
A Comparison of Cell-Cell Interaction Prediction Tools Based on scRNA-seq Data



Abstract
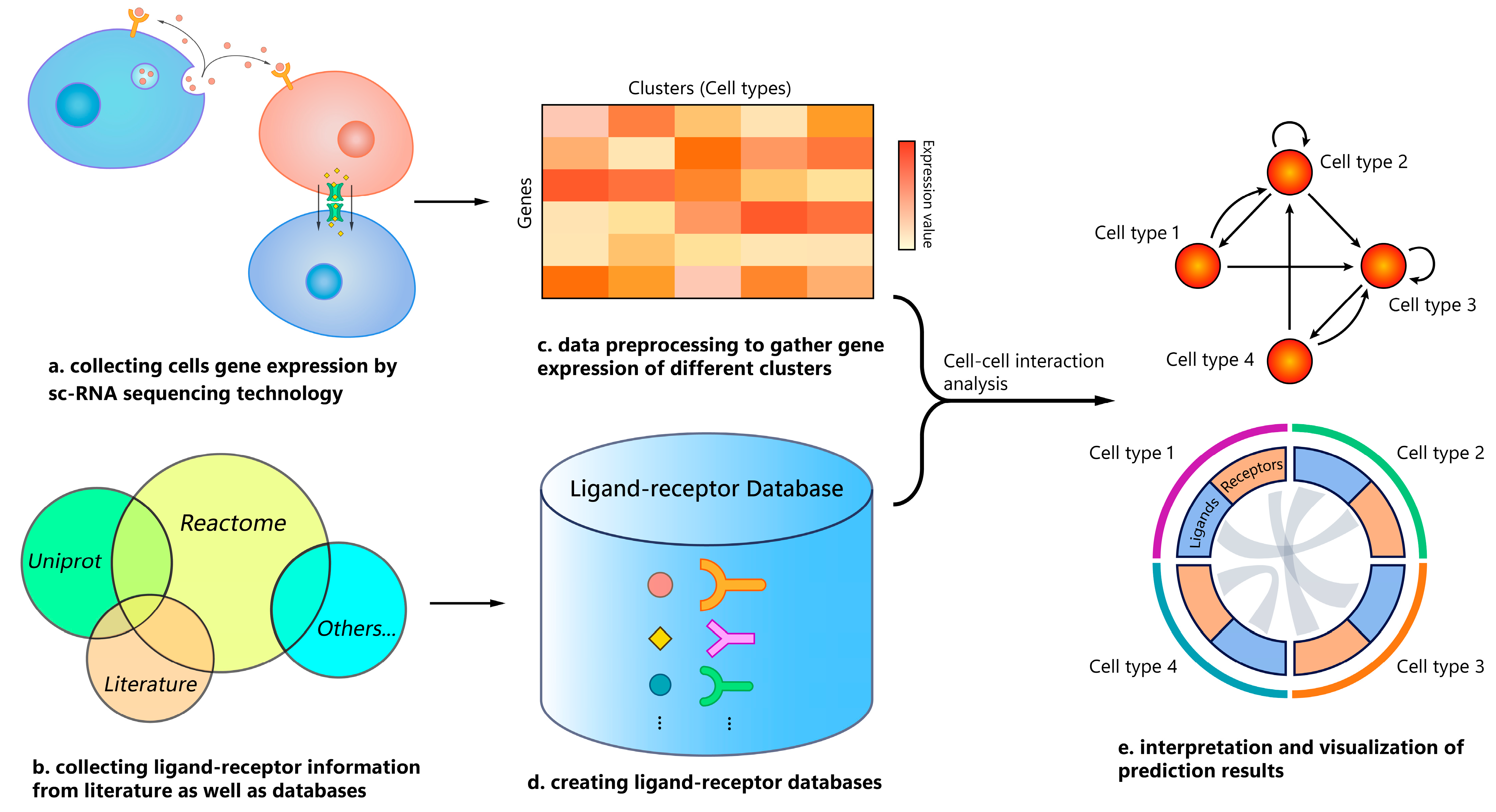
1. Introduction
2. Materials and Methods
2.1. Tool Selection
2.2. Gold Standard
2.3. scRNA-seq Data Pre-Processing
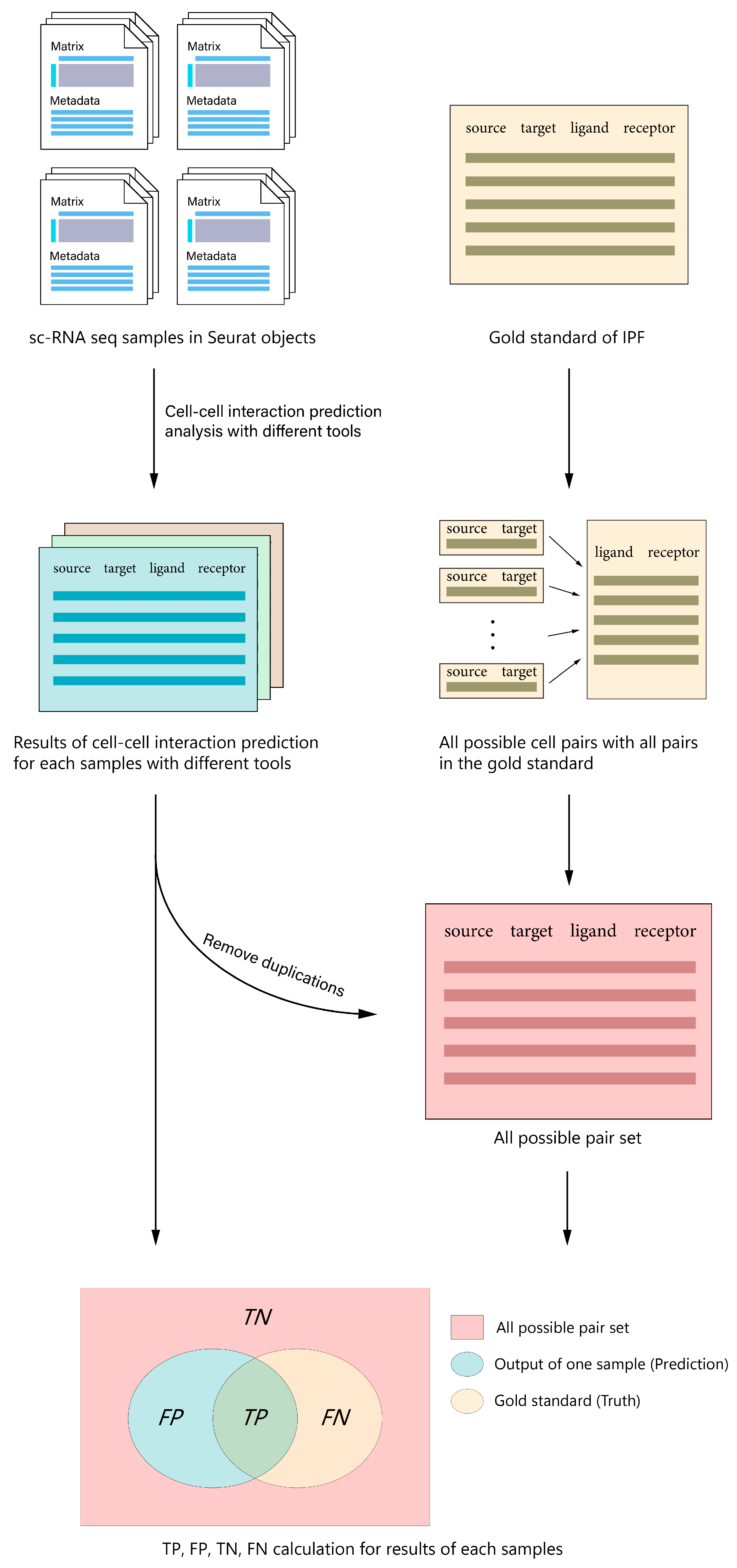
2.4. Benchmark Workflow
2.4.1. Run CCI Prediction Using All Tools, and Collect Prediction Results and Computation Time Information
2.4.2. Convert Different Outputs to a Unified Format
2.4.3. Build Set of All Possible Interaction Pairs
2.4.4. Calculate True and False Positives, and True and False Negatives
2.4.5. Compute Performance Metrics
2.4.6. Hardware and Software
3. Results
3.1. General Setup
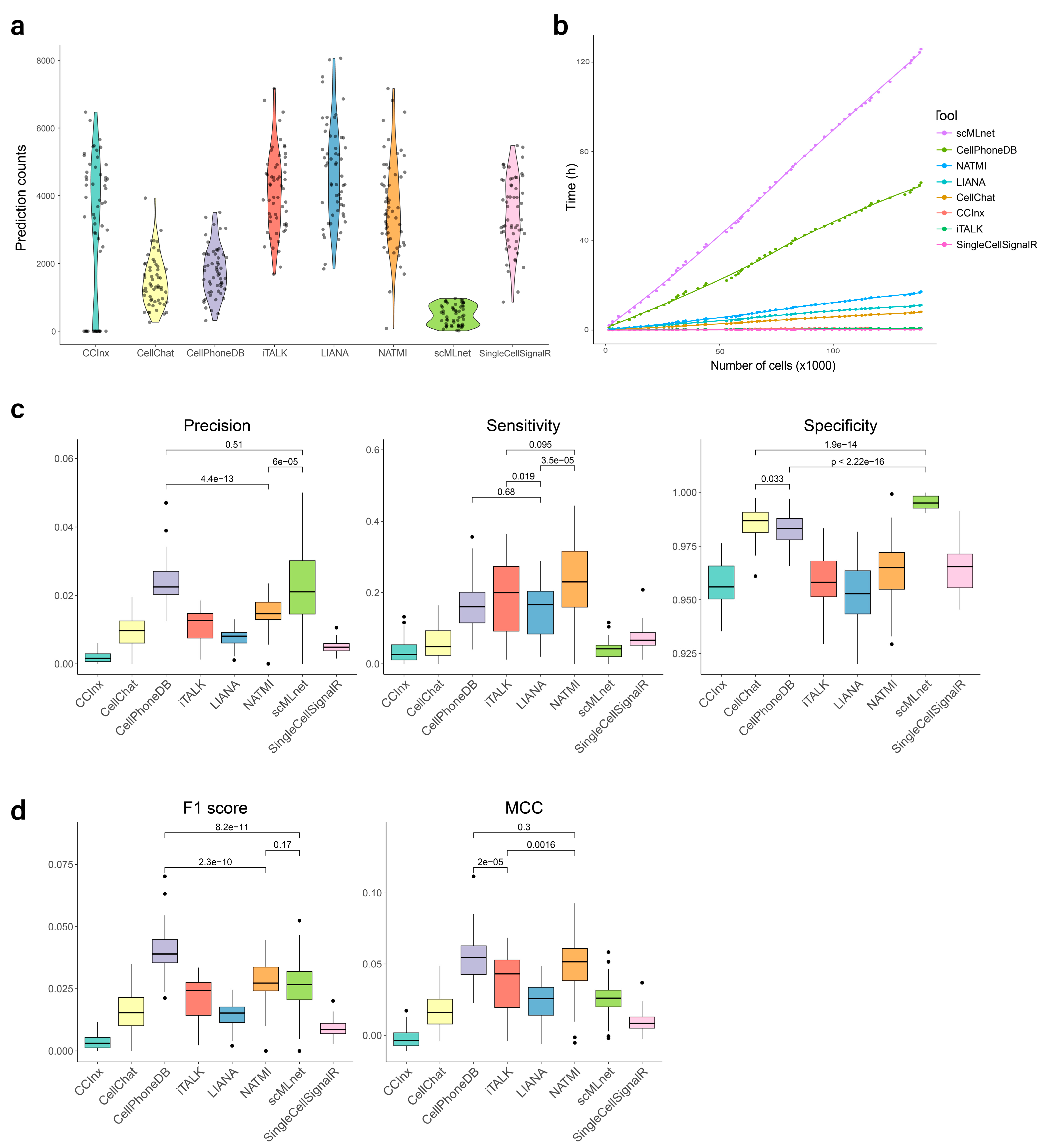
3.2. Predicted Interaction Counts
3.3. Processing Time
3.4. Evaluation of Prediction Performance
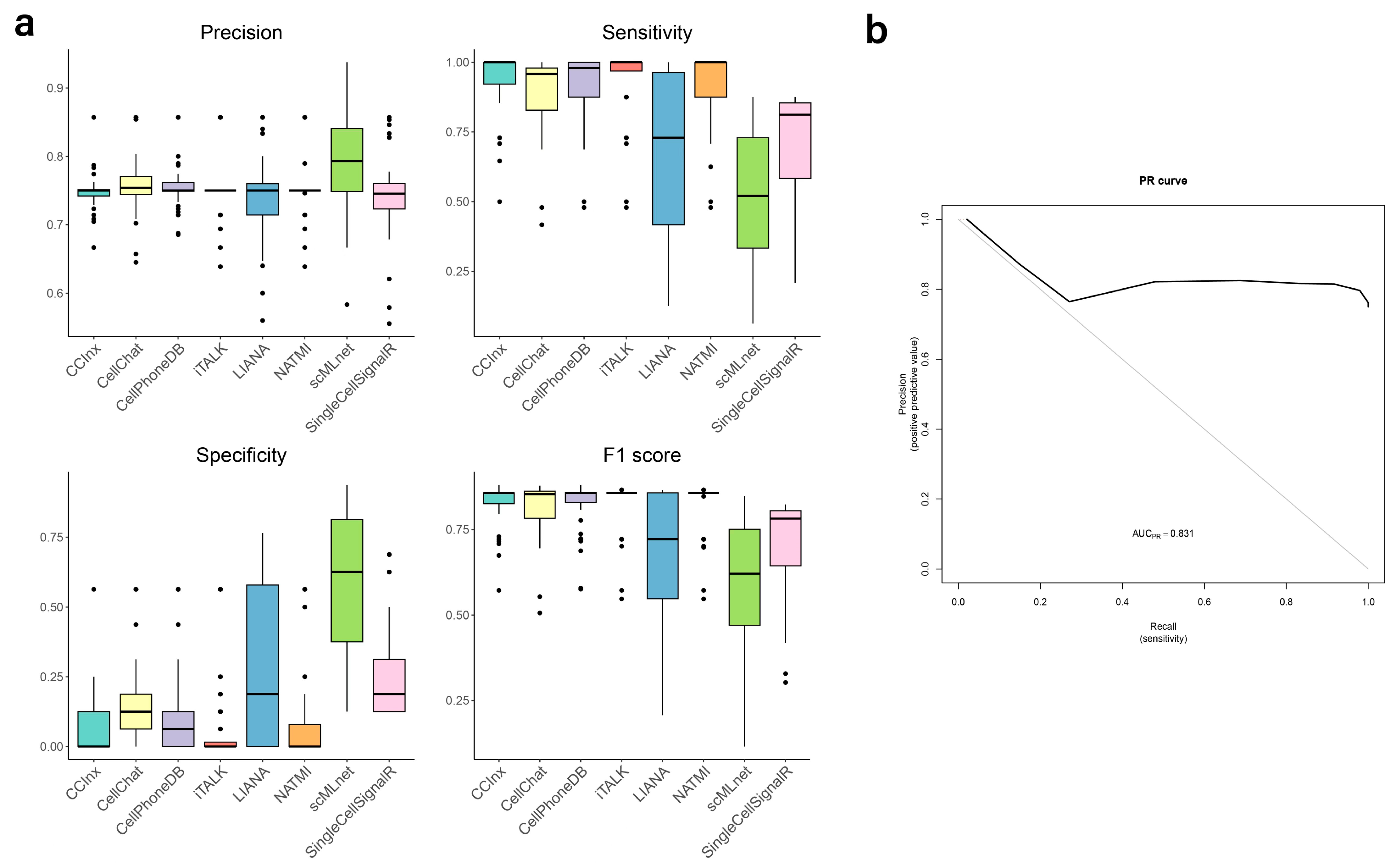
3.5. Validation of Predictions
4. Discussion
4.1. Tool Recommendations
4.2. Comparing Our Study to Other Benchmarks
4.3. General Limitations of the Tools and Alternatives to the scRNA-seq Approach
- (1)
- The fact that neither up-regulation nor expression correlation of the “communicating” molecules necessarily imply that cells are interacting [6].
- (2)
- Communication and structural interactions mainly occur to the protein level, but we use sc-transcriptomics data because sc-proteomics technologies still don’t have the same level of development. However, the assumption that RNA levels reflect protein levels of ligands and receptors is not entirely correct. RNA and protein levels can differ due to multiple post-transcriptional and post-translational processes [1].
- (3)
- Protein-protein interactions are dependent on protein concentration, which is something that most current methods cannot evaluate [1].
- (4)
- Protein abundance is not enough to infer protein-protein interaction strength. Another aspect to consider is the existence of post-translational modifications such as glycosilation. Many ligands and receptors are glycoproteins and glycosylation changes their affinity; therefore, glycomic and other omic data could be added under a multi-omic approach [1,6].
- (5)
- All databases are highly incomplete and biased towards some areas of interest.
- (6)
- (7)
- The previous tools do not consider that cellular communication can also be metabolite-mediated. However, recent papers include detailed reviews of metabolite sensing and signaling [33], single-cell mass spectrometry studies of the metabolic profiles of cell-cell interactions [34], and tools for predicting metabolite-mediated CCIs using scRNA-seq data (based on the expression levels of the metabolite-producing enzymes and the metabolite sensors) [35].
- (8)
- Communicating proteins are limited to ligands and receptors. In the future, proteins involved in juxtacrine interactions, such as cell adhesion proteins and gap junctions, extracellular matrix proteins, endocrine signals, and other proteins involved in cell communication, should also be included [13].
- (9)
- Some CCIs are not as simple as one source, one ligand, one receptor, and one target, but they form complex communication pathways instead.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D.; et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044 e1018. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Zhang, J.; Li, W.; Xiong, Z.; Zhu, J.; Ren, X.; Wang, S.; Kuang, H.; Lin, X.; Mora, A.; Li, X. PDGF-D-induced immunoproteasome activation and cell-cell interactions. Comput. Struct. Biotechnol. J. 2023, 21, 2405–2418. [Google Scholar] [CrossRef]
- Almet, A.A.; Cang, Z.; Jin, S.; Nie, Q. The landscape of cell-cell communication through single-cell transcriptomics. Curr. Opin. Syst. Biol. 2021, 26, 12–23. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Xie, B.; Jiang, Q.; Mora, A.; Li, X. Automatic cell type identification methods for single-cell RNA sequencing. Comput. Struct. Biotechnol. J. 2021, 19, 5874–5887. [Google Scholar] [CrossRef]
- Lv, B.; Xu, X.; Zhang, X.; Qi, L.; He, W.; Wang, L.; Chen, X.; Peng, L.; Xue, J.; Ji, Y.; et al. Activation of Blood Vessel Development in Endometrial Stromal Cells In Vitro Cocultured with Human Peri-Implantation Embryos Revealed by Single-Cell RNA-Seq. Life 2021, 11, 367. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Weber, L.M.; Saelens, W.; Cannoodt, R.; Soneson, C.; Hapfelmeier, A.; Gardner, P.P.; Boulesteix, A.L.; Saeys, Y.; Robinson, M.D. Essential guidelines for computational method benchmarking. Genome Biol. 2019, 20, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, D.; Wang, C. Evaluation of cell-cell interaction methods by integrating single-cell RNA sequencing data with spatial information. Genome Biol. 2022, 23, 218. [Google Scholar] [CrossRef]
- Dimitrov, D.; Turei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Lu, Y.; Guo, H.; Li, D.; Jiang, J.; Yan, L.; Gao, J.; Ren, Y.; Zhao, X.; Hou, L. CITEdb: A manually curated database of cell-cell interactions in human. Bioinformatics 2022, 38, 5144–5148. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G. iTalk: An R package to characterize and illustrate intercellular communication. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cabello-Aguilar, S.; Alame, M.; Kon-Sun-Tack, F.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020, 48, e55. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, J.; Wu, Z.; Sun, X. Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scMLnet with an application to COVID-19. Brief Bioinform. 2021, 22, 988–1005. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Hou, R.; Denisenko, E.; Ong, H.T.; Ramilowski, J.A.; Forrest, A.R.R. Predicting cell-to-cell communication networks using NATMI. Nat. Commun. 2020, 11, 5011. [Google Scholar] [CrossRef]
- Xie, C.; Jauhari, S.; Mora, A. Popularity and performance of bioinformatics software: The case of gene set analysis. BMC Bioinform. 2021, 22, 191. [Google Scholar] [CrossRef]
- Kinoshita, T.; Goto, T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Barkat, M.; Moussa, S. Protein-Protein Negative Interaction Dataset: IEEE DataPort. 2022. Available online: https://ieee-dataport.org/documents/protein-protein-negative-interaction-dataset (accessed on 15 May 2023).
- Russell, R. Negative Protein-Protein Interactions 2012. Available online: http://www.russelllab.org/negatives/ (accessed on 15 May 2023).
- Wang, X.; Lin, L.; Lan, B.; Wang, Y.; Du, L.; Chen, X.; Li, Q.; Liu, K.; Hu, M.; Xue, Y.; et al. IGF2R-initiated proton rechanneling dictates an anti-inflammatory property in macrophages. Sci. Adv. 2020, 6, eabb7389. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, L.; Song, B.; Cui, Z.; Chen, G.; Yu, Z.; Song, B. Insulin-like Growth Factor-2 (IGF-2) in Fibrosis. Biomolecules 2022, 12, 1557. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Wang, Y.P.; Lei, Q.Y. Metabolite sensing and signaling in cell metabolism. Signal Transduct. Target. Ther. 2018, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Peng, Z.; Yang, Z. Metabolomics studies of cell-cell interactions using single cell mass spectrometry combined with fluorescence microscopy. Chem. Sci. 2022, 13, 6687–6695. [Google Scholar] [CrossRef]
- Zheng, R.; Zhang, Y.; Tsuji, T.; Gao, X.; Wagner, A.; Yosef, N. MEBOCOST: Metabolite-mediated Cell Communication Modeling by Single Cell Transcriptome. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Reyes-Robles, T.; Fadeyi, O.O.; Oslund, R.C. Strategies for monitoring cell-cell interactions. Nat. Chem. Biol. 2021, 17, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Murray, C.W.; Linde, I.L.; Kramer, N.J.; Lyu, Z.; Tsai, M.K.; Chen, L.C.; Cai, H.; Gitler, A.D.; Engleman, E.; et al. A versatile system to record cell-cell interactions. eLife 2020, 9, e61080. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef]
- Giladi, A.; Cohen, M.; Medaglia, C.; Baran, Y.; Li, B.; Zada, M.; Bost, P.; Blecher-Gonen, R.; Salame, T.M.; Mayer, J.U.; et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 2020, 38, 629–637. [Google Scholar] [CrossRef]
- Tsuyuzaki, K.; Ishii, M.; Nikaido, I. Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data. bioRxiv 2019, 566182. [Google Scholar]
- Armingol, E.; Ghaddar, A.; Joshi, C.J.; Baghdassarian, H.; Shamie, I.; Chan, J.; Her, H.L.; Berhanu, S.; Dar, A.; Rodriguez-Armstrong, F.; et al. Inferring a spatial code of cell-cell interactions across a whole animal body. PLoS Comput. Biol. 2022, 18, e1010715. [Google Scholar] [CrossRef]
- Tyler, S.R.; Rotti, P.G.; Sun, X.; Yi, Y.; Xie, W.; Winter, M.C.; Flamme-Wiese, M.J.; Tucker, B.A.; Mullins, R.F.; Norris, A.W.; et al. PyMiner finds gene and autocrine-paracrine networks from human islet scRNA-seq. Cell Rep. 2019, 26, 1951–1964, e1958. [Google Scholar] [CrossRef]
- Wang, S.; Karikomi, M.; MacLean, A.L.; Nie, Q. Cell lineage and communication network inference via optimization for single-cell transcriptomics. Nucleic Acids Res. 2019, 47, e66. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Peng, T.; Gao, L.; Tan, K. CytoTalk: De novo construction of signal transduction networks using single-cell transcriptomic data. Sci. Adv. 2021, 7, eabf1356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Hu, X.; Wang, M.; Wang, J.; Zou, B.; Tan, P.; Cui, T.; Dou, Y.; Ning, L.; et al. CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. 2021, 49, 8520–8534. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Nie, Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 2020, 11, 2084. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Language | Method | Database | Output | Reference |
|---|---|---|---|---|---|
| CellChat (v.1.1.3) | R | CCI probabilities are calculated using the law of mass action, based on average expression of ligands/receptors by cell groups and cofactors | Curated ligand/receptor database including subunits and cofactors | CellChat object containing all the inferred CCIs with their probabilities | Jin et al. [15] |
| iTALK (v.0.1.0) | R | CCIs are identified by differentially expressed ligands/receptors between clusters | Manually curated ligand/receptor database | All CCIs with their mean ligand/receptor expression | Wang et al. [16] |
| SingleCellSignalR (v.1.8.0) | R | LRscore, a regularized score, is utilized to assess the confidence in predicted ligand-receptor interactions by controlling false positives | Curated ligand/receptor database with existing sources and manual additions | All CCIs with their regularized LRscore | Cabello-Aguilar et al. [17] |
| CCInx (v.0.5.1) | R | CCIs are predicted using ligand/receptor expression magnitude to rank nodes | Bader Lab’s ligand/receptor database | Node and edge list of all CCIs | https://github.com/BaderLab/CCInx (accessed on 19 November, 2021) |
| scMLnet (v.0.1.0) | R | Fisher’s exact test and correlation | Curated ligand/receptor/TF/target database with prior studies and databases | Ligand-receptor, receptor-TF and TF-target interactions | Cheng et al. [18] |
| CellPhoneDB (v.2.0.0) | Python | CCIs are enriched by empirical shuffling and statistical test | Curated ligand/receptor database (including complex information) with prior studies and databases | Table with information of ligand-receptor pairs by cell pairs, and their p-value | Efremova et al. [19] |
| NATMI | Python | Edge weights of CCIs are calculated by normalized expression level of ligands/receptors | connectomeDB2020 (a database of manually curated ligand-receptor pairs with literature support) | Table of ligand-receptor pairs, their expression level and edge weights | Hou et al. [20] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Z.; Li, X.; Mora, A. A Comparison of Cell-Cell Interaction Prediction Tools Based on scRNA-seq Data. Biomolecules 2023, 13, 1211. https://doi.org/10.3390/biom13081211
Xie Z, Li X, Mora A. A Comparison of Cell-Cell Interaction Prediction Tools Based on scRNA-seq Data. Biomolecules. 2023; 13(8):1211. https://doi.org/10.3390/biom13081211
Chicago/Turabian StyleXie, Zihong, Xuri Li, and Antonio Mora. 2023. "A Comparison of Cell-Cell Interaction Prediction Tools Based on scRNA-seq Data" Biomolecules 13, no. 8: 1211. https://doi.org/10.3390/biom13081211
APA StyleXie, Z., Li, X., & Mora, A. (2023). A Comparison of Cell-Cell Interaction Prediction Tools Based on scRNA-seq Data. Biomolecules, 13(8), 1211. https://doi.org/10.3390/biom13081211