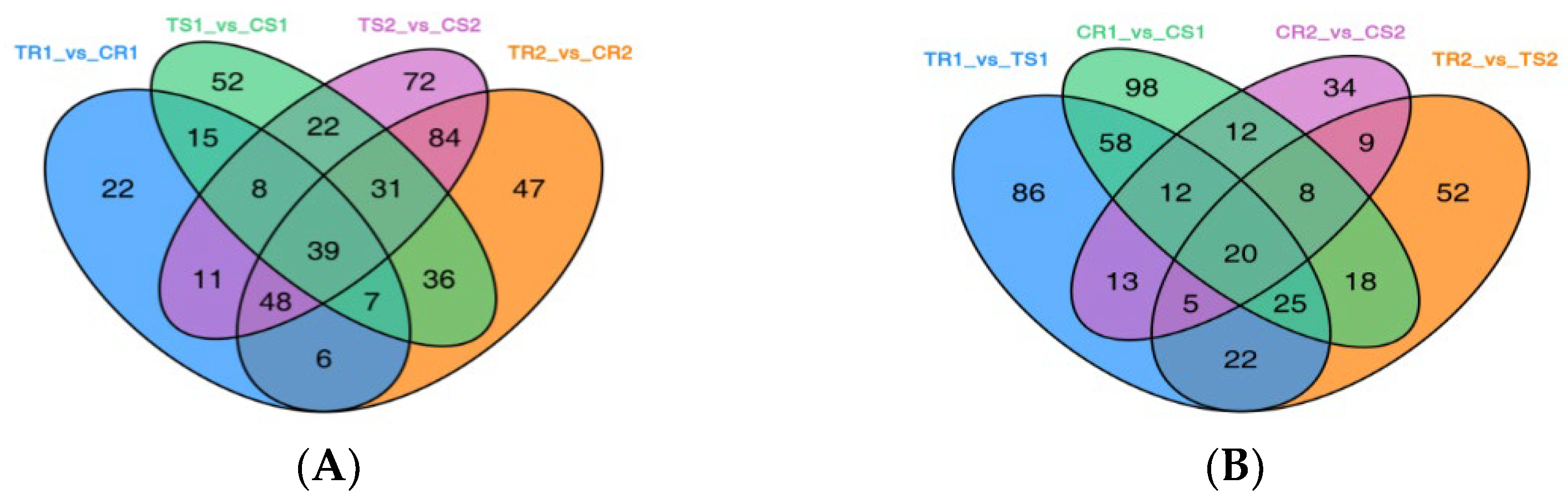
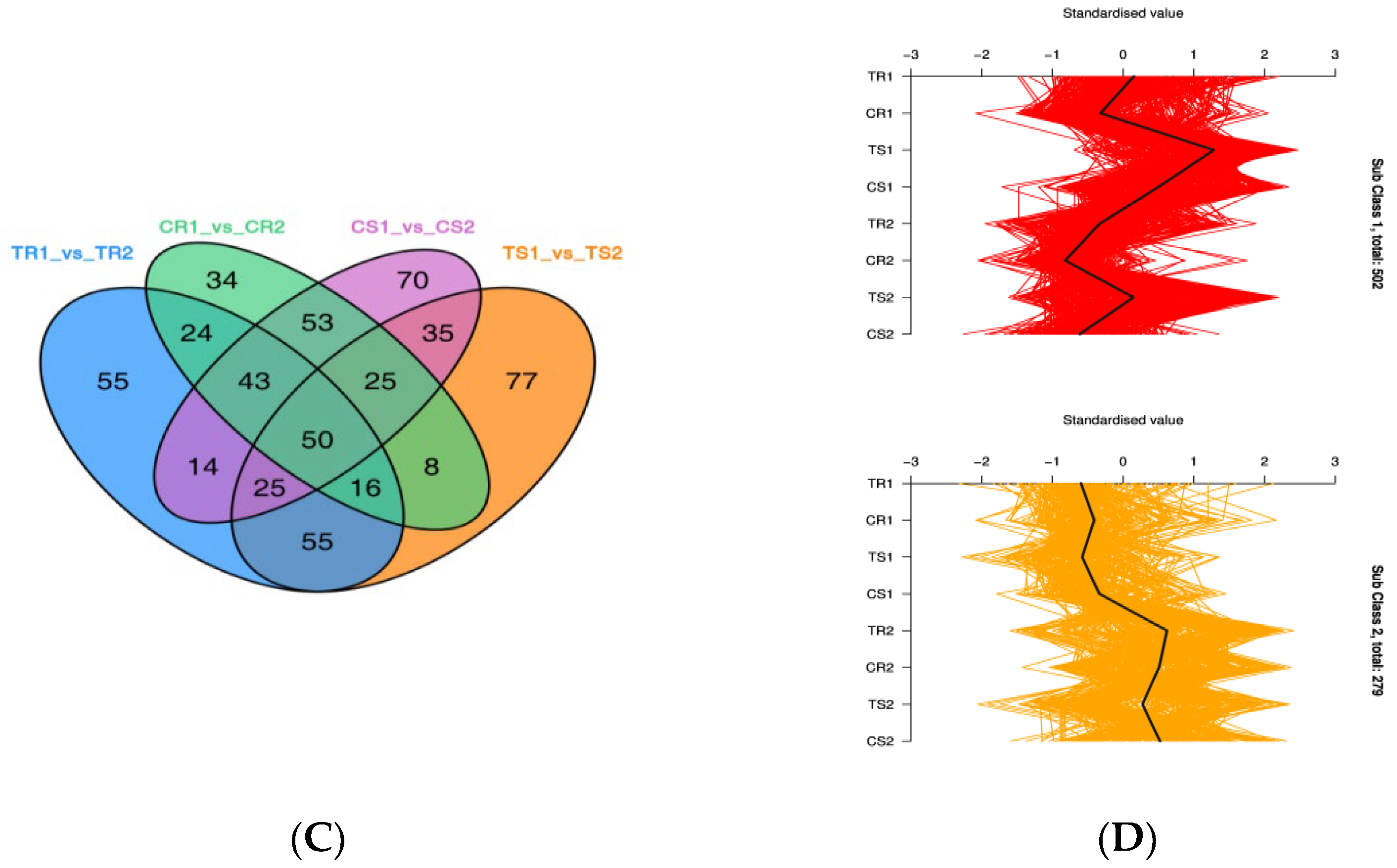
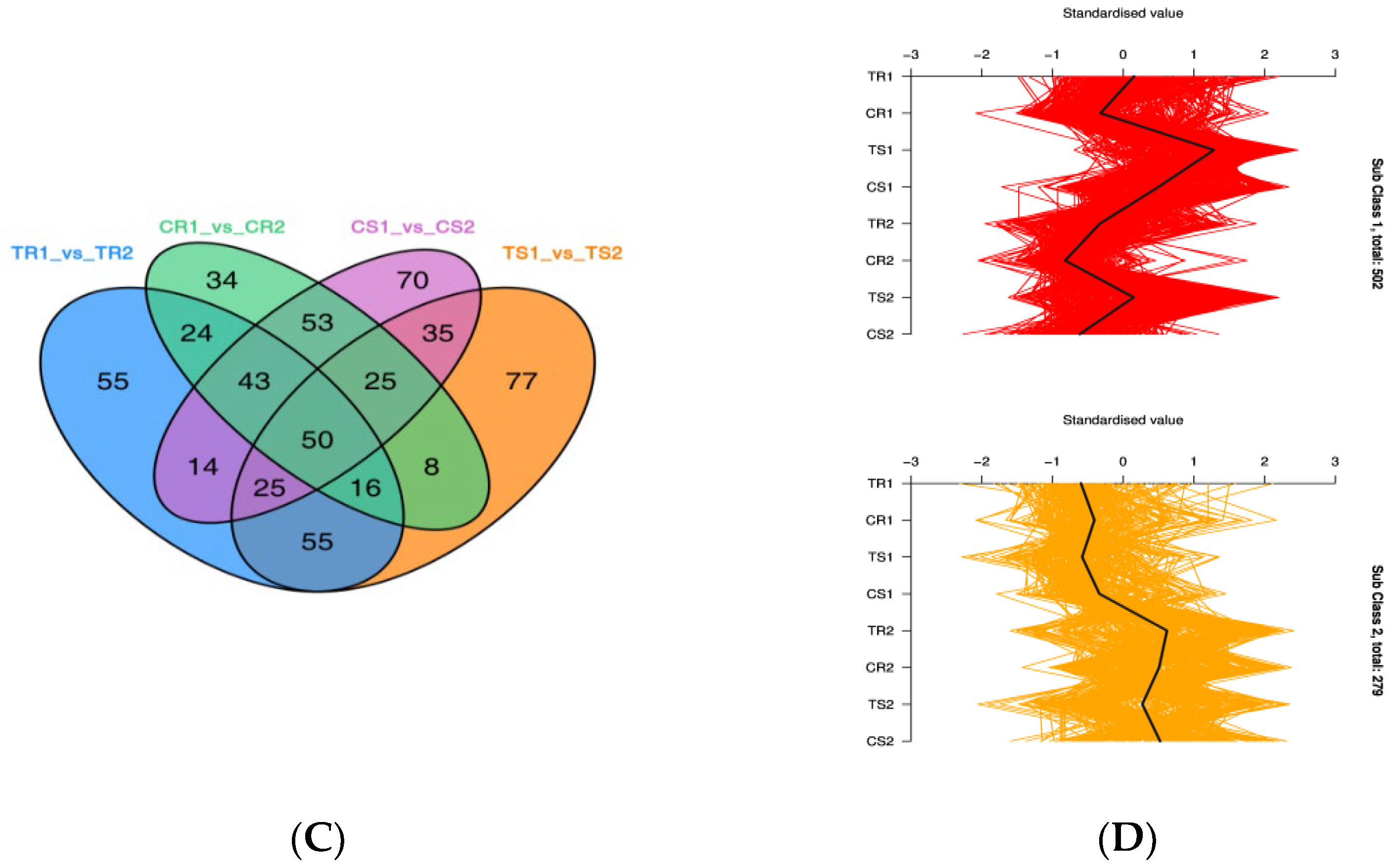
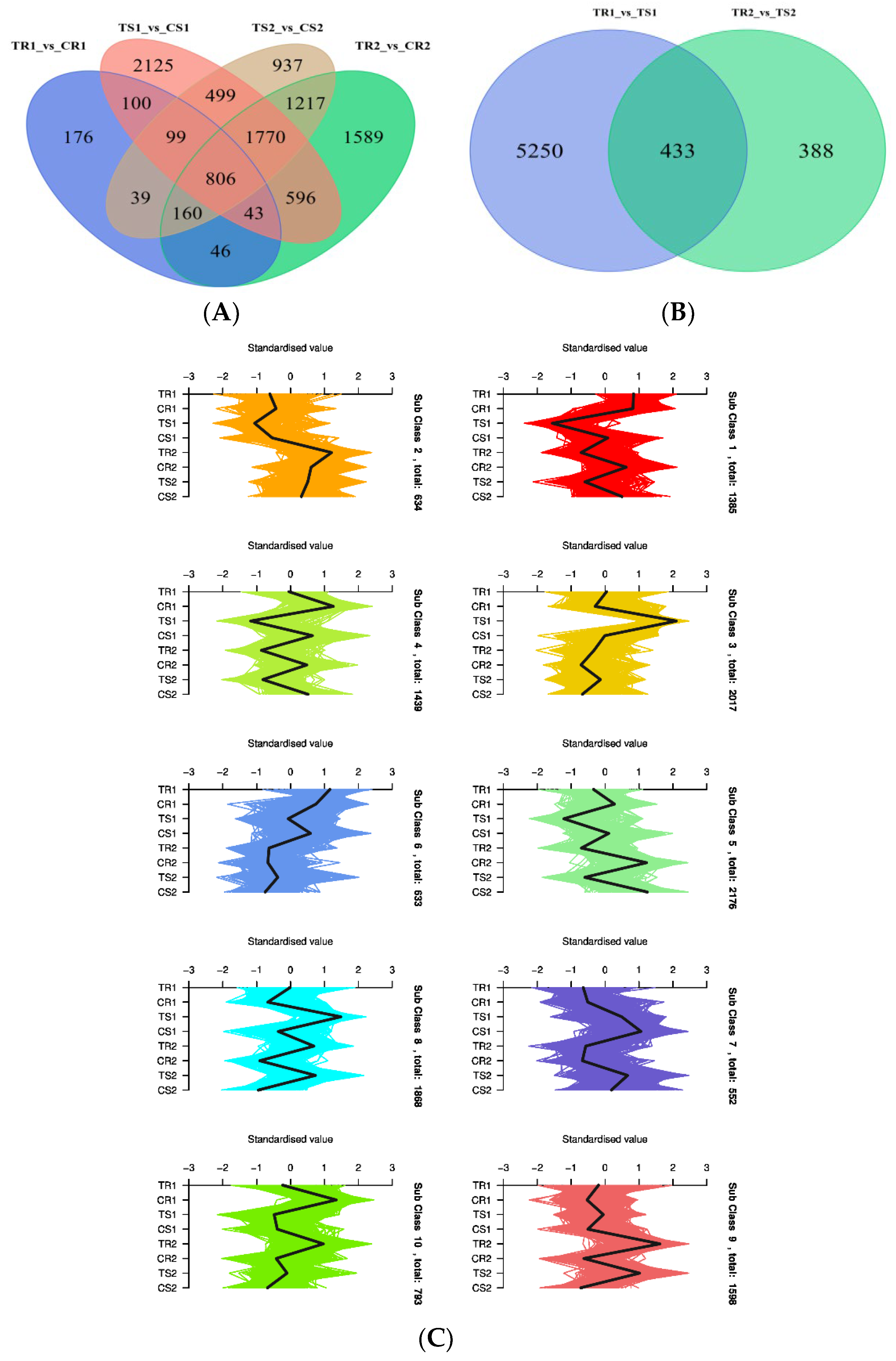
3.4. Significant DAMs and Enrichment Analysis
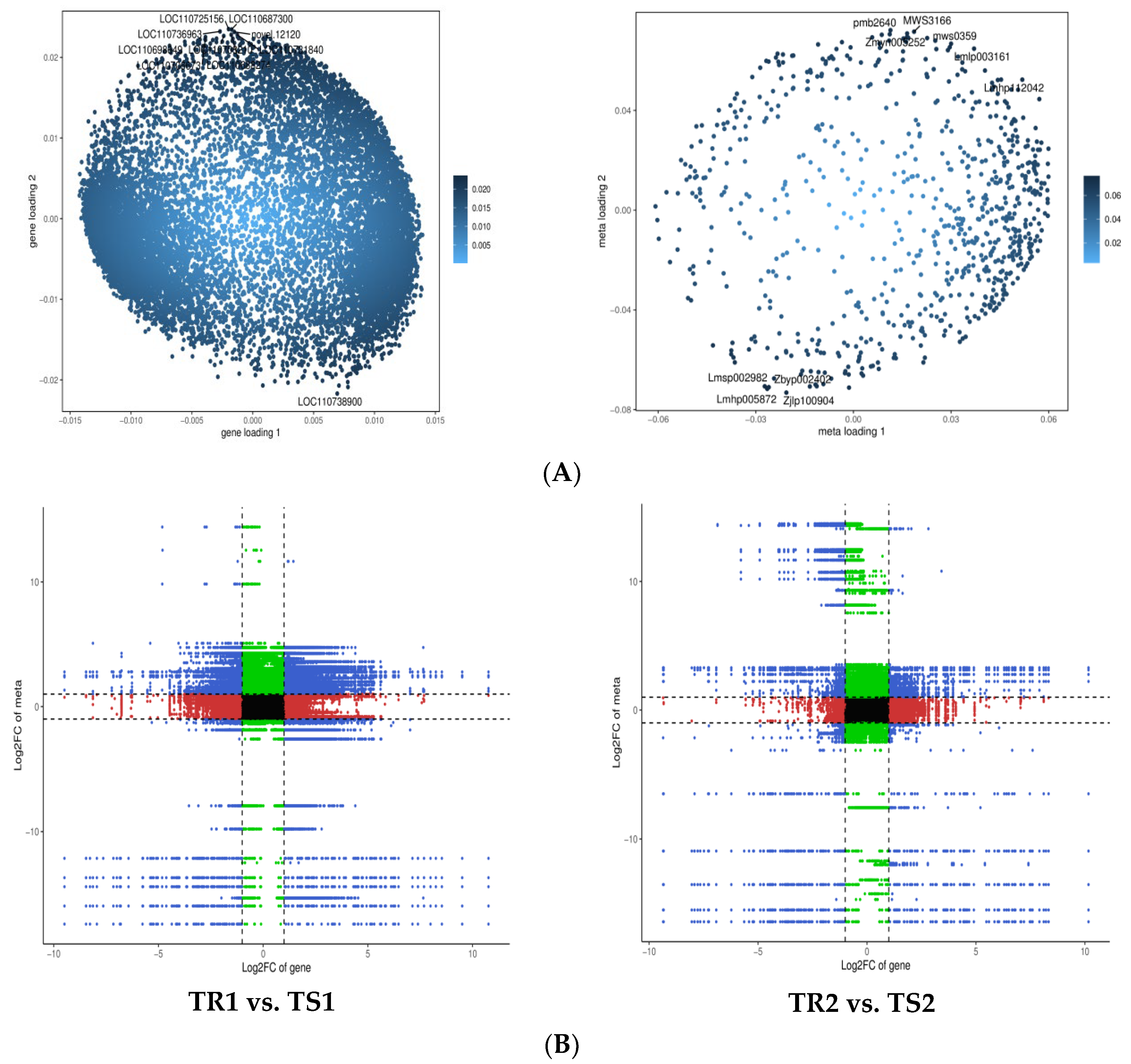
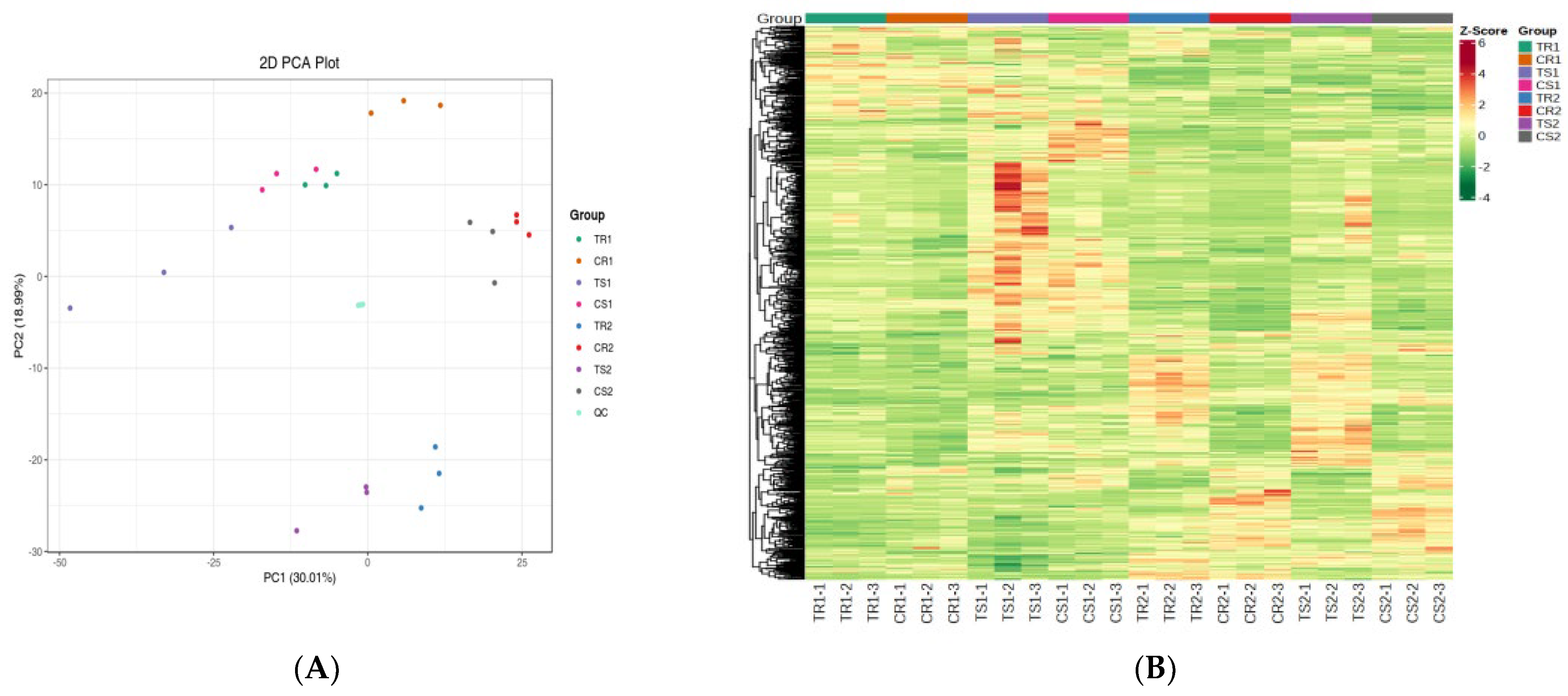
3.5. Transcriptional Profiling of Quinoa Seedlings under High RH Stress
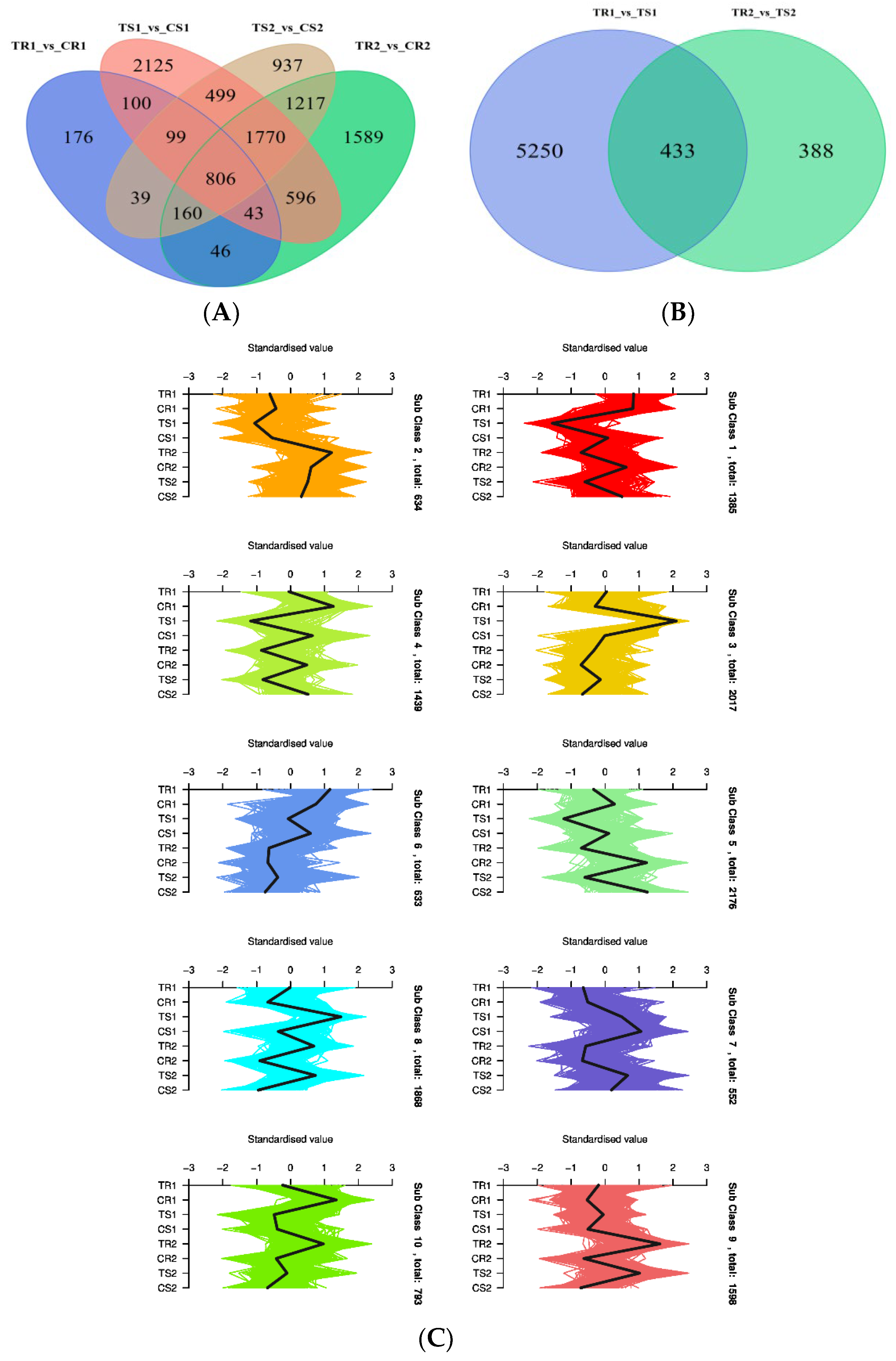
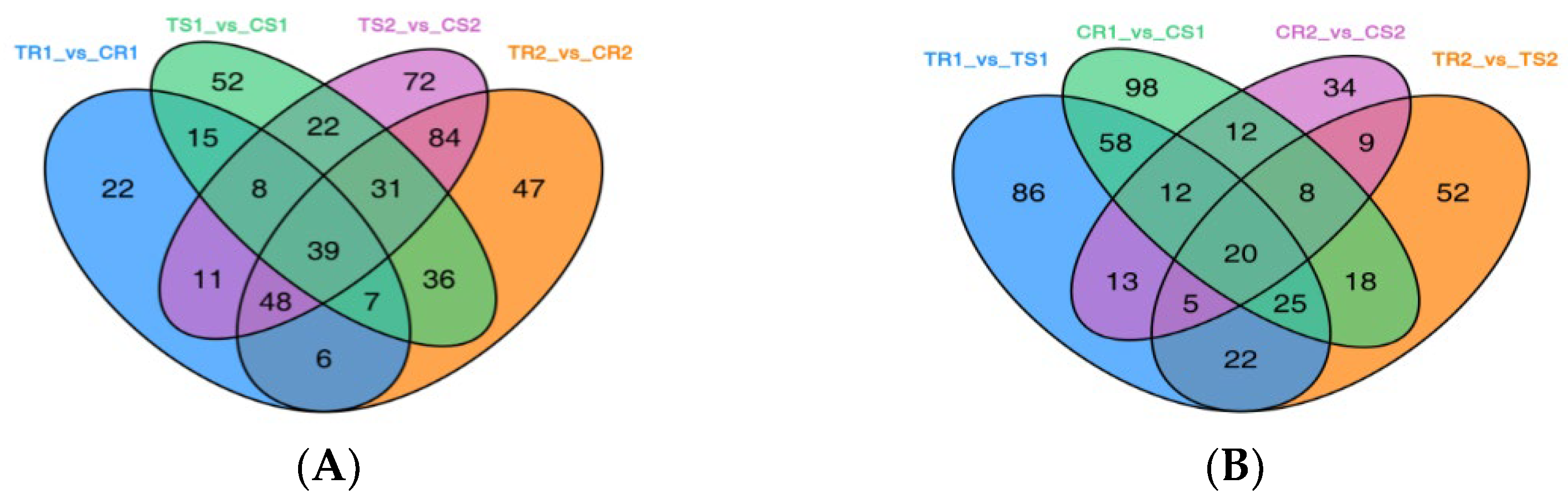
3.6. Identification of DEGs and GO Term and KEGG Pathway Enrichment Analysis
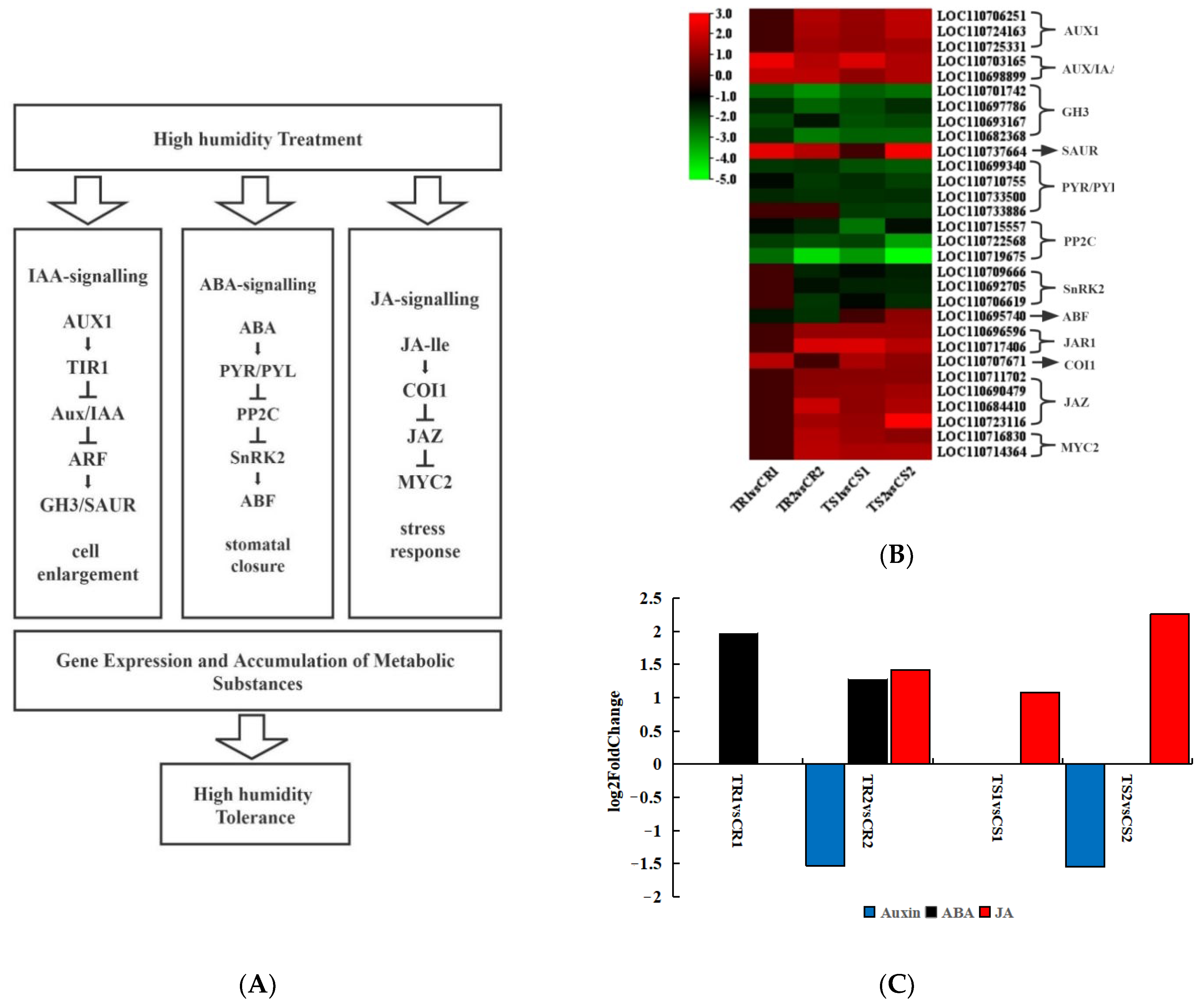
3.7. Changes in Gene Expression and Metabolite Accumulation in Quinoa Seedlings under High RH Stress
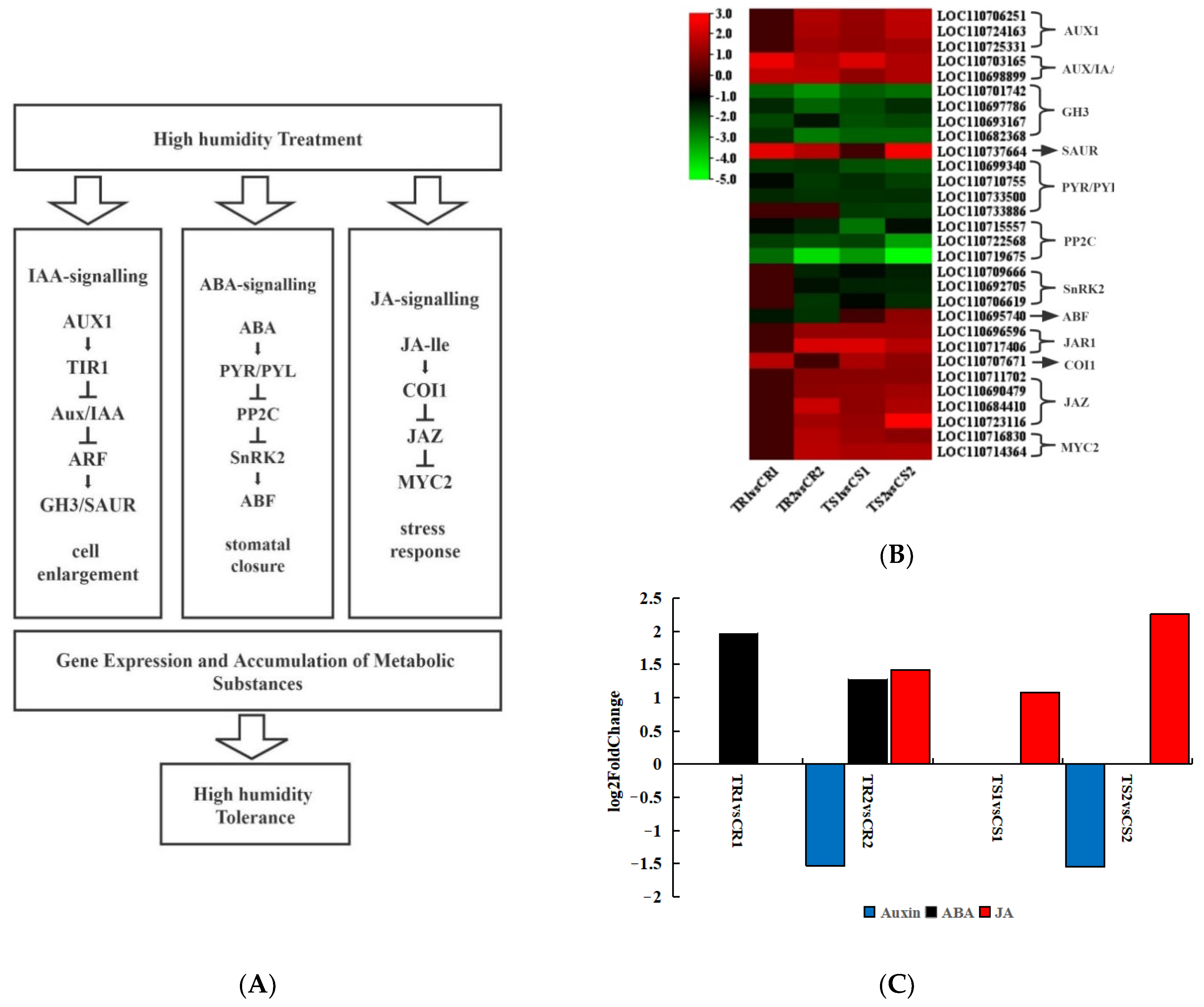
3.8. Response of Plant Hormone Signal Transduction Pathways to High RH Stress
3.9. Differentially Expressed Transcription Factors under High RH Stress
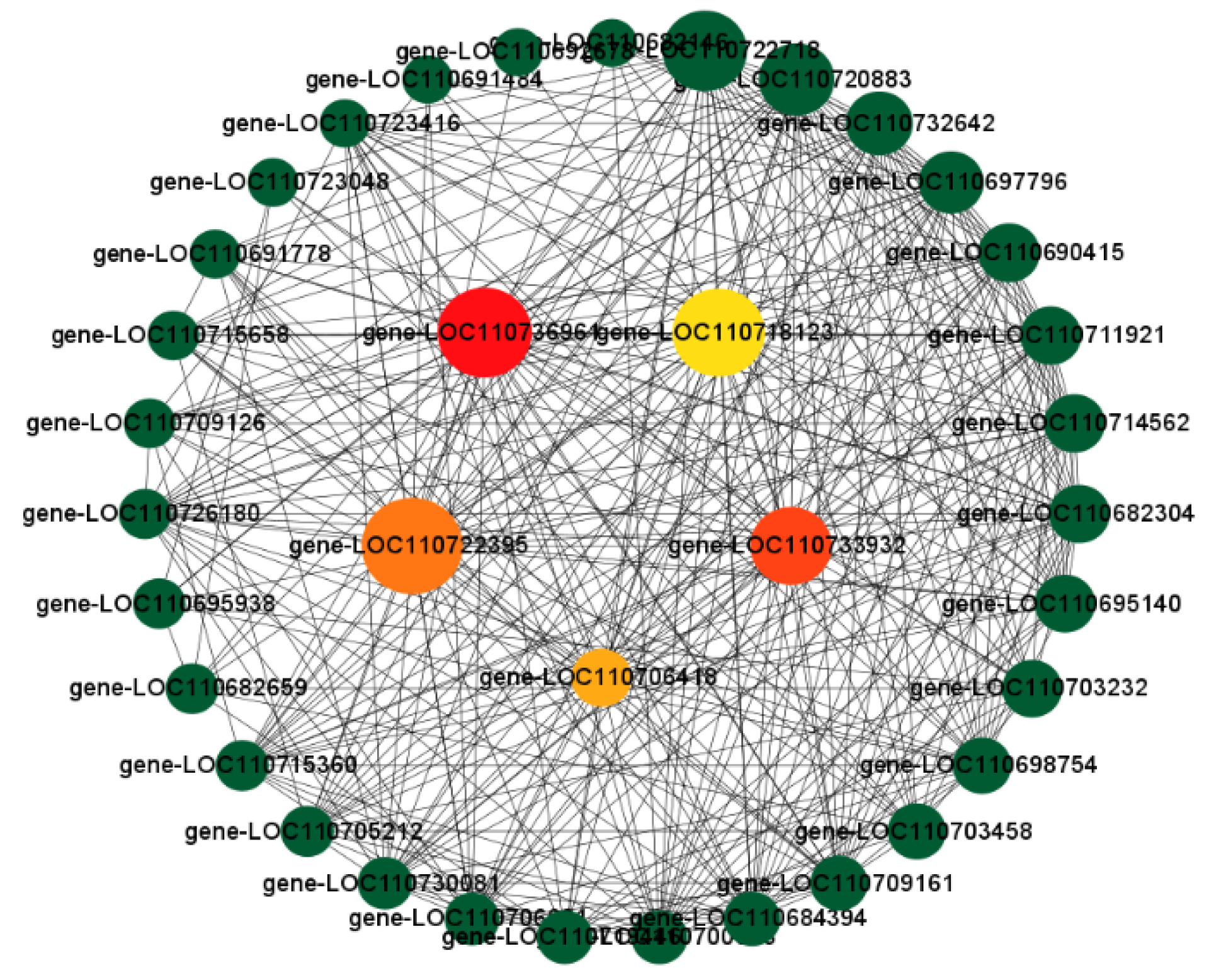
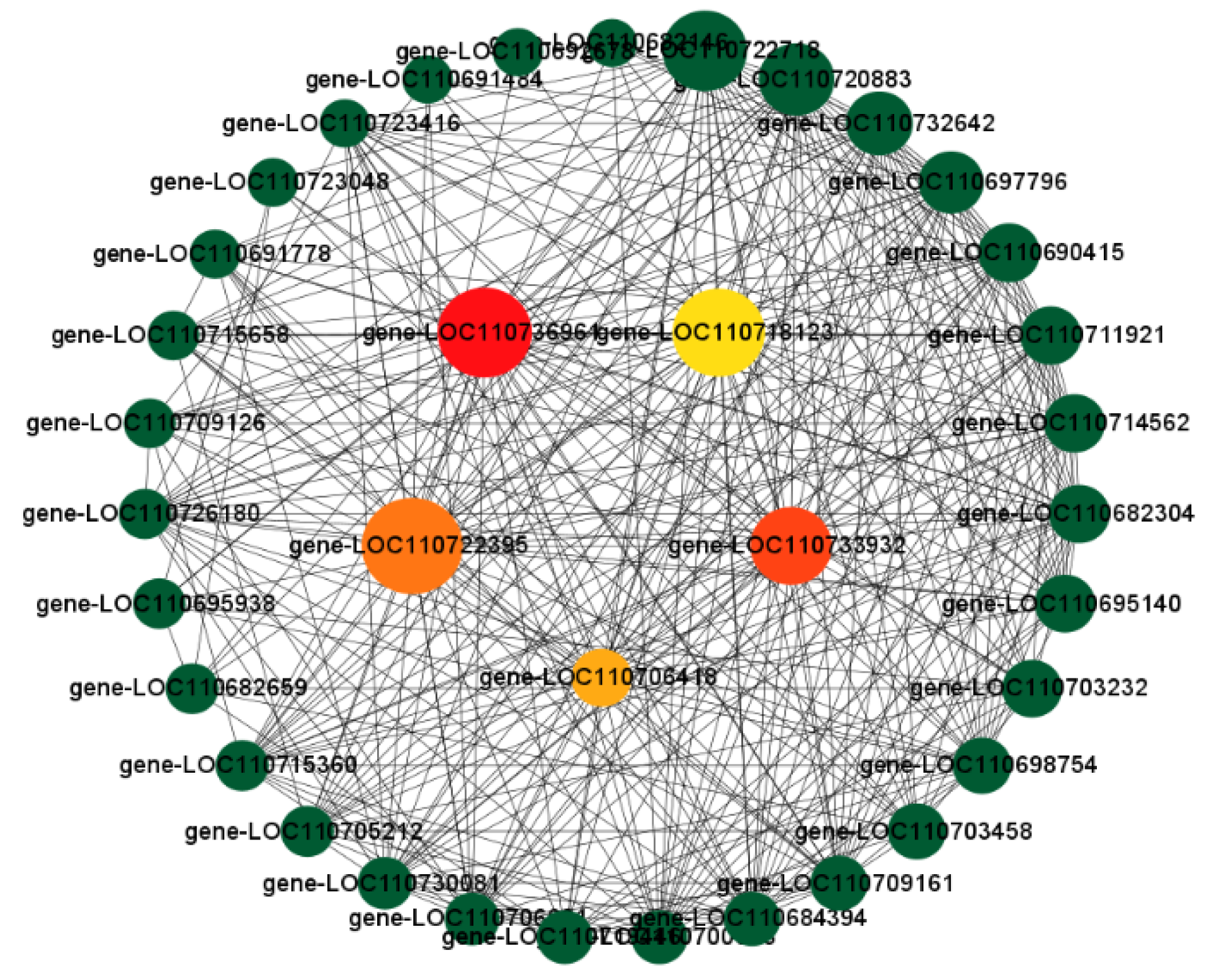
3.10. Combined WGCNA and PPI Analysis to Screen Long-Acting Response Hub Genes in the Highly Resistant Variety
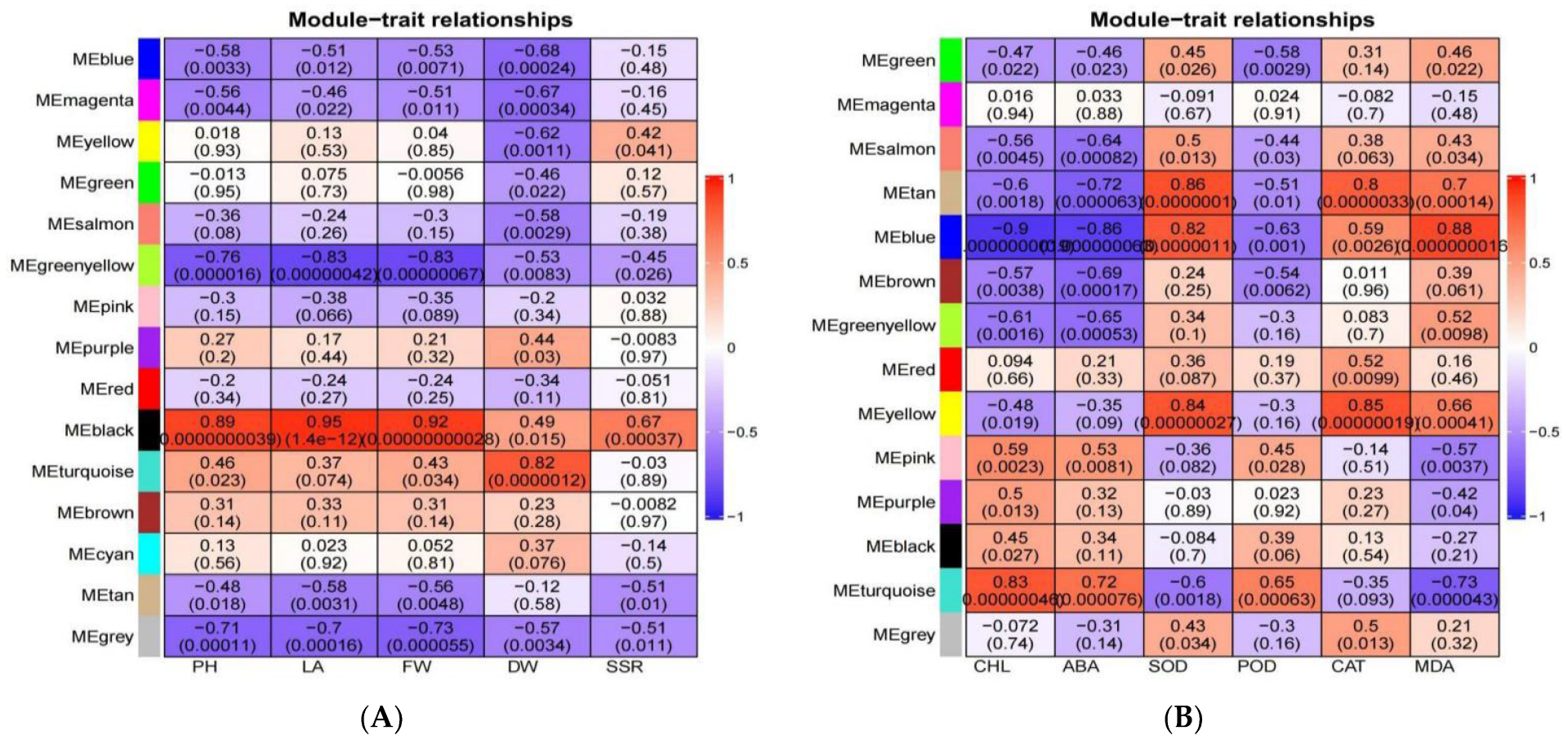
3.11. Screening for Morphologically and Physiologically Important Candidate Genes Using WGCNA
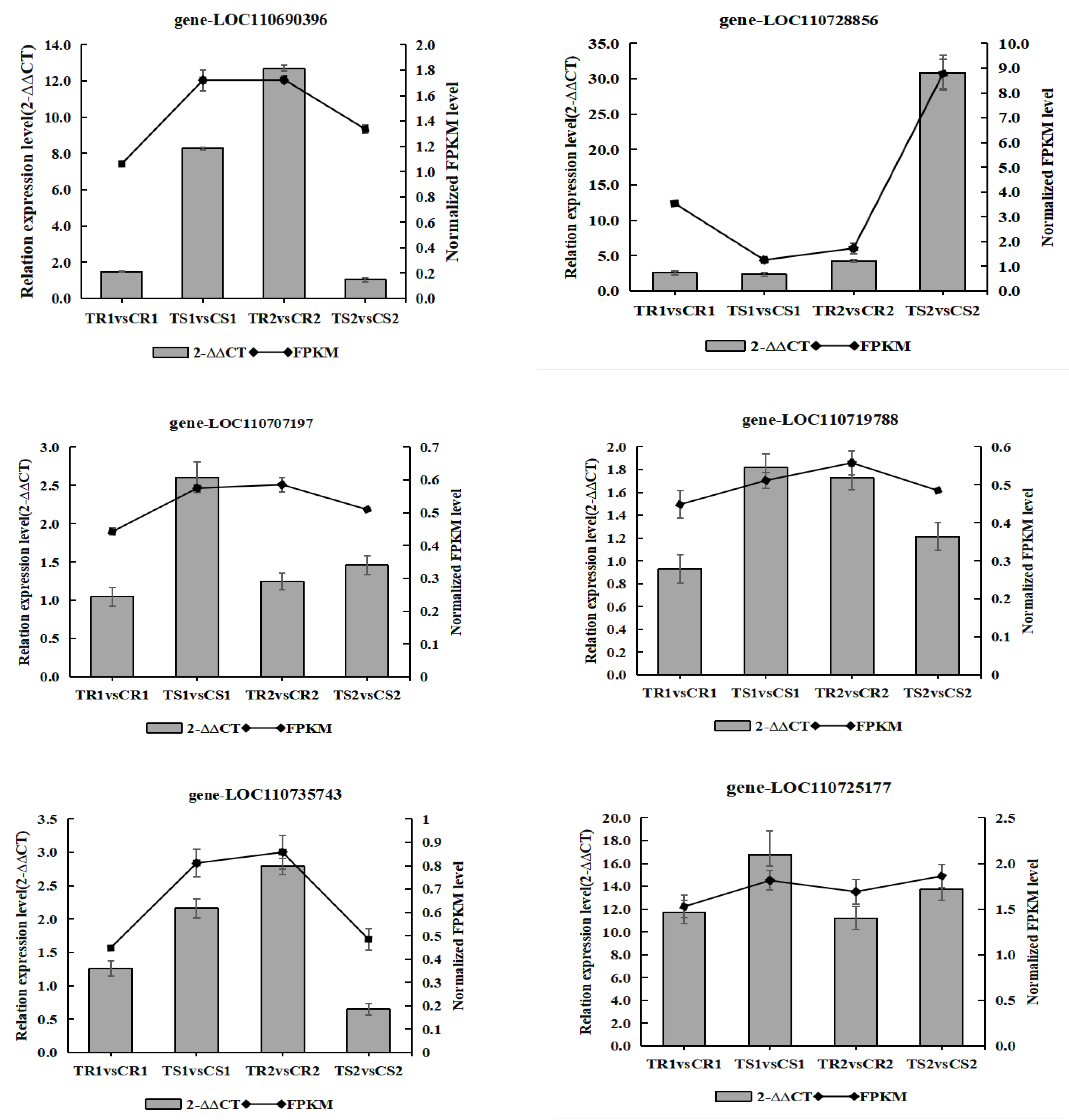
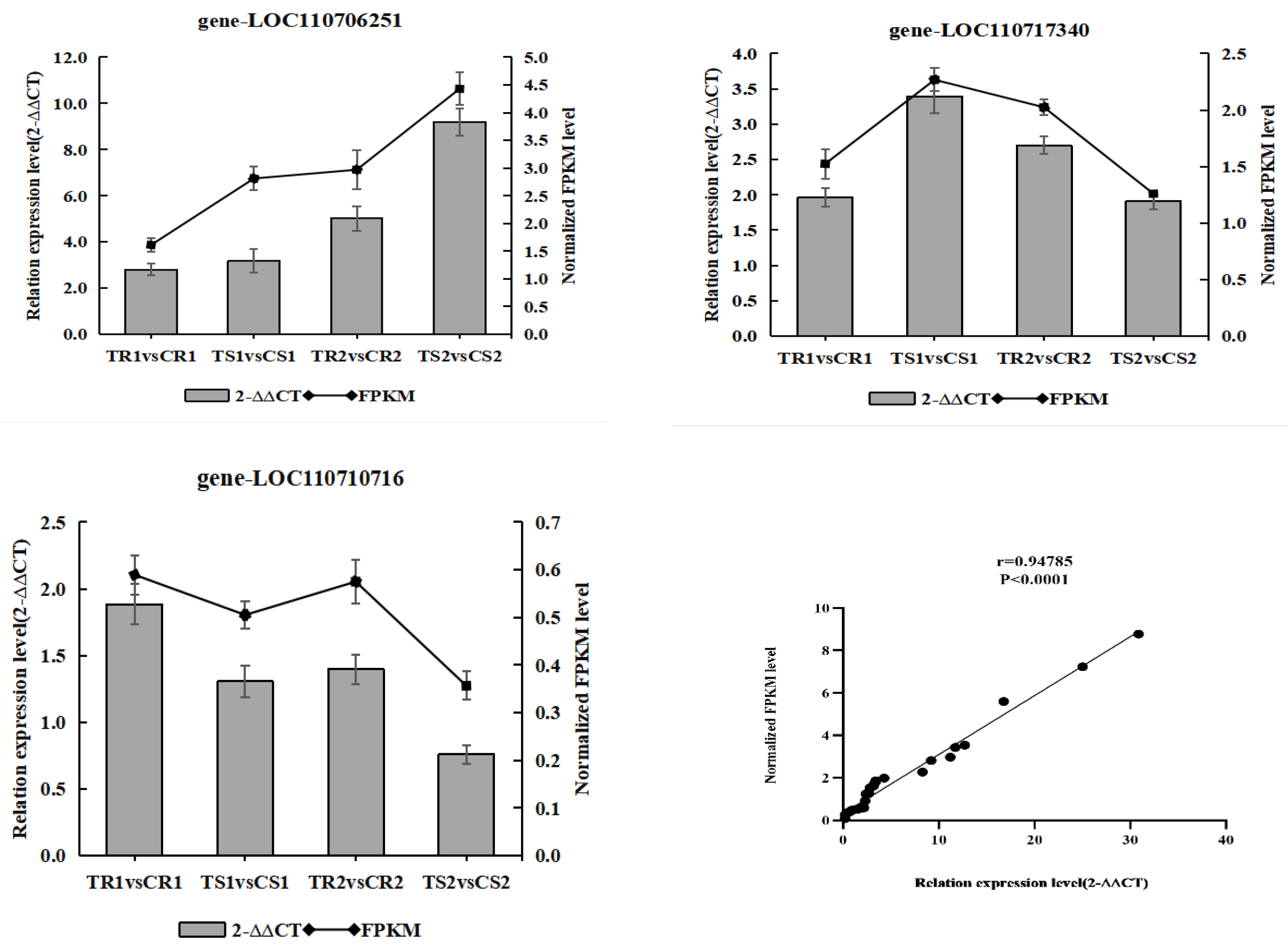
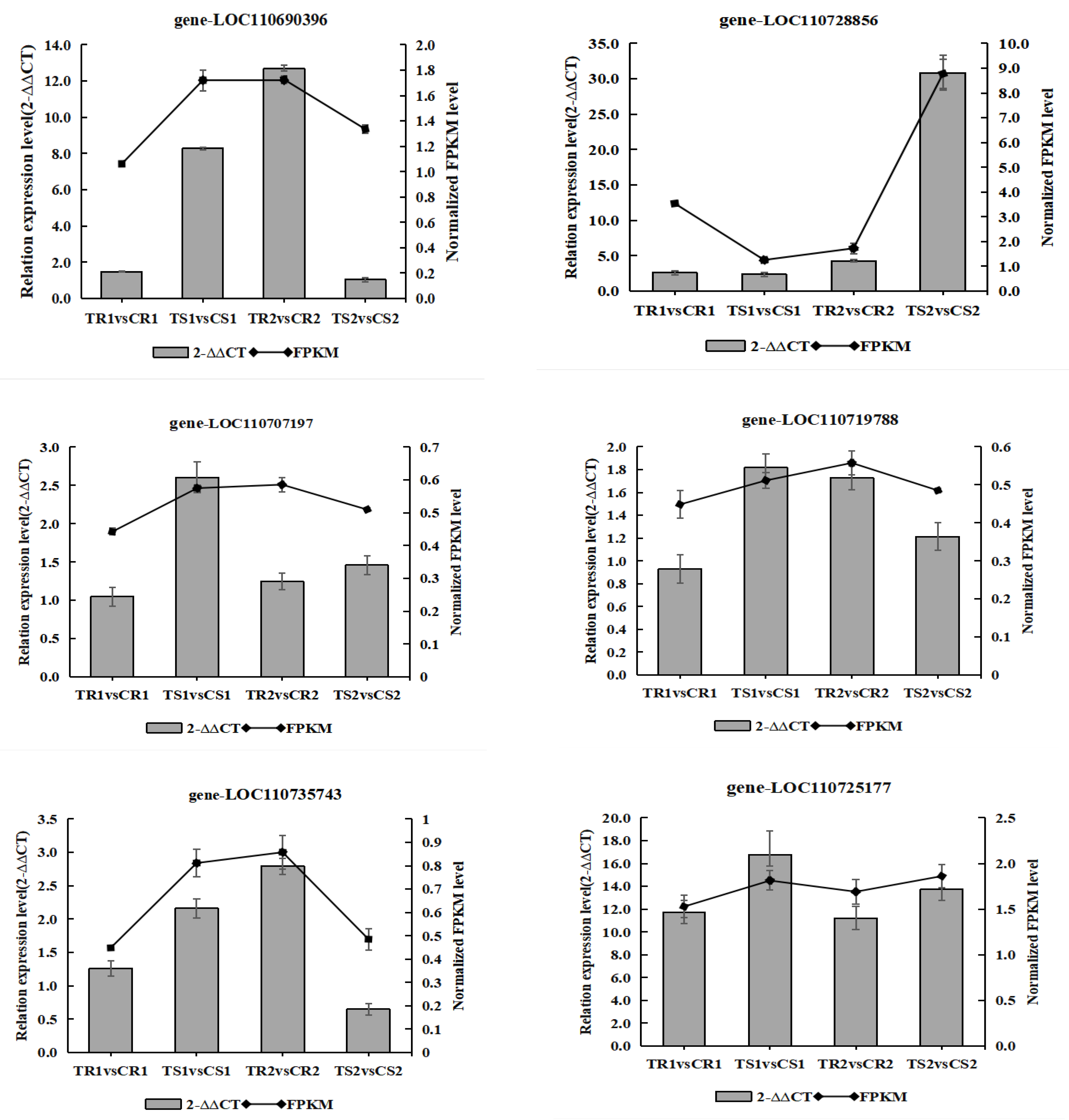
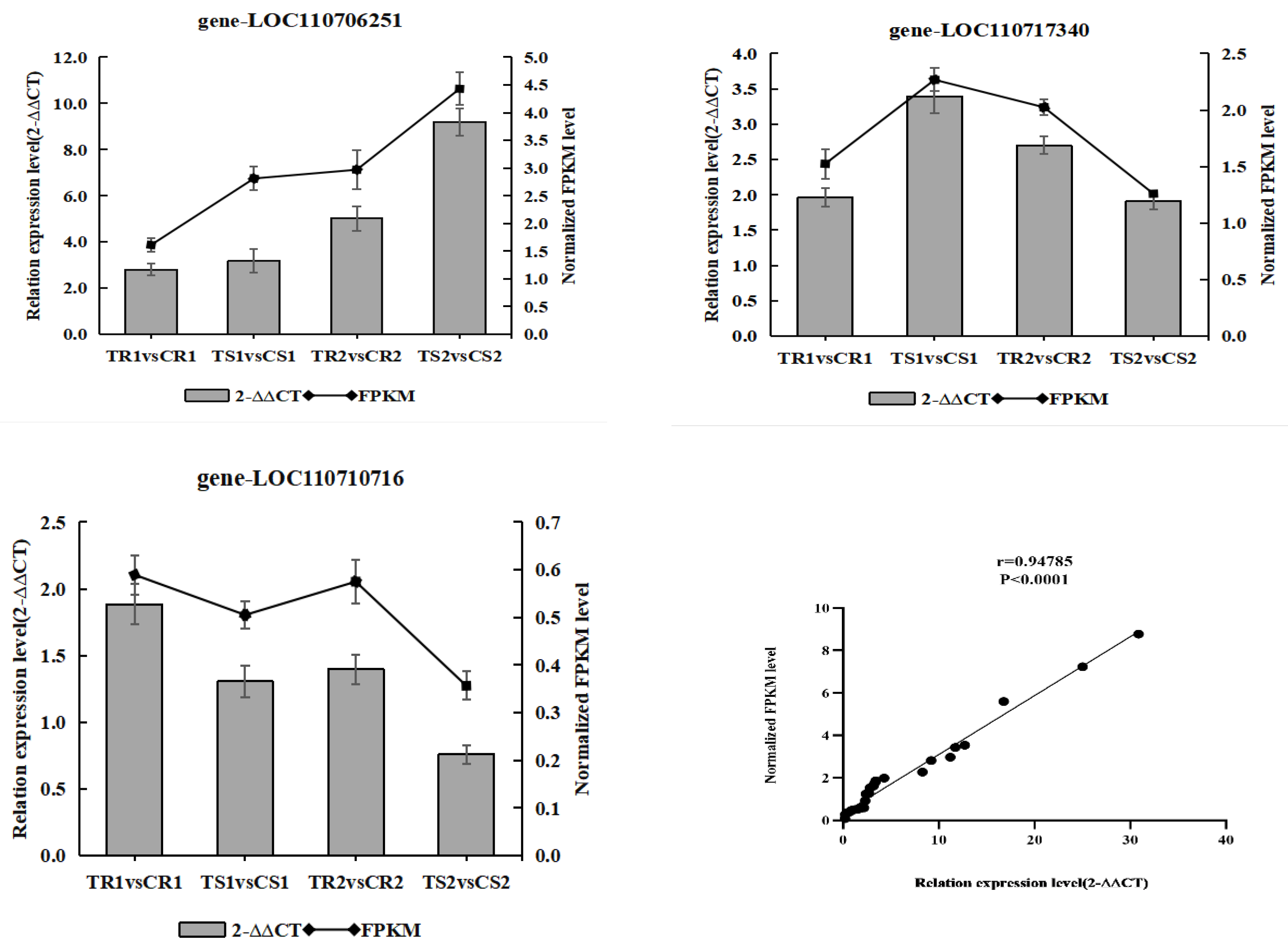
3.12. Validation of the Transcriptome Data by RT-qPCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zurita, S.A.; Fuentes, F.; Zamora, P.; Sven, J.; Andrés, R.S. Breeding quinoa (Chenopodium quinoa Willd.): Potential and perspectives. Mol. Breed. 2014, 34, 13–30. [Google Scholar] [CrossRef]
- Food and Agriculture Organization; Centre de Coopération Internationale en Recherche Agronomique pour le Développement. State of the Art Report of Quinoa in the World in 2013; Bazile, D., Bertero, D., Nieto, C., Eds.; FAO&CIRAD: Roma, Italy, 2015. [Google Scholar]
- Wrisht, K.H.; Pike, O.A.; Fairbanks, D.J. Composition of Atriplex hortensis, sweet and bitter Chenopodium quinoa seeds. J. Food Sci. 2002, 67, 1383–1385. [Google Scholar]
- James, L. Chapter 1 Quinoa (Chenopodium quinoa Willd.). Adv. Food Nut. Res. 2009, 58, 1–31. [Google Scholar]
- Vega, G.A.; Miranda, M.; Vergara, J. Nutrition fact sand functional potential of quinoa (Chenopodium quinoa Willd.), an ancient Andeangrain: A review. J. Sci. Food Agric. 2010, 90, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Amin, B.; Atif, M.J.; Wang, X.; Meng, H.; Ghani, M.I.; Ali, M.; Ding, Y.; Li, X.; Cheng, Z. Effect of low temperature and high humidity stress on physiology of cucumber at different leaf stages. Plant Biol. Stuttg. 2021, 23, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Aboal, J.R.; Jimenez, M.S.; Morales, D.; Hernandez, J.M. Rainfall interception in laurel forest in the Canary Islands. Agric. For. Meteorol. 1999, 97, 73–86. [Google Scholar] [CrossRef]
- Mortensen, L.M.; Gislerd, H.R. Influence of air humidity and lighting period on growth, vase life and water relations of 14 rose cultivars. Sci. Hortic. 1999, 82, 289–298. [Google Scholar] [CrossRef]
- Torre, S.; Fjeld, T.; Gislerød, H.R. Leaf anatomy and stomatal morphology of greenhouse roses grown at moderate or high air humidity. J. Am. Soc. Hor. Sci. 2003, 128, 598–602. [Google Scholar] [CrossRef]
- Fanourakis, D.; Carvalho, S.M.P.; Almeida, D.P.F.; Doorn, W.G.; Heuvelink, E. Postharvest water relations in cut rose cultivars with contrasting sensitivity to high relative air humidity during growth. Post. Biol. Technol. 2012, 64, 64–73. [Google Scholar] [CrossRef]
- Giday, H.; Kjaer, K.H.; Fanourakis, D.; Ottosen, C.O. Smaller stomata require less severe leaf drying to close: A case study in Rosa hydrida. J. Plant Physiol. 2013, 170, 1309–1316. [Google Scholar] [CrossRef]
- Carvalho, D.R.A.; Torre, S.; Kraniotis, D.; Almeida, D.P.F.; Heuvelink, E.; Carvalho, S.M.P. Elevated air movement enhances stomatal sensitivity to abscisicacid in leaves developed at high relative air humidity. Front. Plant Sci. 2015, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Sellin, A.; Rosenvald, K.; Õunapuu-Pikas, E.; Tullus, A.; Ostonen, I.; Lõhmus, K. Elevated air humidity affects hydraulic traits and tree size but not biomass allocation in young silver birches (Betula pendula). Front. Plant Sci. 2015, 6, 860. [Google Scholar] [CrossRef] [PubMed]
- Kupper, P.; Rohula, G.; Inno, L.; Ostonen, I.; Sellin, A.; Sõber, A. Impact of high daytime air humidity on nutrient uptake and night-time water flux in silver birch, a boreal forest tree species. Reg. Environ. Change 2017, 17, 2149–2157. [Google Scholar] [CrossRef]
- Körner, O.; Aaslyng, J.M.; Andreassen, A.U.; Holst, N. Microclimate prediction for dynamic greenhouse climate control. Hort. Sci. 2007, 42, 272–279. [Google Scholar] [CrossRef]
- Giday, H.; Kjaer, K.H.; Ottosen, C.O.; Fanourakis, D. Cultivar differences in plant transpiration rate at high relative air humidity are not related to genotypic variation in stomatal responsiveness. Acta Hort. Sin. 2015, 1064, 99–106. [Google Scholar] [CrossRef]
- Mortensen, L.M.; Fjeld, T. Effects of air humidity, lighting period and lamp type on growth and vase life in roses. Sci. Hortic. 1998, 73, 229–237. [Google Scholar] [CrossRef]
- Torre, S.; Fjeld, T.; Gislerød, H.R. Effects of air humidity and K/Ca ratio in the nutrient supply on growth and postharvest characteristics of cut roses. Sci. Hortic. 2001, 90, 291–304. [Google Scholar] [CrossRef]
- Mortensen, L.M.; Gislerød, H.R. Effect of air humidity variation on powdery mildew and keeping quality of cut roses. Sci. Hortic. 2005, 104, 49–55. [Google Scholar] [CrossRef]
- Torre, S.; Fjeld, T. Water loss and postharvest characteristics of cut roses grown at high or moderate relative humidity. Sci. Hortic. 2001, 89, 217–226. [Google Scholar] [CrossRef]
- Fanourakis, D.; Giday, H.; Hyldgaard, B.; Bouranis, D.; Körner, O.; Ottosen, C.O. Low air humidity during growth promotes stomatal closure ability in roses. Eur. J. Hortic. Sci. 2019, 84, 245–252. [Google Scholar] [CrossRef]
- Gislerød, H.R.; Nelson, P.V. The interaction of the relative air humidity and carbon dioxide enrichment on the growth of Chrysanthemum × morifolium Ramat. Sci. Hortic. 1989, 38, 305–313. [Google Scholar] [CrossRef]
- Mortensen, L.M. Effects of air humidity on growth, flowering, keeping quality and water relations of four short-day greenhouse species. Sci. Hortic. 2000, 86, 299–310. [Google Scholar] [CrossRef]
- Islam, N.; Torre, S.; Wold, A.B.; Gislerod, H.R. Effects of growing conditions on the postharvest quality of herbs. Acta Hortic. 2010, 877, 187–194. [Google Scholar] [CrossRef]
- Giday, H.; Fanourakis, D.; Kjaer, K.H.; Fomsgaard, I.S.; Ottosen, C.O. Threshold response of stomatal closing ability to leaf abscisic acid concentration during growth. J. Exp. Bot. 2014, 65, 4361–4370. [Google Scholar] [CrossRef] [PubMed]
- Sellin, A.; Tullus, A.; Niglas, A.; Õunapuu, E.; Karusion, A.; Lõhmus, K. Humidity driven changes in growth rate, photosynthetic capacity, hydraulic properties and other functional traits in silver birch (Betula pendula). Ecol. Res. 2013, 28, 523–535. [Google Scholar] [CrossRef]
- Sellin, A.; Taneda, H.; Alber, M. Leaf structural and hydraulic adjustment with respect to air humidity and canopy position in silver birch (Betula pendula). J. Plant Res. 2019, 132, 369–381. [Google Scholar] [CrossRef]
- Fanourakis, D.; Carvalho, S.M.P.; Almeida, D.P.F.; Heuvelink, E. Avoiding high relative air humidity during critical stages of leaf ontogeny is decisive for stomatal functioning. Physiol. Plant. 2011, 142, 274–286. [Google Scholar] [CrossRef]
- Carins Murphy, M.R.; Jordan, G.J.; Brodribb, T.J. Acclimation to humidity modifies the link between leaf size and the density of veins and stomata. Plant Cell Environ. 2014, 37, 124–131. [Google Scholar] [CrossRef]
- Xiong, D.; Nadal, M. Linking water relations and hydraulics with photosynthesis. Plant J. 2020, 101, 800–815. [Google Scholar] [CrossRef]
- Rezaei Nejad, A.; Meeteren, U. Stomatal response characteristics of Tradescantia virginiana grown at high relative air humidity. Physiol. Plant. 2005, 125, 324–332. [Google Scholar] [CrossRef]
- Fanourakis, D.; Heuvelink, E.; Carvalho, S.M.P. A comprehensive analysis of the physiological and anatomical components involved in higher water loss rates after leaf development at high humidity. J. Plant Physiol. 2013, 170, 890–898. [Google Scholar] [CrossRef]
- Laur, J.; Hacke, U.G. Transpirational demand affects aquaporin expression in poplar roots. J. Exp. Bot. 2013, 64, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Aliniaeifard, S.; Malcolm Matamoros, P.; Meeteren, U. Stomatal malfunctioning under low VPD conditions: Induced by alterations in stomatal morphology and leaf anatomy or in the ABA signaling? Physiol. Plant. 2014, 152, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.R.A.; Fanourakis, D.; Correia, M.J.; Monteiro, J.A.; Araújo-Alves, J.P.L.; Vasconcelos, M.W.; Almeida, D.P.F.; Heuvelink, E.; Carvalho, S.M.P. Root-to-shoot ABA signaling does not contribute to genotypic variation in stomatal functioning induced by high relative air humidity. Environ. Exp. Bot. 2016, 123, 13–21. [Google Scholar] [CrossRef]
- Fanourakis, D.; Aliniaeifard, S.; Sellin, A.; Giday, H.; Körner, O.; Rezaei Nejad, A.; Delis, C.; Bouranis, D.; Koubouris, G.; Kambourakis, E.; et al. Stomatal behavior following mid or long-term exposure to high relative air humidity: A review. Plant Physiol. Bioch. 2020, 153, 92–105. [Google Scholar] [CrossRef]
- Fanourakis, D.; Bouranis, D.; Giday, H.; Carvalho, D.R.A.; Rezaei Nejad, A.; Ottosen, C.O. Improving stomatal functioning at elevated growth air humidity: A review. J. Plant Physiol. 2016, 207, 51–60. [Google Scholar] [CrossRef]
- Rezaei, N.A.; Meeteren, U. The role of abscisic acid in disturbed stomatal response characteristics of Tradescantia virginiana during growth at high relative air humidity. J. Exp. Bot. 2007, 58, 627–636. [Google Scholar]
- Arve, L.E.; Terfa, M.T.; Gislerod, H.R.; Olsen, J.E.; Torre, S. High relative air humidity and continuous light reduce stomata functionality by affecting the ABA regulation in rose leaves. Plant Cell Environ. 2013, 36, 382–392. [Google Scholar] [CrossRef]
- Arve, L.E.; Torre, S. Ethylene is involved in high air humidity promoted stomatal opening of tomato (Lycopersicon esculentum) leaves. Funct. Plant Biol. 2015, 42, 376–386. [Google Scholar] [CrossRef]
- Bradbury, L.M.; Niehaus, T.D.; Hanson, A.D. Comparative genomics approaches to understanding and manipulating plant metabolism. Curr. Opin. Biotechnol. 2013, 24, 278–284. [Google Scholar] [CrossRef]
- Parkinson, E.I.; Tryon, J.H.; Goering, A.W.; Ju, K.S.; McClure, R.A.; Kemball, J.D.; Zhukovsky, S.; Labeda, D.P.; Homson, R.J.T.; Kelleher, N.L.; et al. Discovery of the tyrobetaine natural products and their biosynthetic gene cluster via metabologenomics. ACS Chem. Biol. 2018, 13, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ren, C.; Dong, S.; Chen, C.; Xian, B.; Wu, Q.; Wang, J.; Pei, J.; Chen, J. Integrated metabolomics and transcriptome analysis of flavonoid biosynthesis in safflower (Carthamus tinctorius L.) with different colors. Front. Plant Sci. 2021, 12, 712–738. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Amin, N.; Ahmad, I.; Ara, G. Impact assessment of leaf pigments in selected landscape plants exposed to roadside dust. Environ. Sci. Pollut. Res. 2018, 25, 23055–23073. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.J. A novel integrated method for large-scale detection, identifification, and quantifification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: A fast spliced aligner with low memory requirements. Nat. Met. 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatic 2015, 30, 923–930. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Chen, C.H.; Chen, Y.; Zhang, H.R.; Thomas, M.H. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rodriguez, P.; Riano-Pachon, D.M.; Correa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Timothy, L.; Bailey, J.J.; Charles, E.; Grant, W.S.N. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar]
- Kohl, S.; Wiese, B. Warscheid Cytoscape: Software for visualization and analysis of biological networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Fanourakis, D.; Hyldgaard, B.; Giday, H.; Bouranis, D.; Rner, O.K.; Nielsen, K.L.; Ottosen, C.O. Differential effects of elevated air humidity on stomatal closing ability of Kalanchoë blossfeldiana between the C3 and CAM states. Environ. Exp. Bot. 2017, 143, 115–124. [Google Scholar] [CrossRef]
- Innes, S.N.; Solhaug, K.A.; Torre, S.; Dodd, I.C. Different ABA efficient mutants show unique morphological and hydraulic responses to high air humidity. Physiol. Plant. 2021, 172, 1795–1807. [Google Scholar] [CrossRef]
- Lu, J.; Shireen, F.; Cheng, F.; Bie, Z. High relative humidity improve chilling tolerance by maintaining leaf water potential in watermelon seedlings. Plant Physiol. Bioch. 2021, 166, 818–826. [Google Scholar] [CrossRef]
- Aliniaeifard, S.; Meeteren, U. Stomatal characteristics and desiccation response of leaves of cut chrysanthemum (Chrysanthemum morifolium) flowers grown at high air humidity. Sci. Hortic. 2016, 205, 84–89. [Google Scholar] [CrossRef]
- Cramer, M.D.; Hawkins, H.J.; Verboom, G.A. The importance of nutritional regulation of plant water flux. Oecologia 2009, 161, 15–24. [Google Scholar] [CrossRef]
- Karbulkova, J.; Schreiber, L.; Macek, P.; Santrucek, J. Differences between water permeability of astomatous and stomatous cuticular membranes: Effects of air humidity in two species of contrasting drought-resistance strategy. J. Exp. Bot. 2008, 59, 3987–3995. [Google Scholar] [CrossRef] [PubMed]
- Chhun, T.; Uno, Y.; Taketa, S.; Azuma, T.; Ichii, M.; Okamoto, T.; Tsurumi, S. Saturated humidity accelerates lateral root development in rice (Oryza sativa L.) seedlings by increasing phloem-based auxin transport. J. Exp. Bot. 2007, 58, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Godbold, D.; Tullus, A.; Kupper, P.; Sõber, J.; Ostonen, I.; Godbold, J.A.; Lukac, M.; Ahmed, I.U.; Smith, A.R. Elevated atmospheric CO2 and humidity delay leaf fall in Betula pendula, but not in Alnus glutinosa or Populus tremula × tremuloides. Ann. For. Sci. 2014, 71, 831–842. [Google Scholar] [CrossRef]
- Roriz, M.; Carvalho, S.M.P.; Vasconcelos, M.W. High relative air humidity influences mineral accumulation and growth in iron deficient soybean plants. Front. Plant Sci. 2014, 5, 726. [Google Scholar] [CrossRef] [PubMed]
- Parts, K.; Tedersoo, L.; Lõhmus, K.; Kupper, P.; Rosenvald, K.; Sõber, A.; Ostonen, I. Increased air humidity and understory composition shape short root traits and the colonizing ectomycorrhizal fungal community in silver birch stands. For. Ecol. Manag. 2013, 310, 720–728. [Google Scholar] [CrossRef]
- Javidi, M.R.; Maali-Amiri, R.; Poormazaheri, H.; Sadeghi, N.M.; Kariman, K. Cold stress-induced changes in metabolism of carbonyl compounds and membrane fatty acid composition in chickpea. 2022. Plant Physiol. Biochem. 2022, 192, 10–19. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, W.; Qin, M.; Li, S.; Qiao, M.; Liu, Z.; Xiang, F. Drought tolerance conferred in soybean (Glycine max. L) by GmMYB84, a novel R2R3-MYB transcription factor. Plant Cell Physiol. 2017, 58, 1764–1776. [Google Scholar] [CrossRef]
- Guo, Q.; Li, X.; Niu, L.; Jameson, P.E.; Zhou, W. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021, 186, 677–695. [Google Scholar] [CrossRef]
- Brown, D.E.; Rashotte, A.M.; Murphy, A.S.; Normanly, J.; Tague, B.W.; Peer, W.A.; Taiz, L.; Muday, G.K. Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis. Plant Physiol. 2001, 126, 524–535. [Google Scholar] [CrossRef]
- Besseau, S.; Hoffmann, L.; Geoffroy, P.; Lapierre, C.; Pollet, B.; Legrand, M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell 2007, 19, 148–162. [Google Scholar] [CrossRef]
- Winter, G.; Todd, C.D.; Trovato, M.; Forlani, G.; Funck, D. Physiological implications of arginine metabolism in plants. Front. Plant Sci. 2015, 6, 534. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C. Seventy-year major research progress in plant hormones by Chinese scholars (in Chinese). Sci. Sin. Vitae. 2019, 49, 1227–1281. [Google Scholar]
- Mao, D.; Xin, Y.; Tan, Y. Natural variation in the HAN1 gene confers chilling tolerance in rice and allowed adaptation to a temperate climate. Proc. Natl. Acad. Sci. USA 2019, 116, 3494–3501. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, L.; Wang, F. Jasmonate regulates the Inducer of cbf expression-c-repeat binding factor/dre binding factor1 cascade and freezing tolerance in Arabidopsis. Plant Cell 2013, 25, 2907–2924. [Google Scholar] [CrossRef]
- Wang, F.; Guo, Z.; Li, H.; Wang, M.; Onac, E.; Zhou, J.; Xia, X.; Shi, K.; Yu, J.; Zhou, Y. Phytochrome A and B function antagonistically to regulate cold tolerance via abscisic acid-dependent jasmonate signaling. Plant Physiol. 2016, 170, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Tanaka, Y.; Abrams, S.R.; Kamiya, Y.; Seki, M.; Nambara, E. High humidity induces abscisic acid 8′-hydroxylase in stomata and vasculature to regulate local and systemic abscisic acid responses in Arabidopsis. Plant Physiol. 2009, 149, 825–834. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H. Multifaceted roles of FHY3 and FAR1 in light signaling and beyond. Trends Plant Sci. 2015, 20, 453–461. [Google Scholar] [CrossRef]
- Feng, K.; Hou, X.L.; Xing, G.M.; Liu, J.X.; Duan, A.Q.; Xu, Z.S.; Li, M.Y.; Zhuang, J.; Xiong, A.S. Advances in AP2/ERF super-family transcription factors in plant. Crit. Rev. Biotechnol. 2020, 40, 750–776. [Google Scholar] [CrossRef]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF Transcription Factor Regulatory Networks in Hormone and Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2019, 28, 228. [Google Scholar] [CrossRef]
- Hussain, Q.; Asim, M.; Zhang, R.; Khan, R.; Farooq, S.; Wu, J. Transcription Factors Interact with ABA through Gene Expression and Signaling Pathways to Mitigate Drought and Salinity Stress. Biomolecules 2021, 5, 1159. [Google Scholar] [CrossRef]
- Ma, L.; Xue, N.; Fu, X.; Zhang, H.; Li, G. Arabidopsis thaliana FAR-RED ELONGATED HYPOCOTYLS3 (FHY3) and FAR-RED-IMPAIRED RESPONSE1 (FAR1) modulate starch synthesis in response to light and sugar. New Phytol. 2017, 213, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Hand, K.A.; Shabek, N. The Role of E3 Ubiquitin Ligases in Chloroplast Function. Int. J. Mol. Sci. 2022, 25, 9613. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Features | Treatment Modalities | Time/Name 1 |
|---|---|---|---|
| Dianli-969 | Sensitive type | Normothermic (25 ± 1 °C); High humidity (90 ± 2%) | 5d/TS1; 14d/TS2 |
| Normothermic (25 ± 1 °C); Normal humidity (50 ± 2%) | 5d/CS1; 14d/CS2 | ||
| Dianli-439 | Highly resistant | Normothermic (25 ± 1 °C); High humidity (90 ± 2%) | 5d/TR1; 14d/TR2 |
| Normothermic (25 ± 1 °C); Normal humidity (50 ± 2%) | 5d/CR1; 14d/CR2 |
| Processing Time (Days) | Group Name | Plant Height (cm) | Leaf Area (mm2) | Fresh Weight (g) | Dry Weight (g) | Root-to-Crown Ratio |
|---|---|---|---|---|---|---|
| 5 | CR1 | 7.83 ± 0.28 a | 281.93 ± 22.81 a | 0.4292 ± 0.0329 a | 0.0602 ± 0.0075 a | 0.0429 ± 0.0022 a |
| TR1 | 6.91 ± 0.86 a | 271.54 ± 22.78 a | 0.3772 ± 0.0287 a | 0.0190 ± 0.0051 b | 0.0490 ± 0.006 a | |
| CS1 | 7.77 ± 1.04 a | 274.69 ± 17.62 a | 0.4133 ± 0.0387 a | 0.0548 ± 0.0048 a | 0.0445 ± 0.0067 a | |
| TS1 | 6.89 ± 0.98 a | 267.57 ± 28.52 a | 0.3818 ± 0.0638 a | 0.0201 ± 0.0109 b | 0.050 ± 0.0008 a | |
| 14 | CR2 | 11.51 ± 1.37 a | 559.65 ± 34.9 a | 0.8699 ± 0.06816 a | 0.1214 ± 0.0071 a | 0.084 ± 0.0234 b |
| TR2 | 10.89 ± 1.16 a | 484.44 ± 92.78 a | 0.7349 ± 0.1530 a | 0.0310 ± 0.0069 a | 0.099 ± 0.0137 a | |
| CS2 | 11.45 ± 0.88 a | 549.67 ± 70.60 a | 0.8785 ± 0.1039 a | 0.1196 ± 0.0157 a | 0.100 ± 0.0164 a | |
| TS2 | 8.38 ± 1.50 b | 421.98 ± 35.12 b | 0.5901 ± 0.0557 b | 0.0236 ± 0.0069 b | 0.067 ± 0.0037 b |
| Time (Days) | Group Name | CHL (μg·mL−1) | ABA (ng·g−1) | SOD (U·mg−1 FW) | POD (U·mg−1 FW) | CAT (U·mg−1 FW) | MDA (μmol·g−1 FW) |
|---|---|---|---|---|---|---|---|
| 5 | CR1 | 37.26 ± 3.985 a | 73.10 ± 16.29 a | 99.28 ± 9.215 c | 1217.8 ± 128.3 c | 3133.0 ± 151.9 b | 8.056 ± 1.3045 c |
| TR1 | 30.48 ± 5.427 b | 54.03 ± 5.635 b | 315.89 ± 29.68 a | 2253.3 ± 242.8 b | 4099.5 ± 102.7 a | 17.48 ± 2.3631 b | |
| CS1 | 38.78 ± 6.916 a | 76.71 ± 8.499 a | 108.11 ± 13.45 c | 1111.6 ± 128.1 c | 2973.6 ± 49.9 b | 9.894 ± 1.8532 c | |
| TS1 | 21.53 ± 2.064 b | 53.69 ± 5.374 b | 244.15 ± 44.57 b | 2536.1 ± 156.9 a | 3289.8 ± 252.2 b | 25.49 ± 3.7486 a | |
| 14 | CR2 | 40.74 ± 5.201 a | 84.23 ± 4.176 b | 125.31 ± 19.45 c | 1446.6 ± 134.4 c | 3256.5 ± 212.6 c | 8.563 ± 2.5638 c |
| TR2 | 27.79 ± 3.165 b | 65.14 ± 22.71 c | 500.93 ± 35.13 a | 2241.2 ± 172.2 b | 4756.2 ± 118.4 a | 25.64 ± 5.383 b | |
| CS2 | 38.91 ± 5.799 a | 93.83 ± 2.724 a | 82.56 ± 6.314 d | 1341.5 ± 122.8 c | 3300.9 ± 173.7 c | 7.478 ± 1.8581 c | |
| TS2 | 19.96 ± 3.931 c | 41.1 ± 12.10 d | 434.71 ± 16.54 b | 3132.9 ± 224.0 a | 4060.8 ± 565.5 b | 37.72 ± 4.861 a |
| Gene Family | Number of DEGs | DEGs Description | Biological Functions |
|---|---|---|---|
| FAR1 | 566 | Far-Red-Impairedresponse1 | Development and stress response |
| AP2/ERF | 130 | APETALA 2/ethylene-responsive element binding factor | Development and stress response |
| bHLH | 202 | Basic helix-loop-helix TFs | Cell development and substance metabolism |
| C2H2 | 127 | C2H2 zinc finger protein | Development and stress response |
| C3H | 94 | C3H zinc finger protein | Embryo formation |
| bZIP | 94 | box-zipper protein | Plant photomorphogenesis and fruit ripening |
| MYB | 147 | MYB TFs | Cell development and flavonoid pathway |
| NAC | 115 | NAC family TFs | Development and stress response |
| WRKY | 102 | WRKY DNA-binding protein | Defense responses and plant development |
| B3 | 155 | B3-domain-containing protein | Plant growth and development |
| Others | 1210 | ||
| Total | 2825 |
| Modules | Module Candidate Hub Genes | Hub Genes’ Description |
|---|---|---|
| Black | LOC110729592 | Belongs to the sterol desaturase family |
| LOC110716937 | Beta-D-xylosidase (BXL3) | |
| LOC110724693 | Belongs to the cytochrome P450 family (CYP83A1) | |
| LOC110736400 | Calreticulin-3-like | |
| LOC110683032 | Belongs to the protein kinase superfamily | |
| LOC110701145 | Wall-associated receptor kinase-like | |
| LOC110712611 | Glycosyl transferase family 17 protein | |
| LOC110706702 | MADS-box protein | |
| LOC110701071 | Calreticulin-3-like | |
| LOC110716012 | Glucose and ribitol | |
| LOC110715868 | Sucrose-cleaving enzyme that provides UDP-glucose and fructose for various Metabolic pathways (SUS1) | |
| LOC110733529 | Amino acid transporter | |
| LOC110695772 | \ | |
| LOC110708285 | Ammonium transporter (AMT3-1) | |
| LOC110717076 | Lob-domain-containing protein | |
| LOC110721833 | Sucrose-cleaving enzyme that provides UDP-glucose and fructose for various metabolic pathways (SUS1) | |
| LOC110709750 | MADS-box protein | |
| LOC110728040 | Cystinosin homolog | |
| LOC110686969 | Protein plant cadmium resistance | |
| LOC110698492 | Partial alpha-/beta-hydrolase lipase region | |
| LOC110684083 | Belongs to the heat shock protein 70 family | |
| LOC110682201 | Major facilitator superfamily protein | |
| LOC110706950 | Serine threonine-protein kinase | |
| LOC110733057 | Universal stress protein | |
| LOC110687281 | Cysteine-rich receptor-like protein kinase | |
| LOC110729587 | Heat shock protein | |
| LOC110696285 | Transposition, RNA-mediated | |
| Blue | LOC110738464 | Transporter |
| LOC110705966 | Glycerophosphoryl diester phosphodiesterase | |
| LOC110731861 | Alcohol dehydrogenase GroES-like domain | |
| LOC110688479 | Protein upstream of | |
| LOC110723960 | Glycerophosphoryl diester phosphodiesterase | |
| LOC110710755 | Abscisic acid receptor | |
| LOC110732716 | Zinc transporter | |
| LOC110686655 | Polyamine oxidase | |
| LOC110723200 | Belongs to the chalcone isomerase family | |
| LOC110718261 | Rho GTPase-activating protein | |
| LOC110695938 | Belongs to the Casparian strip membrane proteins (CASP) family | |
| LOC110685067 | Oligopeptide transporter | |
| LOC110700433 | Belongs to the cytochrome P450 family | |
| LOC110690627 | Belongs to the glycosyl hydrolase 5 (cellulase A) family | |
| LOC110723446 | Belongs to the protein kinase superfamily (SAPK2) | |
| LOC110735654 | Choline kinase | |
| LOC110709626 | Catalyzes xyloglucan endohydrolysis (XEH) and or endotransglycosylation (XET); cleaves and relegates xyloglucan polymers, an essential constituent of the primary cell wall, and thereby participates in cell wall construction of growing tissues | |
| LOC110704465 | Atexp8, atexpa8, athexp alpha 1.11, exp8, expa8 (EXP2) | |
| LOC110733886 | Abscisic acid receptor | |
| LOC110736716 | calcium-dependent protein kinase (CDPK9) | |
| LOC110710508 | GAST1-like | |
| LOC110691154 | phosphoenolpyruvate carboxylase | |
| LOC110739071 | Protein NRT1 PTR FAMILY |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, P.; Liu, J.; Wang, H.; Liu, J.; Li, H.; Xie, H.; Wang, Q.; Li, L.; Zhang, S.; et al. Integrated Metabolomic and Transcriptomic Analysis of the Quinoa Seedling Response to High Relative Humidity Stress. Biomolecules 2023, 13, 1352. https://doi.org/10.3390/biom13091352
Li X, Zhang P, Liu J, Wang H, Liu J, Li H, Xie H, Wang Q, Li L, Zhang S, et al. Integrated Metabolomic and Transcriptomic Analysis of the Quinoa Seedling Response to High Relative Humidity Stress. Biomolecules. 2023; 13(9):1352. https://doi.org/10.3390/biom13091352
Chicago/Turabian StyleLi, Xinyi, Ping Zhang, Jia Liu, Hongxin Wang, Junna Liu, Hanxue Li, Heng Xie, Qianchao Wang, Li Li, Shan Zhang, and et al. 2023. "Integrated Metabolomic and Transcriptomic Analysis of the Quinoa Seedling Response to High Relative Humidity Stress" Biomolecules 13, no. 9: 1352. https://doi.org/10.3390/biom13091352
APA StyleLi, X., Zhang, P., Liu, J., Wang, H., Liu, J., Li, H., Xie, H., Wang, Q., Li, L., Zhang, S., Huang, L., Liu, C., & Qin, P. (2023). Integrated Metabolomic and Transcriptomic Analysis of the Quinoa Seedling Response to High Relative Humidity Stress. Biomolecules, 13(9), 1352. https://doi.org/10.3390/biom13091352