
Exploring the Potential of Sulfur Moieties in Compounds Inhibiting Steroidogenesis

 ,
,  , , , ,
, , , ,  , and
, and
Abstract
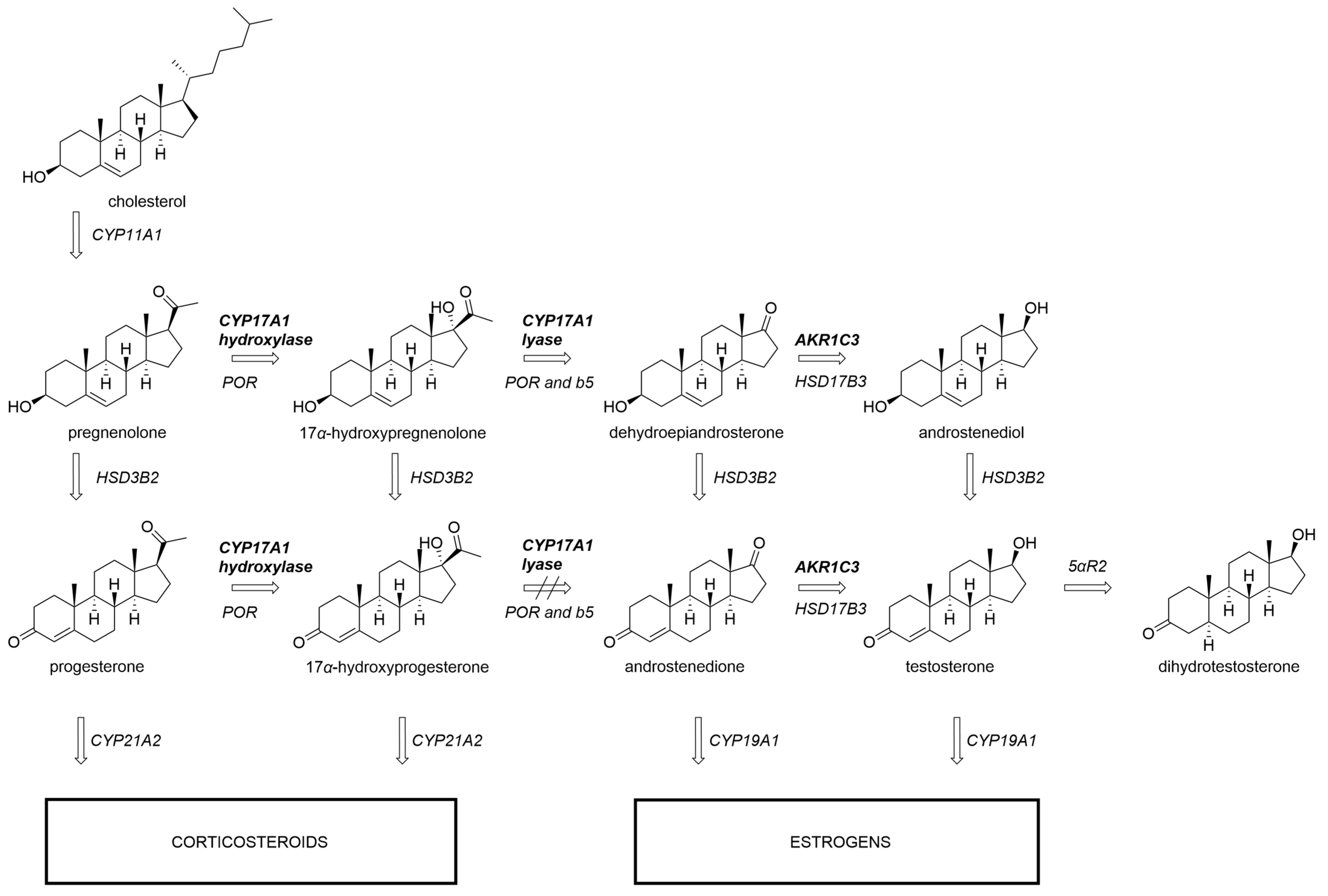
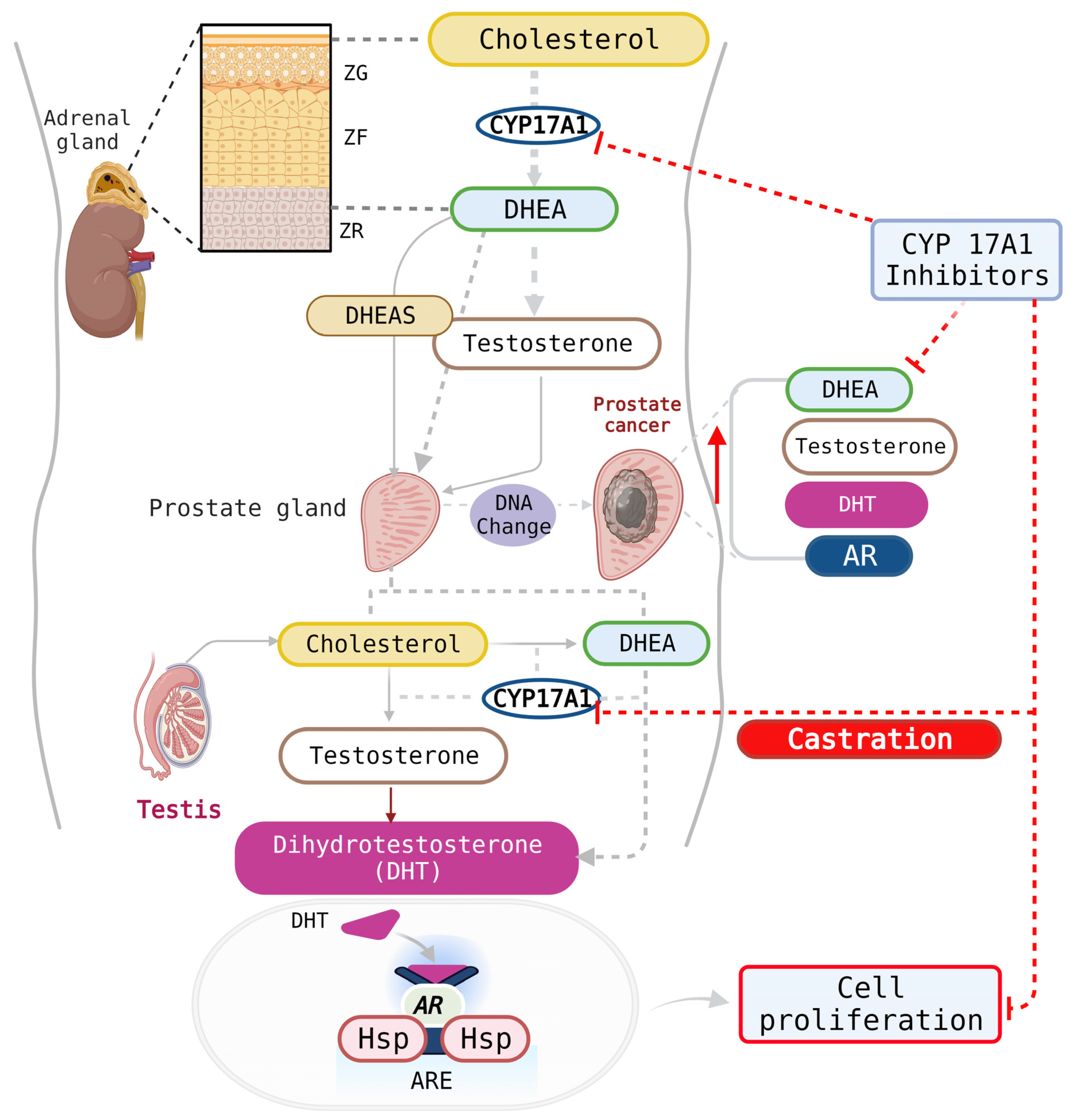
1. Introduction
2. Materials and Methods
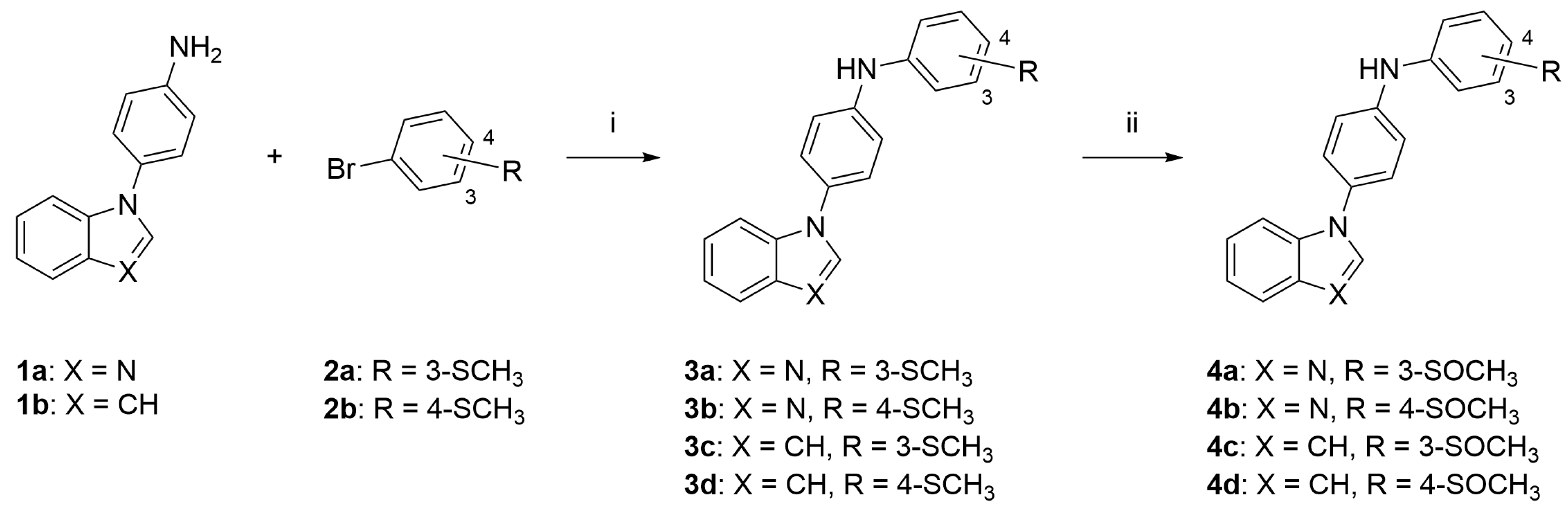
2.1. Chemistry
2.2. General Procedure to Obtain Compounds 3a to 3d
2.2.1. N-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-3-(methylthio)aniline (3a)
2.2.2. N-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-4-(methylthio)aniline (3b)
2.2.3. N-(4-(1H-Indol-1-yl)phenyl)-3-(methylthio)aniline (3c)
2.2.4. N-(4-(1H-Indol-1-yl)phenyl)-4-(methylthio)aniline (3d)
2.3. General Procedure to Obtain Compounds 4a to 4d
2.3.1. N-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-3-(methylsulfinyl)aniline (4a)
2.3.2. N-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-4-(methylsulfinyl)aniline (4b)
2.3.3. N-(4-(1H-Indol-1-yl)phenyl)-3-(methylsulfinyl)aniline (4c)
2.3.4. N-(4-(1H-Indol-1-yl)phenyl)-4-(methylsulfinyl)aniline (4d)
2.4. Biology
2.5. Computational Chemistry
3. Results and Discussion
3.1. Chemistry
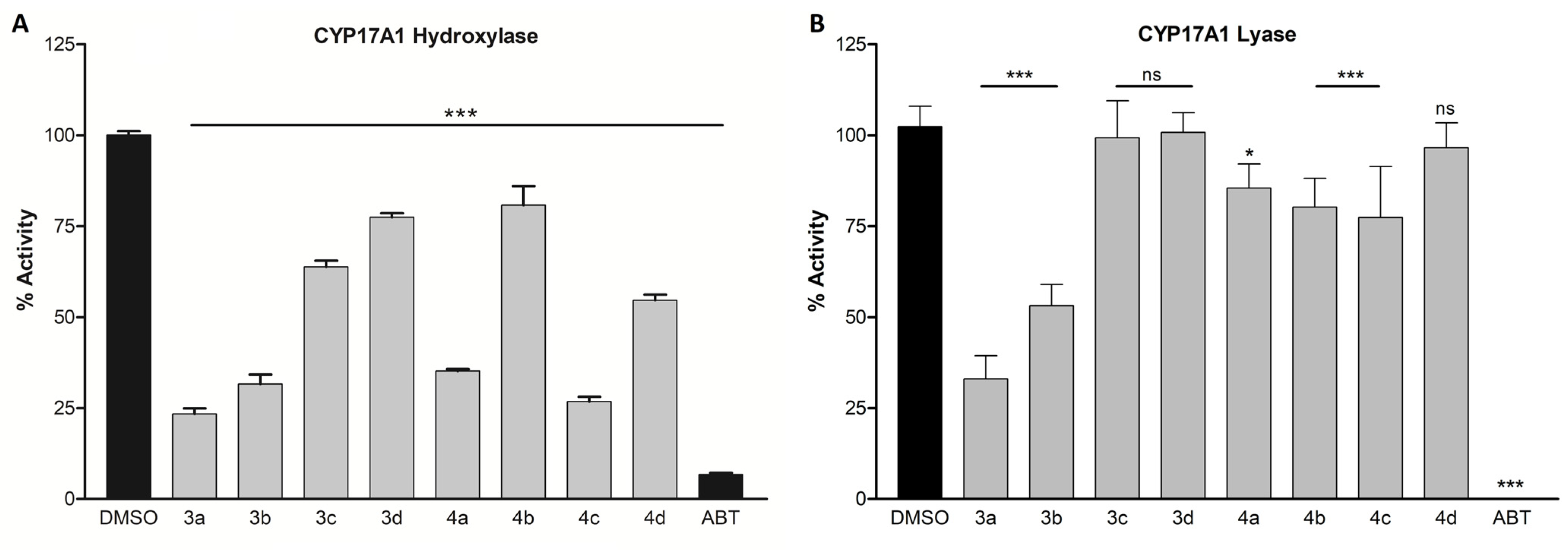
3.2. CYP17A1 Inhibition
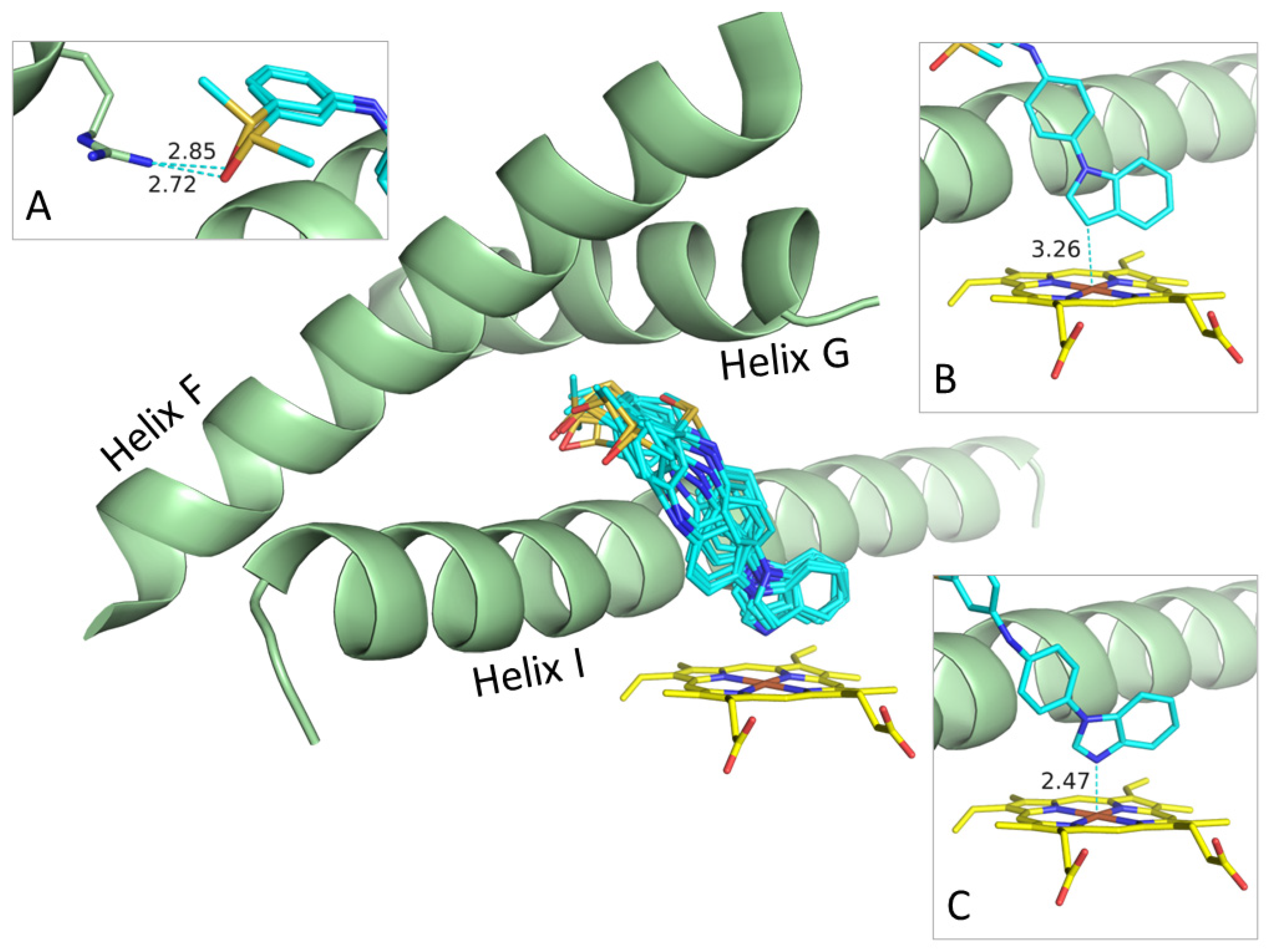
3.3. Possible Binding Modes
3.4. Docking
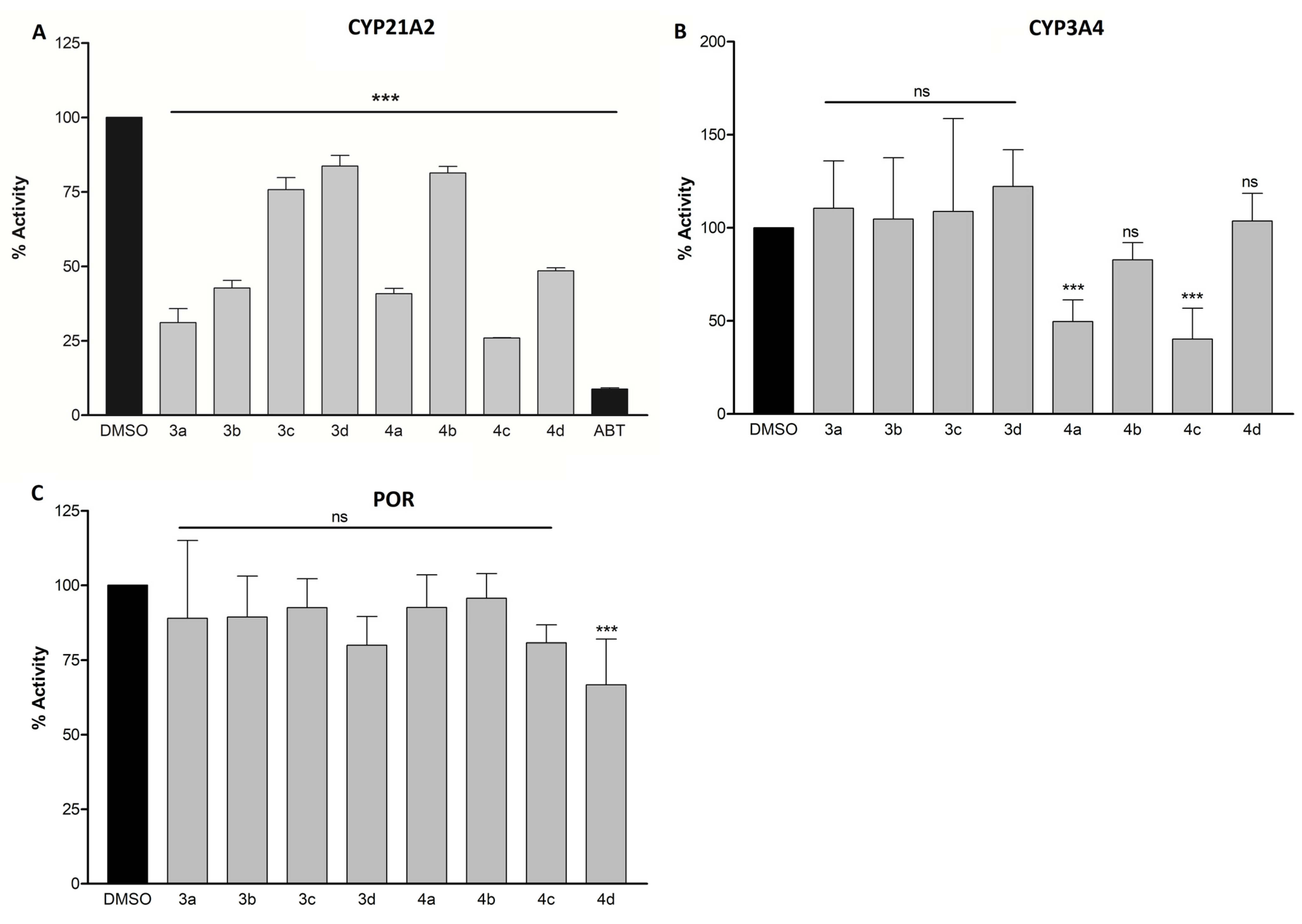
3.5. CYP21A2, CYP3A4, and POR Inhibition
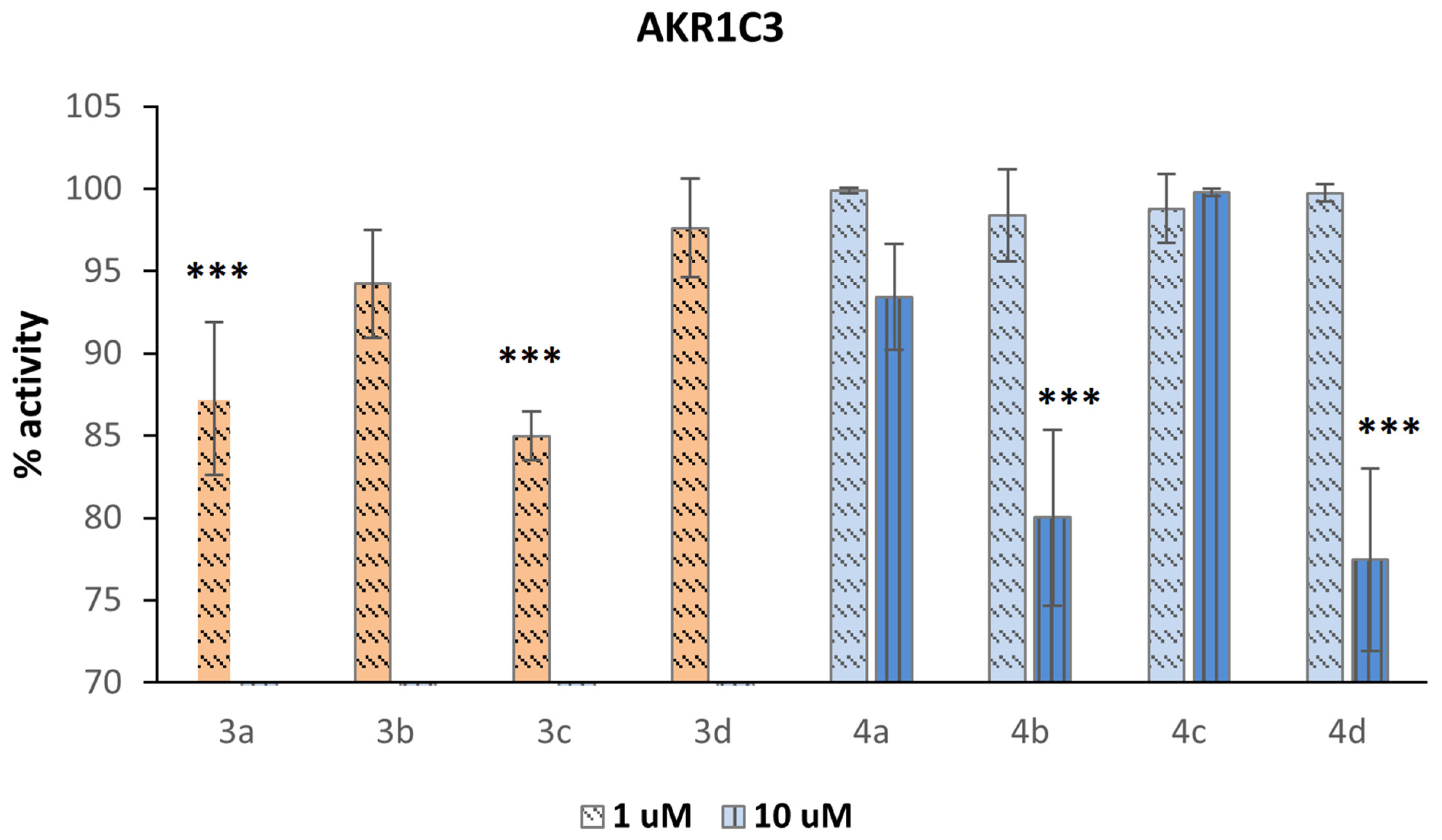
3.6. AKR1C3 Inhibition
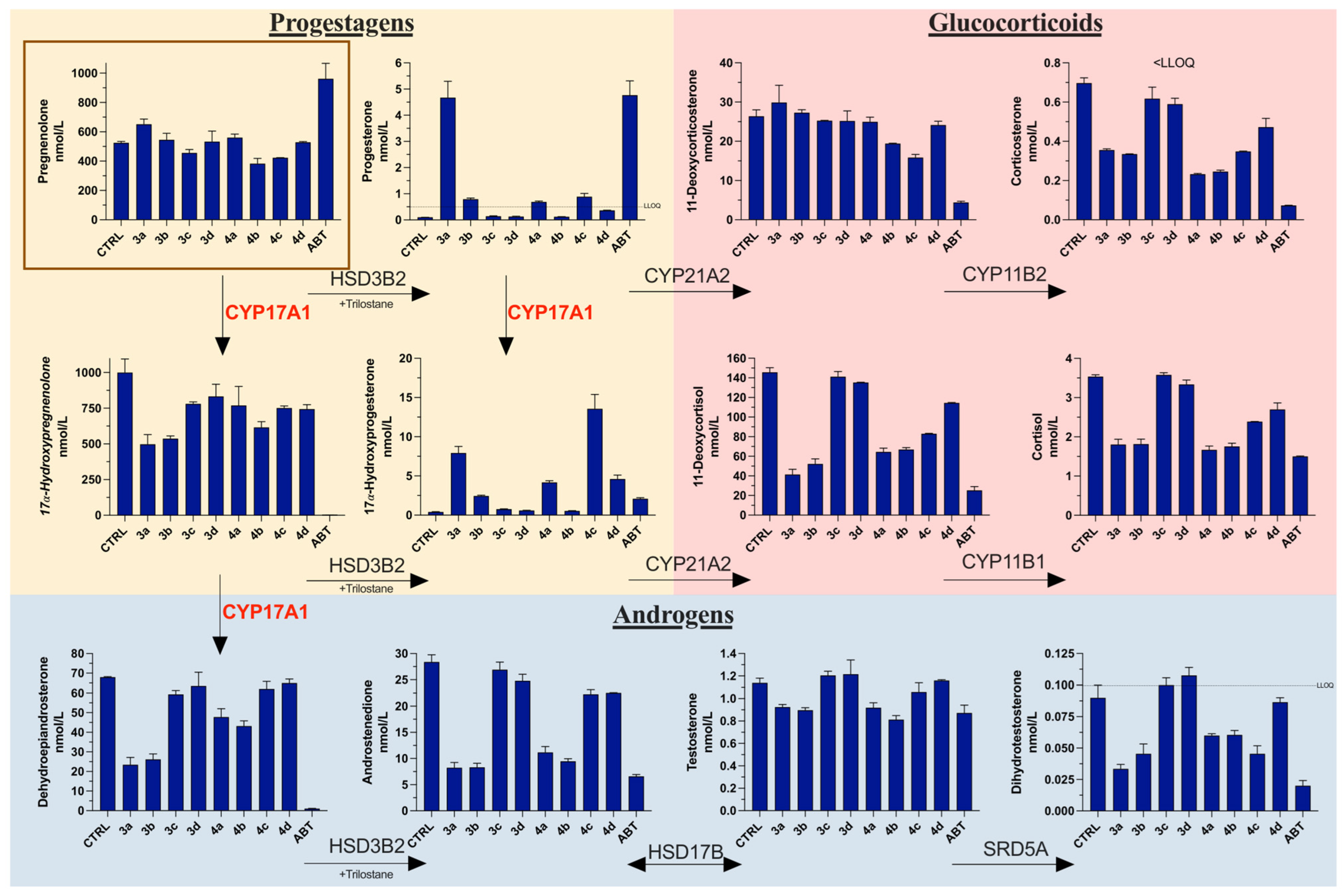
3.7. Steroid Profiling
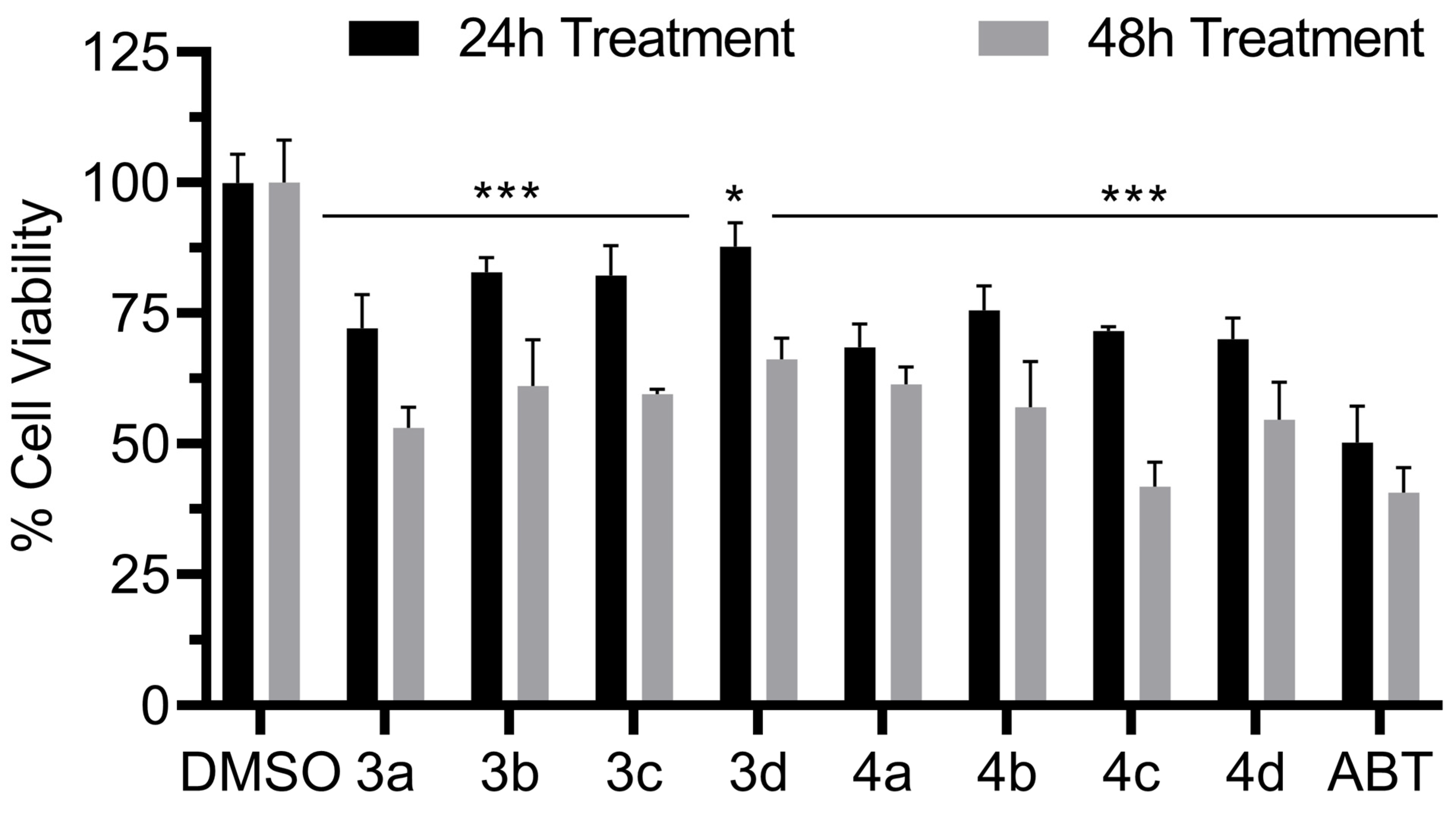
3.8. Antiproliferative Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Muniyan, S.; Li, B.; Batra, S.K. Editorial: Metastatic Castration Resistant Prostate Cancer: Prognosis and Treatment. Front. Oncol. 2022, 12, 913630. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Hu, Q. CYP17 inhibitors—Abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2014, 11, 32–42. [Google Scholar] [CrossRef]
- Wróbel, T.M.; Jørgensen, F.S.; Pandey, A.V.; Grudzińska, A.; Sharma, K.; Yakubu, J.; Björkling, F. Non-steroidal CYP17A1 Inhibitors: Discovery and Assessment. J. Med. Chem. 2023, 66, 6542–6566. [Google Scholar] [CrossRef]
- Maity, S.N.; Titus, M.A.; Gyftaki, R.; Wu, G.; Lu, J.-F.; Ramachandran, S.; Li-Ning-Tapia, E.M.; Logothetis, C.J.; Araujo, J.C.; Efstathiou, E. Targeting of CYP17A1 Lyase by VT-464 Inhibits Adrenal and Intratumoral Androgen Biosynthesis and Tumor Growth of Castration Resistant Prostate Cancer. Sci. Rep. 2016, 6, 35354. [Google Scholar] [CrossRef]
- Hara, T.; Kouno, J.; Kaku, T.; Takeuchi, T.; Kusaka, M.; Tasaka, A.; Yamaoka, M. Effect of a novel 17,20-lyase inhibitor, orteronel (TAK-700), on androgen synthesis in male rats. J. Steroid Biochem. Mol. Biol. 2013, 134, 80–91. [Google Scholar] [CrossRef]
- Fizazi, K.; Jones, R.; Oudard, S.; Efstathiou, E.; Saad, F.; de Wit, R.; De Bono, J.; Cruz, F.M.; Fountzilas, G.; Ulys, A.; et al. Phase III, Randomized, Double-Blind, Multicenter Trial Comparing Orteronel (TAK-700) Plus Prednisone with Placebo Plus Prednisone in Patients with Metastatic Castration-Resistant Prostate Cancer That Has Progressed During or after Docetaxel-Based Therapy: ELM-PC 5. J. Clin. Oncol. 2015, 33, 723–731. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Chen, Y.; Liu, Y.; Feng, F.; Liu, W.; Guo, Q.; Zhao, L.; Sun, H. Overview of AKR1C3: Inhibitor Achievements and Disease Insights. J. Med. Chem. 2020, 63, 11305–11329. [Google Scholar] [CrossRef]
- Shiota, M.; Endo, S.; Blas, L.; Fujimoto, N.; Eto, M. Steroidogenesis in castration-resistant prostate cancer. Urol. Oncol. 2022, 41, 240–251. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.; Evans, C.P.; Gao, A.C. Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Jonnalagadda, S.; Trippier, P.C.; Rižner, T.L. Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev. 2021, 73, 1150–1171. [Google Scholar] [CrossRef]
- Bonomo, S.; Hansen, C.H.; Petrunak, E.M.; Scott, E.E.; Styrishave, B.; Jørgensen, F.S.; Olsen, L. Promising Tools in Prostate Cancer Research: Selective Non-Steroidal Cytochrome P450 17A1 Inhibitors. Sci. Rep. 2016, 6, 29468. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.G.; Kidley, N.J. Quantitatively Interpreted Enhanced Inhibition of Cytochrome P450s by Heteroaromatic Rings Containing Nitrogen. J. Chem. Inf. Model. 2011, 51, 1048–1063. [Google Scholar] [CrossRef]
- Castaño, P.R.; Parween, S.; Pandey, A.V. Bioactivity of Curcumin on the Cytochrome P450 Enzymes of the Steroidogenic Pathway. Int. J. Mol. Sci. 2019, 20, 4606. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.-i.; Itakura, Y.; Goto, R.; Tojima, M.; Egawa, N.; Yoshihama, M. Inhibition of 17α-Hydroxylase/C17,20-Lyase (CYP17) from Rat Testis by Green Tea Catechins and Black Tea Theaflavins. Biosci. Biotechnol. Biochem. 2007, 71, 2325–2328. [Google Scholar] [CrossRef][Green Version]
- Frydenvang, K.; Verkade-Vreeker, M.C.A.; Dohmen, F.; Commandeur, J.N.M.; Rafiq, M.; Mirza, O.; Jorgensen, F.S.; Geerke, D.P. Structural analysis of Cytochrome P450 BM3 mutant M11 in complex with dithiothreitol. PLoS ONE 2019, 14, e0217292. [Google Scholar] [CrossRef]
- Doukov, T.; Li, H.; Sharma, A.; Martell, J.D.; Soltis, S.M.; Silverman, R.B.; Poulos, T.L. Temperature-dependent spin crossover in neuronal nitric oxide synthase bound with the heme-coordinating thioether inhibitors. J. Am. Chem. Soc. 2011, 133, 8326–8334. [Google Scholar] [CrossRef]
- Martell, J.D.; Li, H.; Doukov, T.; Martasek, P.; Roman, L.J.; Soltis, M.; Poulos, T.L.; Silverman, R.B. Heme-coordinating inhibitors of neuronal nitric oxide synthase. Iron-thioether coordination is stabilized by hydrophobic contacts without increased inhibitor potency. J. Am. Chem. Soc. 2010, 132, 798–806. [Google Scholar] [CrossRef][Green Version]
- Jarman, M.; John Smith, H.; Nicholls, P.J.; Simons, C. Inhibitors of enzymes of androgen biosynthesis: Cytochrome P45017α and 5α-steroid reductase. Nat. Prod. Rep. 1998, 15, 495–512. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Oie, H.K.; Shackleton, C.H.; Chen, T.R.; Triche, T.J.; Myers, C.E.; Chrousos, G.P.; Brennan, M.F.; Stein, C.A.; La Rocca, R.V. Establishment and Characterization of a Human Adrenocortical Carcinoma Cell Line That Expresses Multiple Pathways of Steroid Biosynthesis1. Cancer Res. 1990, 50, 5488–5496. [Google Scholar] [PubMed]
- Kurlbaum, M.; Sbiera, S.; Kendl, S.; Martin Fassnacht, M.; Kroiss, M. Steroidogenesis in the NCI-H295 Cell Line Model is Strongly Affected By Culture Conditions and Substrain. Exp. Clin. Endocrinol. Diabetes 2020, 128, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K.; Auchus, R.J. The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J. Steroid Biochem. Mol. Biol. 2015, 151, 52–65. [Google Scholar] [CrossRef]
- Potts, G.O.; Creange, J.E.; Harding, H.R.; Schane, H.P. Trilostane, an orally active inhibitor of steroid biosynthesis. Steroids 1978, 32, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Malikova, J.; Brixius-Anderko, S.; Udhane, S.S.; Parween, S.; Dick, B.; Bernhardt, R.; Pandey, A.V. CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2. J. Steroid Biochem. Mol. Biol. 2017, 174, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Morán, F.M.; VandeVoort, C.A.; Overstreet, J.W.; Lasley, B.L.; Conley, A.J. Molecular Target of Endocrine Disruption in Human Luteinizing Granulosa Cells by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: Inhibition of Estradiol Secretion Due to Decreased 17α-Hydroxylase/17,20-Lyase Cytochrome P450 Expression. Endocrinology 2003, 144, 467–473. [Google Scholar] [CrossRef] [PubMed]
- McManus, J.M.; Bohn, K.; Alyamani, M.; Chung, Y.-M.; Klein, E.A.; Sharifi, N. Rapid and structure-specific cellular uptake of selected steroids. PLoS ONE 2019, 14, e0224081. [Google Scholar] [CrossRef]
- Ahmed, K.E.M.; Frøysa, H.G.; Karlsen, O.A.; Sagen, J.V.; Mellgren, G.; Verhaegen, S.; Ropstad, E.; Goksøyr, A.; Kellmann, R. LC-MS/MS based profiling and dynamic modelling of the steroidogenesis pathway in adrenocarcinoma H295R cells. Toxicol. Vitro 2018, 52, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Wudy, S.A.; Schuler, G.; Sánchez-Guijo, A.; Hartmann, M.F. The art of measuring steroids: Principles and practice of current hormonal steroid analysis. J. Steroid Biochem. Mol. Biol. 2018, 179, 88–103. [Google Scholar] [CrossRef]
- Andrieu, T.; du Toit, T.; Vogt, B.; Mueller, M.D.; Groessl, M. Parallel targeted and non-targeted quantitative analysis of steroids in human serum and peritoneal fluid by liquid chromatography high-resolution mass spectrometry. Anal. Bioanal. Chem. 2022, 414, 7461–7472. [Google Scholar] [CrossRef]
- Pippione, A.C.; Giraudo, A.; Bonanni, D.; Carnovale, I.M.; Marini, E.; Cena, C.; Costale, A.; Zonari, D.; Pors, K.; Sadiq, M.; et al. Hydroxytriazole derivatives as potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors discovered by bioisosteric scaffold hopping approach. Eur. J. Med. Chem. 2017, 139, 936–946. [Google Scholar] [CrossRef] [PubMed]
- DeVore, N.M.; Scott, E.E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Kirton, S.B.; Murray, C.W.; Verdonk, M.L.; Taylor, R.D. Prediction of binding modes for ligands in the cytochromes P450 and other heme-containing proteins. Proteins 2005, 58, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, T.M.; Rogova, O.; Andersen, K.L.; Yadav, R.; Brixius-Anderko, S.; Scott, E.E.; Olsen, L.; Jørgensen, F.S.; Björkling, F. Discovery of Novel Non-Steroidal Cytochrome P450 17A1 Inhibitors as Potential Prostate Cancer Agents. Int. J. Mol. Sci. 2020, 21, 4868. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Wróbel, T.M.; Rogova, O.; Sharma, K.; Rojas Velazquez, M.N.; Pandey, A.V.; Jørgensen, F.S.; Arendrup, F.S.; Andersen, K.L.; Björkling, F. Synthesis and Structure-Activity Relationships of Novel Non-Steroidal CYP17A1 Inhibitors as Potential Prostate Cancer Agents. Biomolecules 2022, 12, 165. [Google Scholar] [CrossRef]
- Xu, W.L.; Li, Y.Z.; Zhang, Q.S.; Zhu, H.S. A Selective, Convenient, and Efficient Conversion of Sulfides to Sulfoxides. Synthesis 2004, 2004, 227–232. [Google Scholar] [CrossRef]
- Fehl, C.; Vogt, C.D.; Yadav, R.; Li, K.; Scott, E.E.; Aubé, J. Structure-Based Design of Inhibitors with Improved Selectivity for Steroidogenic Cytochrome P450 17A1 over Cytochrome P450 21A2. J. Med. Chem. 2018, 61, 4946–4960. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Pippione, A.C.; Carnovale, I.M.; Bonanni, D.; Sini, M.; Goyal, P.; Marini, E.; Pors, K.; Adinolfi, S.; Zonari, D.; Festuccia, C.; et al. Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: Application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur. J. Med. Chem. 2018, 150, 930–945. [Google Scholar] [CrossRef]
- Pippione, A.C.; Kilic-Kurt, Z.; Kovachka, S.; Sainas, S.; Rolando, B.; Denasio, E.; Pors, K.; Adinolfi, S.; Zonari, D.; Bagnati, R.; et al. New aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the hydroxytriazole scaffold. Eur. J. Med. Chem. 2022, 237, 114366. [Google Scholar] [CrossRef]
- Kikuchi, A.; Furutani, T.; Azami, H.; Watanabe, K.; Niimi, T.; Kamiyama, Y.; Kuromitsu, S.; Baskin-Bey, E.; Heeringa, M.; Ouatas, T.; et al. In vitro and in vivo characterisation of ASP9521: A novel, selective, orally bioavailable inhibitor of 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5; AKR1C3). Investig. New Drugs 2014, 32, 860–870. [Google Scholar] [CrossRef]
- Sun, M.; Zhou, Y.; Zhuo, X.; Wang, S.; Jiang, S.; Peng, Z.; Kang, K.; Zheng, X.; Sun, M. Design, Synthesis and Cytotoxicity Evaluation of Novel Indole Derivatives Containing Benzoic Acid Group as Potential AKR1C3 Inhibitors. Chem. Biodivers. 2020, 17, e2000519. [Google Scholar] [CrossRef]
- Hou, Z.; Huang, S.; Mei, Z.; Chen, L.; Guo, J.; Gao, Y.; Zhuang, Q.; Zhang, X.; Tan, Q.; Yang, T.; et al. Inhibiting 3βHSD1 to eliminate the oncogenic effects of progesterone in prostate cancer. Cell Rep. Med. 2022, 3, 100561. [Google Scholar] [CrossRef]
- Sobel, R.E.; Sadar, M.D. Cell Lines Used in Prostate Cancer Research: A Compendium of Old and New Lines—Part 1. J. Urol. 2005, 173, 342–359. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking Score a | R/S Difference b | Fe-X3 Distance c [A] | X3 d |
|---|---|---|---|---|
| 3a | 47.0 | 2.3 | N | |
| 3b | 45.9 | 2.5 | N | |
| 3c | 43.4 | 3.2 | CH | |
| 3d | 44.0 | 3.3 | CH | |
| R-4a | 47.9 | 2.4 | N | |
| S-4a | 49.9 | −2.1 | 2.5 | N |
| R-4b | 47.8 | 2.5 | N | |
| S-4b | 46.4 | 1.4 | 2.2 | N |
| R-4c | 45.3 | 3.4 | CH | |
| S-4c | 46.2 | −0.9 | 3.3 | CH |
| R-4d | 44.1 | 2.7 | CH | |
| S-4d | 43.4 | 0.7 | 3.5 | CH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróbel, T.M.; Sharma, K.; Mannella, I.; Oliaro-Bosso, S.; Nieckarz, P.; Du Toit, T.; Voegel, C.D.; Rojas Velazquez, M.N.; Yakubu, J.; Matveeva, A.; et al. Exploring the Potential of Sulfur Moieties in Compounds Inhibiting Steroidogenesis. Biomolecules 2023, 13, 1349. https://doi.org/10.3390/biom13091349
Wróbel TM, Sharma K, Mannella I, Oliaro-Bosso S, Nieckarz P, Du Toit T, Voegel CD, Rojas Velazquez MN, Yakubu J, Matveeva A, et al. Exploring the Potential of Sulfur Moieties in Compounds Inhibiting Steroidogenesis. Biomolecules. 2023; 13(9):1349. https://doi.org/10.3390/biom13091349
Chicago/Turabian StyleWróbel, Tomasz M., Katyayani Sharma, Iole Mannella, Simonetta Oliaro-Bosso, Patrycja Nieckarz, Therina Du Toit, Clarissa Daniela Voegel, Maria Natalia Rojas Velazquez, Jibira Yakubu, Anna Matveeva, and et al. 2023. "Exploring the Potential of Sulfur Moieties in Compounds Inhibiting Steroidogenesis" Biomolecules 13, no. 9: 1349. https://doi.org/10.3390/biom13091349
APA StyleWróbel, T. M., Sharma, K., Mannella, I., Oliaro-Bosso, S., Nieckarz, P., Du Toit, T., Voegel, C. D., Rojas Velazquez, M. N., Yakubu, J., Matveeva, A., Therkelsen, S., Jørgensen, F. S., Pandey, A. V., Pippione, A. C., Lolli, M. L., Boschi, D., & Björkling, F. (2023). Exploring the Potential of Sulfur Moieties in Compounds Inhibiting Steroidogenesis. Biomolecules, 13(9), 1349. https://doi.org/10.3390/biom13091349