
Autophagy/Mitophagy in Airway Diseases: Impact of Oxidative Stress on Epithelial Cells
Abstract
1. Introduction
2. Oxidative Stress and Dysfunctional Mechanisms of Autophagy/Mitophagy
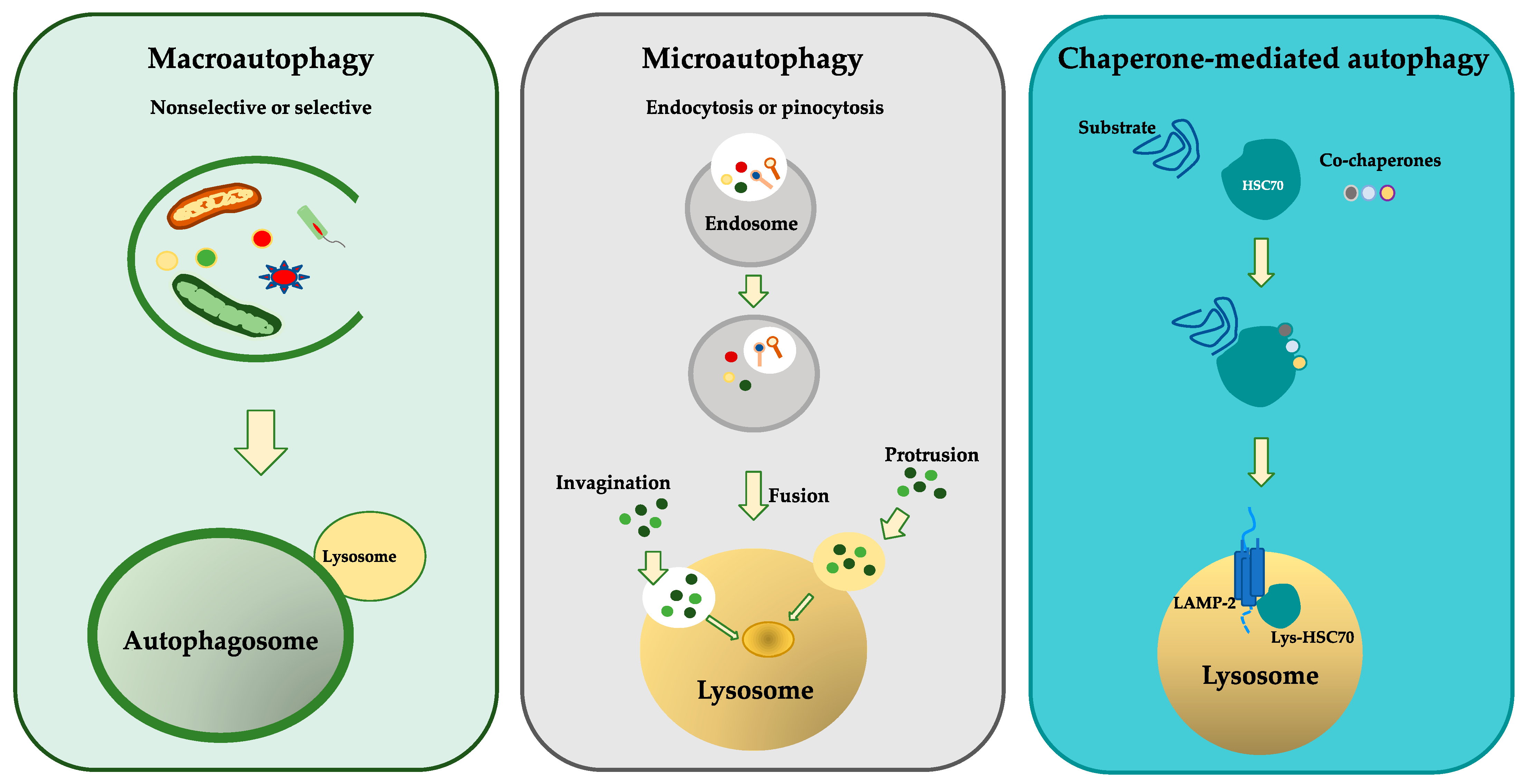
3. Molecular Mechanisms of Autophagy
4. Molecular Mechanisms of Mitophagy
5. Epithelial Cells in Airway Diseases
6. COPD and Autophagy/Mitophagy
7. Asthma and Autophagy/Mitophagy
8. Pharmacological Approach in Airway Diseases
9. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasarmidi, E.; Sarantoulaki, S.; Trachalaki, A.; Margaritopoulos, G.; Bibaki, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K. Investigation of key autophagy-and mitophagy-related proteins and gene expression in BALF cells from patients with IPF and RA-ILD. Mol. Med. Rep. 2018, 18, 3891–3897. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Porras, M.A.P.; Choi, A.M.K. Autophagy in Pulmonary Diseases. Am. J. Respir. Crit. Care Med. 2016, 194, 1196–1207. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef]
- Vainshtein, A.; Grumati, P. Selective Autophagy by Close Encounters of the Ubiquitin Kind. Cells 2020, 9, 2349. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Autophagy in asthma and chronic obstructive pulmonary disease. Clin. Sci. 2022, 136, 733–746. [Google Scholar] [CrossRef]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7–18. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2013, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-X.; Sun, P.-P.; Gu, Y.-H.; Rao, X.-M.; Zhang, L.-Y.; Ou-Yang, Y. Autophagy and pulmonary disease. Ther. Adv. Respir. Dis. 2019, 13, 1753466619890538. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2011, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kuwano, K. Role of chaperone-mediated autophagy in the pathophysiology including pulmonary disorders. Inflamm. Regen. 2021, 41, 29. [Google Scholar] [CrossRef]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2015, 71, 5–14. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.-M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef]
- Marsh, T.; Tolani, B.; Debnath, J. The pleiotropic functions of autophagy in metastasis. J. Cell Sci. 2021, 134, jcs247056. [Google Scholar] [CrossRef]
- Beljanski, V.; Grinnemo, K.-H.; Österholm, C. Pleiotropic roles of autophagy in stem cell–based therapies. Cytotherapy 2019, 21, 380–392. [Google Scholar] [CrossRef]
- Hwang, J.-W.; Chung, S.; Sundar, I.K.; Yao, H.; Arunachalam, G.; McBurney, M.W.; Rahman, I. Cigarette smoke-induced autophagy is regulated by SIRT1–PARP-1-dependent mechanism: Implication in pathogenesis of COPD. Arch. Biochem. Biophys. 2010, 500, 203–209. [Google Scholar] [CrossRef]
- Soodaeva, S.; Kubysheva, N.; Klimanov, I.; Nikitina, L.; Batyrshin, I. Features of Oxidative and Nitrosative Metabolism in Lung Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1689861. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Albano, G.D.; Gagliardo, R.P.; Montalbano, A.M.; Profita, M. Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases. Antioxidants 2022, 11, 2237. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Rogers, L.K.; Cismowski, M.J. Oxidative stress in the lung—The essential paradox. Curr. Opin. Toxicol. 2017, 7, 37–43. [Google Scholar] [CrossRef]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef]
- Rajendran, P.; Nandakumar, N.; Rengarajan, T.; Palaniswami, R.; Gnanadhas, E.N.; Lakshminarasaiah, U.; Gopas, J.; Nishigaki, I. Antioxidants and human diseases. Clin. Chim. Acta 2014, 436, 332–347. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses 2022, 2, 256–274. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Cabrera, S.; Maciel, M.; Herrera, I.; Nava, T.; Vergara, F.; Gaxiola, M.; López-Otín, C.; Selman, M.; Pardo, A. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy 2015, 11, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1–Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [PubMed]
- Schläfli, A.M.; Adams, O.; Galván, J.A.; Gugger, M.; Savic, S.; Bubendorf, L.; Schmid, R.A.; Becker, K.-F.; Tschan, M.P.; Langer, R.; et al. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 2016, 7, 39544–39555. [Google Scholar] [CrossRef]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Choi, A.M.; Choi, M.E. Autophagy in chronic lung disease. Prog. Mol. Biol. Transl. Sci. 2020, 172, 135–156. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Fairlie, W.D.; Lee, E.F. BECLIN1: Protein Structure, Function and Regulation. Cells 2021, 10, 1522. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, Y.; Zhang, P.; Lin, P.; Xie, N.; Wu, M. Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases. Cells 2019, 8, 123. [Google Scholar] [CrossRef]
- Pavlinov, I.; Salkovski, M.; Aldrich, L.N. Beclin 1–ATG14L Protein–Protein Interaction Inhibitor Selectively Inhibits Autophagy through Disruption of VPS34 Complex I. J. Am. Chem. Soc. 2020, 142, 8174–8182. [Google Scholar] [CrossRef]
- Mathur, P.; Santos, C.D.B.; Lachuer, H.; Patat, J.; Latgé, B.; Radvanyi, F.; Goud, B.; Schauer, K. Transcription factor EB regulates phosphatidylinositol-3-phosphate levels that control lysosome positioning in the bladder cancer model. Commun. Biol. 2023, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Theofani, E.; Xanthou, G. Autophagy: A Friend or Foe in Allergic Asthma? Int. J. Mol. Sci. 2021, 22, 6314. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To Be or Not to Be? How Selective Autophagy and Cell Death Govern Cell Fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Simon, A.G.; Tolkach, Y.; Esser, L.K.; Ellinger, J.; Stöhr, C.; Ritter, M.; Wach, S.; Taubert, H.; Stephan, C.; Hartmann, A.; et al. Mitophagy-associated genes PINK1 and PARK2 are independent prognostic markers of survival in papillary renal cell carcinoma and associated with aggressive tumor behavior. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Zhang, F.; Yu, L.-P.; Mu, J.-K.; Yang, Y.-Q.; Yu, J.; Yang, X.-X. Targeting PINK1 Using Natural Products for the Treatment of Human Diseases. BioMed Res. Int. 2021, 2021, 4045819. [Google Scholar] [CrossRef]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hou, H.; Ma, G.; Ma, Q.; Li, N.; Zhang, L.; Dong, C.; Cao, M.; Tam, K.Y.; Ying, Z.; et al. The interaction between E3 ubiquitin ligase Parkin and mitophagy receptor PHB2 links inner mitochondrial membrane ubiquitination to efficient mitophagy. J. Biol. Chem. 2022, 298, 102704. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ahmad, S.; Ahmad, T.; Ali, S.; Syed, M.A. Mitochondrial dynamics and mitophagy in lung disorders. Life Sci. 2021, 284, 119876. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of Serines 17 and 24 in the LC3-interacting Region of Bnip3 Determines Pro-survival Mitophagy versus Apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Y.; Hou, X.; Tao, Z.; Ren, H.; Wang, G. Dependence of PINK1 accumulation on mitochondrial redox system. Aging Cell 2020, 19, e13211. [Google Scholar] [CrossRef]
- Li, G.; Li, J.; Shao, R.; Zhao, J.; Chen, M. FUNDC1: A Promising Mitophagy Regulator at the Mitochondria-Associated Membrane for Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 788634. [Google Scholar] [CrossRef]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Oyang, L.; Li, J.; Jiang, X.; Lin, J.; Xia, L.; Yang, L.; Tan, S.; Wu, N.; Han, Y.; Yang, Y.; et al. The function of prohibitins in mitochondria and the clinical potentials. Cancer Cell Int. 2022, 22, 343. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Bhandari, V. Targeting mitochondrial dysfunction in lung diseases: Emphasis on mitophagy. Front. Physiol. 2013, 4, 384. [Google Scholar] [CrossRef]
- Carlier, F.M.; de Fays, C.; Pilette, C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front. Physiol. 2021, 12, 691227. [Google Scholar] [CrossRef]
- Herminghaus, A.; Kozlov, A.V.; Szabó, A.; Hantos, Z.; Gylstorff, S.; Kuebart, A.; Aghapour, M.; Wissuwa, B.; Walles, T.; Walles, H.; et al. A Barrier to Defend—Models of Pulmonary Barrier to Study Acute Inflammatory Diseases. Front. Immunol. 2022, 13, 895100. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.A.; Fisher, A.J.; Borthwick, L.A. The Role of Epithelial Damage in the Pulmonary Immune Response. Cells 2021, 10, 2763. [Google Scholar] [CrossRef] [PubMed]
- Güney, T.; Herranz, A.M.; Mumby, S.; Dunlop, I.E.; Adcock, I.M. Epithelial–stromal cell interactions and extracellular matrix mechanics drive the formation of airway-mimetic tubular morphology in lung organoids. iScience 2021, 24, 103061. [Google Scholar] [CrossRef]
- Abohalaka, R. Crosstalk of Airway Smooth Muscle and Epithelial Cells in Chronic Lung Diseases. Preprints 2022, 2022120153. [Google Scholar] [CrossRef]
- Parker, D.; Prince, A. Innate Immunity in the Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and Function of the Tight Junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight Junction Pore and Leak Pathways: A Dynamic Duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [PubMed]
- Rusu, A.D.; Georgiou, M. The multifarious regulation of the apical junctional complex. Open Biol. 2020, 10, 190278. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, H.; Ocalan, M.; Yilmaz, O. E-Cadherin: An Important Functional Molecule at Respiratory Barrier Between Defence and Dysfunction. Front. Physiol. 2021, 12, 720227. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Akimoto, K.; Homma, T.; Tanaka, A.; Sagara, H. Airway Epithelial Dysfunction in Asthma: Relevant to Epidermal Growth Factor Receptors and Airway Epithelial Cells. J. Clin. Med. 2020, 9, 3698. [Google Scholar] [CrossRef]
- Lillehoj, E.P.; Kato, K.; Lu, W.; Kim, K.C. Cellular and molecular biology of airway mucins. Int. Rev. Cell Mol. Biol. 2013, 303, 139–202. [Google Scholar] [CrossRef]
- Wagner, C.; Wheeler, K.; Ribbeck, K. Mucins and Their Role in Shaping the Functions of Mucus Barriers. Annu. Rev. Cell Dev. Biol. 2018, 34, 189–215. [Google Scholar] [CrossRef]
- Bayarri, M.A.; Milara, J.; Estornut, C.; Cortijo, J. Nitric Oxide System and Bronchial Epithelium: More Than a Barrier. Front. Physiol. 2021, 12, 687381. [Google Scholar] [CrossRef]
- Sears, P.R.; Thompson, K.; Knowles, M.R.; Davis, C.W. Human airway ciliary dynamics. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L170–L183. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Roth, M. Grundlagen von Asthma und COPD [Fundamentals of chronic inflammatory lung diseases (asthma, COPD, fibrosis)]. Ther. Umsch. 2014, 71, 258–261. (In German) [Google Scholar] [CrossRef]
- Ghigo, A.; Prono, G.; Riccardi, E.; De Rose, V. Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 1952. [Google Scholar] [CrossRef]
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N.; Hegedűs, K.; Takáts, S.; Boda, A.; Jipa, A.; Nagy, P.; Varga, K.; et al. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Davies, M.P.A.; Wilson, C.M. The Role of Autophagy in Lung Disease. In Autophagy in Current Trends in Cellular Physiology and Pathology; Intech: Vienna, Austria, 2016. [Google Scholar] [CrossRef]
- Christenson, S.A.; Smith, B.M.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R. Pharmacological Therapy of COPD. Chest 2018, 154, 1404–1415. [Google Scholar] [CrossRef]
- Postma, D.S.; Bush, A.; van den Berge, M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet 2015, 385, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Q.; Zheng, R. The interplay between oxidative stress and autophagy in chronic obstructive pulmonary disease. Front. Physiol. 2022, 13, 1004275. [Google Scholar] [CrossRef]
- Wen, W.; Yu, G.; Liu, W.; Gu, L.; Chu, J.; Zhou, X.; Liu, Y.; Lai, G. Silencing FUNDC1 alleviates chronic obstructive pulmonary disease by inhibiting mitochondrial autophagy and bronchial epithelium cell apoptosis under hypoxic environment. J. Cell. Biochem. 2019, 120, 17602–17615. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Sun, J.; Mohammadtursun, N.; Hu, Z.; Li, Q.; Zhao, Z.; Zhang, H.; Dong, J. Dual role of autophagy/mitophagy in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2019, 56, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nature 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nature 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Bodas, M.; Silverberg, D.; Walworth, K.; Brucia, K.; Vij, N. Augmentation of S-Nitrosoglutathione Controls Cigarette Smoke-Induced Inflammatory–Oxidative Stress and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis by Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function. Antioxid. Redox Signal. 2017, 27, 433–451. [Google Scholar] [CrossRef]
- Bodas, M.; Patel, N.; Silverberg, D.; Walworth, K.; Vij, N.; Brucia, K.; Leitner, L.M.; Wilson, R.J.; Yan, Z.; Gödecke, A.; et al. Master Autophagy Regulator Transcription Factor EB Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease–Emphysema Pathogenesis. Antioxid. Redox Signal. 2017, 27, 150–167. [Google Scholar] [CrossRef]
- Bodas, M.; Vij, N. Augmenting autophagy for prognosis based intervention of COPD-pathophysiology. Respir. Res. 2017, 18, 83. [Google Scholar] [CrossRef]
- Shivalingappa, P.C.; Hole, R.; Van Westphal, C.; Vij, N.; Bisio, H.; Bonilla, M.; Manta, B.; Graña, M.; Salzman, V.; Aguilar, P.S.; et al. Airway Exposure to E-Cigarette Vapors Impairs Autophagy and Induces Aggresome Formation. Antioxid. Redox Signal. 2016, 24, 186–204. [Google Scholar] [CrossRef]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of Cigarette Smoke–Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease–Emphysema Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Chichger, H.; Rounds, S.; Harrington, E.O. Endosomes and Autophagy: Regulators of Pulmonary Endothelial Cell Homeostasis in Health and Disease. Antioxid. Redox Signal. 2019, 31, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Fu, Z. Role of autophagy in lung diseases and ageing. Eur. Respir. Rev. 2022, 31, 220134. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. 2015, 4, 215–225. [Google Scholar] [CrossRef]
- Yeganeh, B.; Lee, J.; Ermini, L.; Lok, I.; Ackerley, C.; Post, M. Autophagy is required for lung development and morphogenesis. J. Clin. Investig. 2019, 129, 2904–2919. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 18. [Google Scholar] [CrossRef]
- Ryter, S.W.; Rosas, I.O.; Owen, C.A.; Martinez, F.J.; Choi, M.E.; Lee, C.G.; Elias, J.A.; Choi, A.M.K. Mitochondrial Dysfunction as a Pathogenic Mediator of Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. S4), S266–S272. [Google Scholar] [CrossRef]
- Schellens, J.P.; Vreeling-Sindelárová, H.; Plomp, P.J.; Meijer, A.J. Hepatic autophagy and intracellular ATP a morphometric study. Exp. Cell Res. 1988, 177, 103–108. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowsk, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Erratum: Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1608. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Leist, M.; Single, B.; Castoldi, A.F.; Kühnle, S.; Nicotera, P. Intracellular Adenosine Triphosphate (ATP) Concentration: A Switch in the Decision Between Apoptosis and Necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhou, J.-S.; Huang, H.-Q.; Li, Z.-Y.; Xu, X.-C.; Lai, T.-W.; Hu, Y.; Zhou, H.-B.; Chen, H.-P.; et al. MTOR Suppresses Cigarette Smoke–Induced Epithelial Cell Death and Airway Inflammation in Chronic Obstructive Pulmonary Disease. J. Immunol. 2018, 200, 2571–2580. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, G.; Yuan, S.; Tan, C.; Xie, J.; Ding, Y.; Lian, P.; Fu, L.; Hou, Q.; Xu, B.; et al. 14,15-Epoxyeicosatrienoic acid suppresses cigarette smoke condensate-induced inflammation in lung epithelial cells by inhibiting autophagy. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L970–L980. [Google Scholar] [CrossRef]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6–mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef]
- Zhou, J.-S.; Zhao, Y.; Zhou, H.-B.; Wang, Y.; Wu, Y.-F.; Li, Z.-Y.; Xuan, N.-X.; Zhang, C.; Hua, W.; Ying, S.-M.; et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L1042–L1052. [Google Scholar] [CrossRef]
- Ning, W.; Li, C.-J.; Kaminski, N.; Feghali-Bostwick, C.A.; Alber, S.M.; Di, Y.P.; Otterbein, S.L.; Song, R.; Hayashi, S.; Zhou, Z.; et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2004, 101, 14895–14900. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hu, J.; Wang, T.; Zhang, X.; Liu, L.; Wang, H.; Wu, Y.; Xu, D.; Wen, F. Silymarin attenuates cigarette smoke extract-induced inflammation via simultaneous inhibition of autophagy and ERK/p38 MAPK pathway in human bronchial epithelial cells. Sci. Rep. 2016, 6, 37751. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Lam, H.C.; Jin, Y.; Kim, H.-P.; Cao, J.; Lee, S.-J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M.K. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef]
- van Rijt, S.H.; Keller, I.E.; John, G.; Kohse, K.; Yildirim, A.; Eickelberg, O.; Meiners, S. Acute cigarette smoke exposure impairs proteasome function in the lung. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L814–L823. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Colley, T.; Baker, J.R.; Vuppussetty, C.; Kono, Y.; Clarke, C.; Tooze, S.; Johansen, T.; Barnes, P.J. Bicaudal D1 impairs autophagosome maturation in chronic obstructive pulmonary disease. FASEB BioAdv. 2019, 1, 688–705. [Google Scholar] [CrossRef] [PubMed]
- An, C.H.; Wang, X.M.; Lam, H.C.; Ifedigbo, E.; Washko, G.R.; Ryter, S.W.; Choi, A.M.K.; Haw, T.J.; Starkey, M.R.; Pavlidis, S.; et al. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L748–L757. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Kim, H.P.; Sciurba, F.C.; Lee, S.-J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 Regulates Autophagy in Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, H.; Strulovici-Barel, Y.; Al-Hijji, M.; Ou, X.; Salit, J.; Walters, M.S.; Staudt, M.R.; Kaner, R.J.; Crystal, R.G. Role of OSGIN1 in mediating smoking-induced autophagy in the human airway epithelium. Autophagy 2017, 13, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.-H.; Cheng, S.-L.; Chung, K.-P.; Kuo, M.Y.-P.; Yeh, C.-C.; Chang, B.-E.; Lu, H.-H.; Wang, H.-C.; Yu, C.-J. Elastase induces lung epithelial cell autophagy through placental growth factor. Autophagy 2014, 10, 1509–1521. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, X.; Hu, D. Mitochondrial alterations during oxidative stress in chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 13, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Samitas, K.; Zervas, E.; Gaga, M. T2-low asthma. Curr. Opin. Pulm. Med. 2017, 23, 48–55. [Google Scholar] [CrossRef]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-N.; Suh, D.-H.; Trinh, H.K.T.; Chwae, Y.-J.; Park, H.-S.; Shin, Y.S. The role of autophagy in allergic inflammation: A new target for severe asthma. Exp. Mol. Med. 2016, 48, e243. [Google Scholar] [CrossRef]
- Lv, X.; Li, K.; Hu, Z. Asthma and autophagy. Adv. Exp. Med. Biol. 2020, 1207, 581–584. [Google Scholar]
- Suzuki, Y.; Aono, Y.; Akiyama, N.; Horiike, Y.; Naoi, H.; Horiguchi, R.; Shibata, K.; Hozumi, H.; Karayama, M.; Furuhashi, K.; et al. Involvement of autophagy in exacerbation of eosinophilic airway inflammation in a murine model of obese asthma. Autophagy 2022, 18, 2216–2228. [Google Scholar] [CrossRef]
- Murai, H.; Okazaki, S.; Hayashi, H.; Kawakita, A.; Hosoki, K.; Yasutomi, M.; Sur, S.; Ohshima, Y. Alternaria extract activates autophagy that induces IL-18 release from airway epithelial cells. Biochem. Biophys. Res. Commun. 2015, 464, 969–974. [Google Scholar] [CrossRef]
- Ghavami, S.; Cunnington, R.H.; Gupta, S.; Yeganeh, B.; Filomeno, K.L.; Freed, D.H.; Chen, S.; Klonisch, T.; Halayko, A.J.; Ambrose, E.; et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015, 6, e1696. [Google Scholar] [CrossRef]
- McAlinden, K.D.; Deshpande, D.A.; Ghavami, S.; Xenaki, D.; Sohal, S.S.; Oliver, B.G.; Haghi, M.; Sharma, P. Autophagy Activation in Asthma Airways Remodeling. Am. J. Respir. Cell Mol. Biol. 2019, 60, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gupta, J.; Jyothula, S.S.S.K.; Kovacic, M.B.; Myers, J.M.B.; Patterson, T.L.; Ericksen, M.B.; He, H.; Gibson, A.M.; Baye, T.M.; et al. Functional Variant in the Autophagy-Related 5 Gene Promotor is Associated with Childhood Asthma. PLoS ONE 2012, 7, e33454. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.H.; Chouiali, F.; Tse, S.M.; Litonjua, A.A.; Hussain, S.N.; Baglole, C.J.; Eidelman, D.H.; Olivenstein, R.; Martin, J.G.; Weiss, S.T.; et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J. Allergy Clin. Immunol. 2012, 129, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Le Pham, D.; Kim, S.-H.; Losol, P.; Yang, E.-M.; Shin, Y.S.; Ye, Y.-M.; Park, H.-S. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. Korean J. Intern. Med. 2016, 31, 375–385. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Poon, A.H.; Choy, D.F.; Chouiali, F.; Ramakrishnan, R.K.; Mahboub, B.; Audusseau, S.; Mogas, A.; Harris, J.M.; Arron, J.R.; Laprise, C.; et al. Increased Autophagy-Related 5 Gene Expression Is Associated with Collagen Expression in the Airways of Refractory Asthmatics. Front. Immunol. 2017, 8, 355. [Google Scholar] [CrossRef]
- Weng, C.-M.; Wang, C.-H.; Lee, M.-J.; He, J.-R.; Huang, H.-Y.; Chao, M.-W.; Chung, K.F.; Kuo, H.-P. Aryl hydrocarbon receptor activation by diesel exhaust particles mediates epithelium-derived cytokines expression in severe allergic asthma. Allergy 2018, 73, 2192–2204. [Google Scholar] [CrossRef]
- Dickinson, J.D.; Alevy, Y.; Malvin, N.P.; Patel, K.K.; Gunsten, S.P.; Holtzman, M.J.; Stappenbeck, T.S.; Brody, S.L. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy 2016, 12, 397–409. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Sachdeva, K.; Do, D.C.; Zhang, Y.; Hu, X.; Chen, J.; Gao, P. Environmental Exposures and Asthma Development: Autophagy, Mitophagy, and Cellular Senescence. Front. Immunol. 2019, 10, 2787. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial Dysfunction in Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef] [PubMed]
- Malsin, E.S.; Kamp, D.W. The mitochondria in lung fibrosis: Friend or foe? Transl. Res. 2018, 202, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Fan, P.; Xie, X.-H.; Chen, C.-H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.-T. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol. 2019, 38, 10–22. [Google Scholar] [CrossRef]
- Wang, H.-T.; Lin, J.-H.; Yang, C.-H.; Haung, C.-H.; Weng, C.-W.; Lin, A.M.-Y.; Lo, Y.-L.; Chen, W.-S.; Tang, M.-S. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget 2017, 8, 70406–70421. [Google Scholar] [CrossRef]
- Qu, J.; Do, D.C.; Zhou, Y.; Luczak, E.; Mitzner, W.; Anderson, M.E.; Gao, P. Oxidized CaMKII promotes asthma through the activation of mast cells. J. Clin. Investig. 2017, 2, e90139. [Google Scholar] [CrossRef]
- Qu, J.; Li, Y.; Zhong, W.; Gao, P.; Hu, C. Recent developments in the role of reactive oxygen species in allergic asthma. J. Thorac. Dis. 2017, 9, E32–E43. [Google Scholar] [CrossRef]
- Sanders, P.N.; Koval, O.M.; Jaffer, O.A.; Prasad, A.M.; Businga, T.R.; Scott, J.A.; Hayden, P.J.; Luczak, E.D.; Dickey, D.D.; Allamargot, C.; et al. CaMKII Is Essential for the Proasthmatic Effects of Oxidation. Sci. Transl. Med. 2013, 5, 195ra97. [Google Scholar] [CrossRef]
- Abdala-Valencia, H.; Earwood, J.; Bansal, S.; Jansen, M.; Babcock, G.; Garvy, B.; Wills-Karp, M.; Cook-Mills, J.M.; Campbell, J.; Morales-Nebreda, L.; et al. Nonhematopoietic NADPH oxidase regulation of lung eosinophilia and airway hyperresponsiveness in experimentally induced asthma. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L1111–L1125. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: A Target for Drugs and Natural Products With Effects on Both Diabetes and Cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR Signaling in Tissue Regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Morris, M.; Hockey, H.-U.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Schlemmer, F.; Boyer, L.; Soumagne, T.; Ridoux, A.; Chouaid, C.; Maitre, B.; Lanone, S.; Adnot, S.; Audureau, E.; Boczkowski, J. Beclin1 circulating levels and accelerated aging markers in COPD. Cell Death Dis. 2018, 9, 156. [Google Scholar] [CrossRef]
- Knobloch, J.; Sibbing, B.; Jungck, D.; Lin, Y.; Urban, K.; Stoelben, E.; Strauch, J.; Koch, A. Resveratrol Impairs the Release of Steroid-Resistant Inflammatory Cytokines from Human Airway Smooth Muscle Cells in Chronic Obstructive Pulmonary Disease. Experiment 2010, 335, 788–798. [Google Scholar] [CrossRef]
- Wood, L.G.; Wark, P.A.; Garg, M.L.; Wang, D.; Li, S.-P.; Fu, J.-S.; Bai, L.; Guo, L.; Sadarani, B.N.; Majumdar, A.S.; et al. Antioxidant and Anti-Inflammatory Effects of Resveratrol in Airway Disease. Antioxid. Redox Signal. 2010, 13, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Nediani, C.; Ruzzolini, J.; Romani, A.; Calorini, L. Oleuropein, a Bioactive Compound from Olea europaea L., as a Potential Preventive and Therapeutic Agent in Non-Communicable Diseases. Antioxidants 2019, 8, 578. [Google Scholar] [CrossRef] [PubMed]
- Son, E.S.; Kim, S.-H.; Ryter, S.W.; Yeo, E.-J.; Kyung, S.Y.; Kim, Y.J.; Jeong, S.H.; Lee, C.S.; Park, J.-W. Quercetogetin protects against cigarette smoke extract-induced apoptosis in epithelial cells by inhibiting mitophagy. Toxicol. Vitr. 2018, 48, 170–178. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, W.; Wang, J.; Xie, Y.; Wang, W. Puerarin inhibits FUNDC1-mediated mitochondrial autophagy and CSE-induced apoptosis of human bronchial epithelial cells by activating the PI3K/AKT/mTOR signaling pathway. Aging 2022, 14, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.Y.; Kim, Y.J.; Son, E.S.; Jeong, S.H.; Park, J.-W. The Phosphodiesterase 4 Inhibitor Roflumilast Protects against Cigarette Smoke Extract-Induced Mitophagy-Dependent Cell Death in Epithelial Cells. Tuberc. Respir. Dis. 2018, 81, 138–147. [Google Scholar] [CrossRef]
- Sunata, K.; Kabata, H.; Kuno, T.; Takagi, H.; So, M.; Masaki, K.; Fukunaga, K. The effect of statins for asthma. A systematic review and meta-analysis. J. Asthma 2021, 59, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Modulatory effects of statins on the autophagy: A therapeutic perspective. J. Cell. Physiol. 2019, 235, 3157–3168. [Google Scholar] [CrossRef]
- Thomson, N.C.; Charron, C.E.; Chaudhuri, R.; Spears, M.; Ito, K.; McSharry, C. Atorvastatin in combination with inhaled beclometasone modulates inflammatory sputum mediators in smokers with asthma. Pulm. Pharmacol. Ther. 2015, 31, 1–8. [Google Scholar] [CrossRef]
- Maneechotesuwan, K.; Kasetsinsombat, K.; Wongkajornsilp, A.; Barnes, P.J. Role of autophagy in regulating interleukin-10 and the responses to corticosteroids and statins in asthma. Clin. Exp. Allergy 2021, 51, 1553–1565. [Google Scholar] [CrossRef]
- Maneechotesuwan, K.; Wongkajornsilp, A.; Adcock, I.M.; Barnes, P.J. Simvastatin Suppresses Airway IL-17 and Upregulates IL-10 in Patients With Stable COPD. Chest 2015, 148, 1164–1176. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DRUG | Autophagy Target |
|---|---|
| 3-methyladenine (3-MA) | PI3K inhibition |
| Rapamicyn | mTORC1 inhibition |
| Metformin | mTORC1 inhibition |
| Carbamazepine | PI3K inhibition |
| Chloroquine and hydroxychloroquine | inhibitor of toll-like receptors |
| Hydroxychloroquine | inhibitor of toll-like receptors |
| Tat-beclin1 | PI3K CIII complex |
| Resveratrol | AMPK activation |
| Oleuropein | AMPK activation |
| Quercetogetin | phospho-DRP-1 inhibition |
| Puerarin | FUNDC1 inhibition |
| Statin | Not clear |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albano, G.D.; Montalbano, A.M.; Gagliardo, R.; Profita, M. Autophagy/Mitophagy in Airway Diseases: Impact of Oxidative Stress on Epithelial Cells. Biomolecules 2023, 13, 1217. https://doi.org/10.3390/biom13081217
Albano GD, Montalbano AM, Gagliardo R, Profita M. Autophagy/Mitophagy in Airway Diseases: Impact of Oxidative Stress on Epithelial Cells. Biomolecules. 2023; 13(8):1217. https://doi.org/10.3390/biom13081217
Chicago/Turabian StyleAlbano, Giusy Daniela, Angela Marina Montalbano, Rosalia Gagliardo, and Mirella Profita. 2023. "Autophagy/Mitophagy in Airway Diseases: Impact of Oxidative Stress on Epithelial Cells" Biomolecules 13, no. 8: 1217. https://doi.org/10.3390/biom13081217
APA StyleAlbano, G. D., Montalbano, A. M., Gagliardo, R., & Profita, M. (2023). Autophagy/Mitophagy in Airway Diseases: Impact of Oxidative Stress on Epithelial Cells. Biomolecules, 13(8), 1217. https://doi.org/10.3390/biom13081217