1. Introduction
Synthetic messenger ribonucleic acid (mRNA) vaccines have become a promising alternative to conventional vaccine approaches because they can be quickly designed, tested, and mass produced [
1]. mRNA contains genetic information that encodes a specific protein of interest for expression by the organism [
2]. The use of mRNA to express specific proteins is a promising vaccine approach that avoids many safety issues associated with viral- or DNA-based systems. For example, when using a DNA vaccine there is always a risk it can cause a permanent change to the cell’s natural DNA sequence [
3]. Early studies using mRNA were difficult due to the relative instability of the molecule and the preponderance of mRNA degrading enzymes (RNAses) in the natural environment [
2]. Advances in RNA stability enhancement using modified bases and configurations indicate the greater applicability of mRNA for the purposes of gene therapy, vaccinations, and intra-cellular therapeutics [
4].
Human immunodeficiency virus (HIV) is one area where the potential for use of mRNA has been suggested [
5]. Although many strategies have been attempted for HIV vaccine development, none has been successfully established. The big challenges for HIV vaccine development are due to the vast diversity of HIV-1 and the formation of latent reservoir in cells that makes it very difficult to clear the virus using antiretroviral or immune therapy [
6]. Now, scientists aim to harness the power of mRNA technology to fight HIV [
7]. mRNA–based vaccine approaches have several advantages compared to classic protein-based vaccines. They are effectively expressed in the cytoplasm of a cell and can be targeted for any of the biological processes requiring mRNA as well as antigen protein production via HIV-specific mRNA. mRNAs are also non-specific activators of the innate immune system acting via Toll-like receptors (TLR) [
8]. Alternatively, extensive evidence indicates that permanent control of HIV is mediated not simply by antibodies but primarily by effective HIV-specific CD8+ T-cells. Induced HIV-specific CD8+ T-cell responses could limit both the transmission and establishment of persistent viral reservoirs [
9]. As a result, a T-cell-based HIV prevention strategy combined with ongoing B-cell vaccine approaches will be the primary consideration for future vaccine development. Recent technological advances have now overcome some of the issues with HIV vaccination and the delivery of mRNA–based vaccines. For example, the lipid nanoparticle (LNP)–based delivery of mRNA against infectious diseases and several types of cancer has demonstrated encouraging results in both animal models and in human use [
10].
Despite the advantages of the LNP delivery of mRNA vaccines, challenges remain, including how to increase the payload of mRNA within LNPs, how to ensure uptake by cells and efficient expression in the cytoplasm, how to reduce the LNP toxicity, and more. As a result, there is a necessity to formulate mRNA vaccines with effective delivery systems that have negligible toxicity but provide functional vaccination. In this research paper, we report on a short carbon nanotube-based novel delivery platform (NanoVac) [
11] that can deliver or co-deliver a broad range of mRNAs and encoding HIV-1 glycoproteins/peptides to antigen-presenting cells of the immune system via either intramuscular (IM) or intranasal (IN) routes. The NanoVac delivered vaccine candidates demonstrate robust systemic and cellular immune responses and potentially target specific mucosal sites, such as vaginal tissue, and thus could induce frontline immunity at the site of HIV-1 entry. This could help prevent the establishment and dissemination of an infection.
The HIV-1 vaccine candidate, RV144, is the only Phase 2b/3 clinical vaccine trial to date to demonstrate modest but significant efficacy in preventing HIV infection [
11]. Subsequent studies of specimens from RV144 volunteers indicated that the only primary, independent correlate of reduced risk was a robust level of non-neutralizing antibodies (Abs) binding to a recombinant protein containing the first and second variable regions (V1V2) of gp120, a domain in the envelope (Env) glycoprotein of HIV-1 [
12]. These data demonstrated that a V1V2-targeting vaccine has advantages over the imprecise targeting of SIV/SHIV infections and over whole Env-based immunization regimens for inducing a more focused functional V2p– and V2i–specific Ab response. Based on these studies, we used V1V2 (ZM53)-2F5K [
13] as the target immunogen for the NanoVac delivery of its encoding mRNA in this work.
2. Materials and Methods
2.1. Short Carbon Nanotube Preparation
The process of synthesizing short carbon nanotubes (SCNT) was described in our previous publication and is briefly summarized here [
13]. As such, purchased multiwalled carbon nanotubes (MWCNTs, SES Research, Houston, TX, USA) were suspended in concentrated H
2SO
4/HNO
3 solution (3:1
v/
v) and sonicated in a water bath for 16 h at 40 °C to obtain carboxyl functionalities on the surface. This process also “cuts” the CNTs into shorter lengths to obtain short carbon nanotubes (SCNTs). Purification and multiple filtration/separation steps were performed to obtain different size ranges of short MWCNTs. The size of CNTs collected from each purification stage was determined using scanning electron microscopy (SEM) and dynamic light scattering (DLS) methods.
2.2. Short Carbon Nanotube Surface Modification for HIV-1 Glycoprotein V1V2 Loading
For the surface modification of SCNTs, detailed methods are again provided in our prior publication [
13]. Briefly, a short chain PEG linker (NH
2-PEG-COOH, MW, 2 K, Sigma-Aldrich, St. Louis, MO, USA) was selected for conjugation through the EDC/Sulfo-NHS method. The EDC/NHS–activated SCNT was conjugated with a PEG linker. The resultant PEG-SCNT was again separated and purified using a centrifuge filter (molecular weight cutoff of 10 KDa, Millipore, Burlington, VT, USA) three times to remove excess PEG. PEG-SCNT products were dried under vacuum for analysis and conjugation with V1V2 proteins via an EDC/NHS reaction. Thermal Gravimetric Analysis (TGA) curves of PEG-SCNT and SCNT alone were used to confirm and quantify the coating of functionalities (linkers) on the SCNT surfaces. The size of CNT samples was measured using DLS and SEM to ensure the obtention of proper dimensional SCNTs.
2.3. mRNA Production
The antigen V1V2 (ZM53)-2F5K [
14] –encoded mRNA sequence was submitted to Tri-Link for production. To stabilize the mRNA, the 5′ and 3′ UTR elements flanking the coding sequence profoundly influenced the stability and translation of mRNA, both of which are critical concerns for vaccine production. This HIV V1V2 sequence was capped with CleanCap AG (a fully mature, Cap1 structure), phosphatase-treated to remove any 5′ triphosphates, which has a synthetic 5′ UTR with a strong Kozak sequence, murine alpha-globin 3′ UTR, and a 120 mer poly A tail. Further, all uridines were substituted with N1-methylpseudouridine (Ψ) to increase translation and stability.
2.4. Short Carbon Nanotube Surface Modification for mRNA Loading
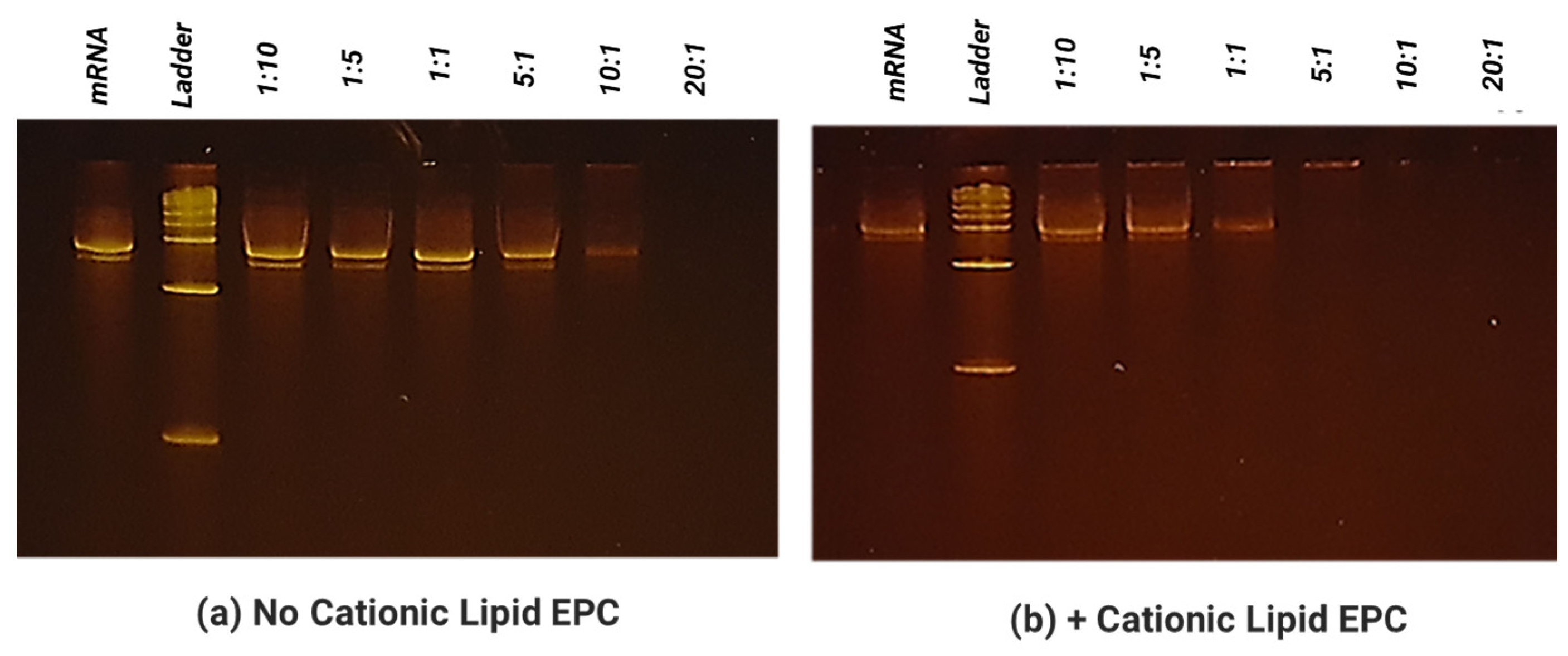
To obtain SCNT–mRNA conjugates, varying MWs (600–25 K) of polyethyleneimine (PEI) were first conjugated on the SCNT surface using the EDC/Sulfo-NHS method. To prepare different loading amounts of mRNA on the SCNT surface, SCNT-PEI was first formulated with 1,2-distearoyl-sn-glycero-3-ethylphosphocholine (EPC, Avanti Polar Lipids, Alabaster, AL, USA) in a weight ratio of 1:2 for 1 h via water bath sonication. Excess PEI and EPC were removed by centrifugation. Aliquots of SCNT-PEI–EPC were then mixed with EGFP-mRNA (a model green fluorescent protein encoding mRNA) or V1V2 encoding mRNA at mRNA:CNT mass ratios of 10:1, 5:1, 1:1, 1:10; 1:20 via pipetting and then incubated at room temperature for 10 min before tests.
2.5. Cell Uptake and Simulation of DC Cell Maturation
THP-1 (American Type Culture Collection, TIB-202, Manassas, VA, USA) –derived immature dendritic cells (DCs) were harvested and counted for the SCNT uptake assay; the SCNT and SCNT-modified antigen samples were prepared at twice the concentration desired for the final volume/concentration. Briefly, 250 µL was removed from each well containing cells and replaced using 250 µL of the prepared samples. Media containing GM-CSF (0.5 µg/mL), IL-4 (1 µg/mL), TNF-α (0.1 µg/mL), and ionomycin (200 ng/mL) to induce maturation were added to the wells containing the positive controls. The wells were inspected under the microscope at 5, 15, 30, 60 min, and then 18 h and 5 days for uptake and cell maturation. The cells containing the Cy3™ dye labeled SCNT antigen was observed using fluorescent microscopy (ZEISS Axio Vert. A1 inverted microscope, ZEN 2.6 lite Blue Edition, North Chesterfield, VA, USA) with a red filter (Cy3/TRITC; excitation, 545 nm/emission, 605 nm). On the fifth day after SCNT exposure, the presence or absence of the surface marker for mature dendritic cells, including CD-83, was tested using a FITC-labeled monoclonal antibody. The anti-CD-83 antibody was added in a 1:10 dilution per the manufacturer’s recommendation and incubated for 30 min at 37 °C with 5% CO2. The free antibody was removed via aspiration and the cells were washed twice with fresh complete media. After the second wash, the cells were added with the fresh media and inspected via fluorescent microscopy using a green filter (FTIC/Cy2; excitation, 470 nm/emission, 525 nm). Following the initial inspection for the presence of CD-83-positive cells, the samples containing dye were re-examined for the co-localization of the CD-83 label with the SCNT label, indicating that the SCNTs may aid in the maturation of mature dendritic cells from immature dendritic cells.
2.6. Electrophoretic Mobility Shift Assay for Evaluation of mRNA Binding on SCNT
The ability of SCNTs to bind and form a complex with mRNA (at different mass ratio between 10:1–1:20 as indicated in the above section) was determined by a mobility shift assay. The total mRNA amount was held constant at 0.25 µg per sample. To each sample, 7.5 µL of 2× RNA dye was added. Each sample was brought up to a final volume of 15 µL with RNAse-free water. The samples were then heated to 70 °C for 5 min then chilled for 3 min. The samples were then loaded onto a 6% TBE-Urea gel and run with a 1% TBE buffer at 180 mV at ~120 mA for 50 min. The gels were washed 3× with diH2O, five minutes each. The gels were then stained using the SYBR safe stain (Invitrogen ca# S33102, Waltham, MA, USA) at a 1:10,000 dilution in diH2O for 2 h in the dark. The gels were then washed again 3× with diH2O, 5 min each, and finally imaged under a UV light box.
2.7. In Vitro Transfection
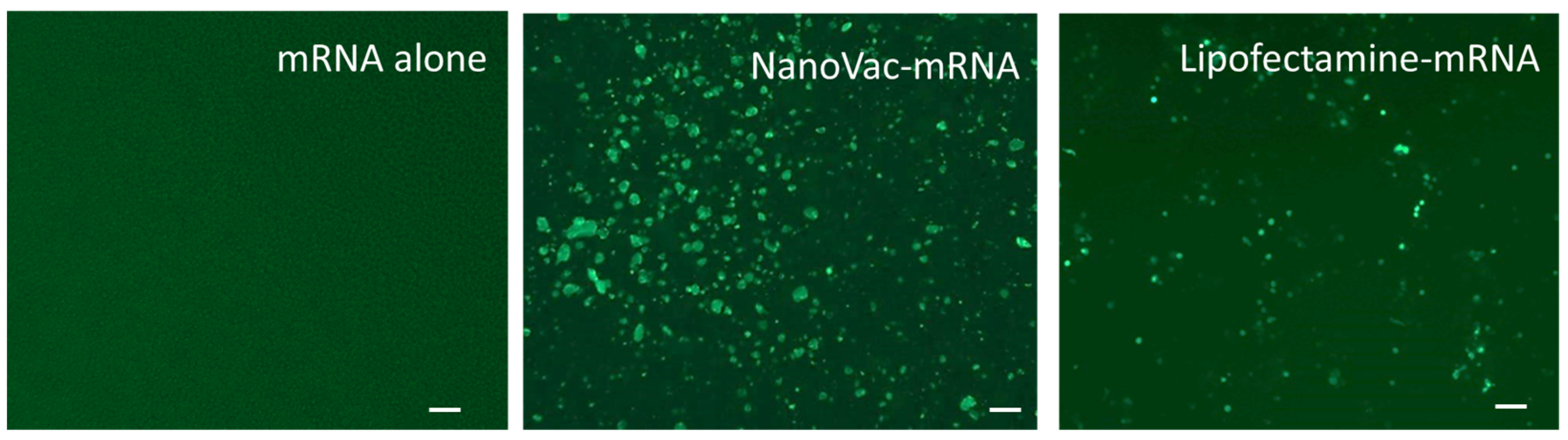
THP-1 cells (American Type Culture Collection, TIB-202, Manassas, VA, USA) were maintained in T75 tissue culture flasks with growth medium RPMI-1640 (ATCC#30-2001, Manassas, VA, USA) containing 10% fetal bovine Serum, and 0.05 mM 2-Mercaptoethanol for about 3–4 days. The flask was kept at 37 °C with 5% CO2 to an immature dendritic phenotype prior to experimentation. For plating, the cells were collected via centrifugation and counted using a hemacytometer. The needed number of cells were separated and adjusted to 200,000 cells/mL in induction media (growth media, via the addition of growth factors hIL-4 and hGM-CSF at a final concentration of 100 ng/mL of media). Briefly, 100 µL of the cells in the induction media were added to each well of a 96-well plate and were maintained at 37 °C with 5% CO2 with media changes as needed (every 2–3 days). Prior to transfection, SCNT: EGFP-mRNA in a mass ratio of 1:1, 5:1 and 10:1 was added to SCNT solutions, as well as positive control lipofectamine (Invitrogen, #L3000008, Waltham, MA, USA), and all were allowed to incubate for 10 min at room temperature. Then, SCNT–mRNA complex solutions were loaded into each well, in triplicate, to yield final SCNT concentration of 5, 25 and 50 µg/mL. The positive controls comprised 0.2 µL of lipofectamine and 0.5 µg of EGFP–mRNA per well. All wells were monitored at 48 h.
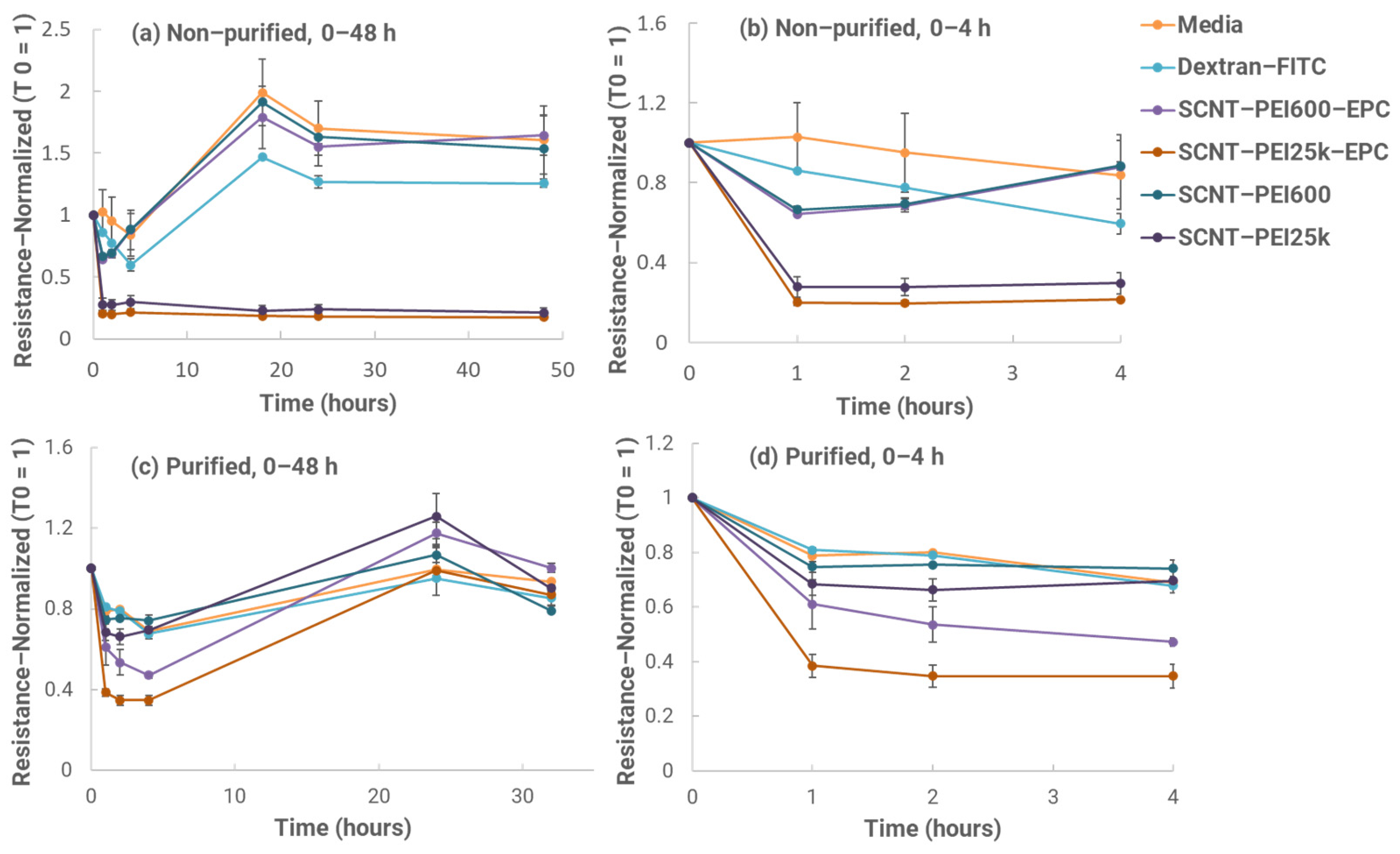
2.8. In Vitro Mucosal Penetration Test
To test mucosal penetration in vitro, Calu-3 cells (American Type Culture Collection, HTB-55, Manassas, VA, USA) were seeded onto the apical side of 12–well transwell inserts at 3.0 × 10
5 viable cells/insert. Trans-epithelial electrical resistance (TEER) measurements were taken approximately every other day to confirm cell confluency and tight junction formation. After thirteen days in culture, average insert resistance was higher than 1000 ohms, indicating that tight junctions had formed. Testing was then initiated the next day. Formulations with or without EPC were compared for performance. SCNT-PEI samples with and without EPC were prepared and sonicated for 1 h at ambient temperature. After sonication, samples were diluted in a ratio of 1:10 in complete culture media (EMEM, 10% FBS, ATCC, Manassas, VA, USA) bringing their concentration to 100 μg/mL. Fluorescein isothiocyanate dextran (Dextran-FITC) is a 3–5 kDa marker used to measure tight junction permeability [
11,
13,
15]. Additionally, Dextran-FITC was added to all samples except the only control media at a final concentration of 100 μg/mL. Before adding the samples, time 0 TEER measurements were taken for each well. The basal chamber of each well was replaced with 1 mL of fresh media, and 500 μL of each prepared sample solution along with media only and media with Dextran-FITC only–controls were added to the apical chamber of each insert in duplicate. Samples were then returned to an incubator (37 °C, 5.0% CO
2) for 1 h. TEER measurements were taken again before removing the full 1 mL of the basal chamber media to measure the FITC fluorescence (excitation, 487/10 nm; emission, 528/10 nm) contained in each basal chamber. Wells were replaced with 1 mL of fresh media in the basal chamber before returning to the incubator. These steps were repeated at the 2, 4, 18, 24 and 48 h time points. A Dextran-FITC standard curve using complete culture media was created to estimate Dextran-FITC concentration amounts in the basal chamber media for all samples at each time point. TEER values were normalized based on their T = 0 values for each individual insert.
2.9. Stability Test for mRNA Payload
A pair of primers was successfully designed on V1V2 HIV-1 glycoprotein sequences to amplify the cDNA produced from any RNA present in each sample. The primers were tested using a cDNA made from pure V1V2 mRNA that act as the positive control for the qPCR assay. To begin the stability study, 10 μL aliquots containing 4 µg of mRNA (at 1.5 mg/mL) with or without NanoVac were prepared. For NanoVac–mRNA samples, NanoVac formulations (4 µg of mRNA: 4 µg of SCNT-PEI: 8 µg of EPC) were incubated for 15 min at room temperature. The Zeta potential was measured to confirm that the complex was stable before testing. Samples were prepared for 7 timepoints in triplicate and stored at 4 °C for the duration of the study. At each time point, 5 μL per replicate was examined via gel electrophoresis while the remaining 5 μL was used to prepare cDNA using RT2 Easy First Strand Kit (Qiagen, 330421, Germantown, MD, USA). The resulting cDNA reactions were then stored at −20 °C. This step was performed at Day 0, 1, 7, 14, 23 and 30, and at three months (Day 90). Once all timepoints were collected, these cDNA samples and a positive cDNA control were used to run a qPCR assay. cDNA samples were diluted in a ratio of 1:100 in nuclease-free water, and 1μL of this dilution was then added to each 20 μL qPCR reaction along with the qPCR master mix (NEB, M3003S) and the forward and reverse primers (0.25 µM each). For the sample dilutions, each replicate was measured in duplicate wells (6 wells total for each timepoint sample). The positive cDNA control was diluted 10-fold and 1 μL of each dilution was used as the template for the standard curve with each dilution measured in triplicate wells. The qPCR thermocycling program included an initial denaturation step at 95 °C for 60 s, followed by 40 cycles each containing a 95 °C denaturation step for 15 s, a 60 °C extension step for 30 s and a plate read step.
2.10. Rabbit Immunogenicity Studies
Female New Zealand white rabbits 6–8 weeks old (~2 kg) each received three boosters. A total of 5 female New Zealand white rabbits in each group were dosed with 50 µg of NanoVac–mRNA at a volume of 100 µL via intramuscular administration. The rabbits were immunized via NanoVac–mRNA at weeks 0, 2, and 4 with blood collected at 2 (pre-bleed), 4 and 8 weeks (the end point of the experiment). Serum samples were collected to analyze the kinetics of the antibody response. Serum was separated from whole blood and stored at −20 °C until assays were performed. Body fluids (vaginal washes) were collected post-mortem. These analyses were specifically developed to compare both the breadth and potency of the antibody responses induced by mRNA and the encoding protein. At the end of the study, animals were euthanized via non-inhaled barbiturate injection under anesthesia. For the ELISA assay, 96-well flat-bottom plates were coated overnight at 4 °C with a HIV-1 V1V2 (ZM53)-2F5K immunogen at 1 μg/mL. The plates were washed with PBS containing 0.05% Tween-20 (PBST), at pH 7.4. Non-specific binding sites were blocked with PBS containing 3% BSA for 1 h. The plates were then washed once with PBST and subsequently 100 µL of rabbit serum was added and incubated for 1 h. Serum samples were diluted in a ratio of 1:10 for antibody detection. After incubation, the plates were washed with PBST. The bound antibodies were detected by incubating the with goat anti-rabbit IgG conjugated with alkaline phosphatase diluted in a ratio of 1:2000 in PBS. Finally, the plates were washed with PBST, and the bound alkaline phosphatase enzyme activity was revealed by adding 100 µL/well of the substrate solution. Absorbance at 405 nm in each well was measured using a microplate reader.
2.11. Immunogenicity Studies Using HIS Mice
The immunogenicity of Luna Labs vaccine candidates was measured in humanized immune system (HIS) mice. NOD.Cg-B2m
tm1Unc Prkdc
scid Il2rg
tm1Wjl/SzJ (NSG-B2m) triple mutant mice, which combined the features of severe combined immune deficiency mutation with IL-2 receptor γ chain and β2-microglobulin (β2m) deficiencies, were purchased from The Jackson Laboratories. Two–weeks after the adeno-associated virus serotype 9 (AAV9)–mediated transfer of HLA-A2/DR4 and human cytokine genes (IL-3, Il-4, IL-6, IL-7, IL-15 and GM-CSF), the NSG-B2m KO mice (4-5 weeks old) received a sub-lethal dose (150 cGy) of X-ray irradiation to myeloablate the remaining murine immune cells present in NSG-B2m KO mice [
16]. A few hours later, the NSG-B2m KO mice were engrafted intravenously with 1 × 10
5 human hematopoietic stem cells (HSCs), which were purified by CD34+ beads (Miltenyi, San Diego, CA, USA) from human cord blood cells. Fifteen weeks after the HSC engraftment, the percentages of human CD45+ cell repopulation, as well as the reconstitution status of various human immune competent cells, in the peripheral blood of AAV9-transduced NSG-B2m KO mice were determined via a flow cytometric analysis. As shown in the
Appendix A Table A1, more than 60% of PBMCs of all the HIS-A2/DR4 mice were human CD45+ cells with a similar proportion of human CD3+ T-cells, CD4+ T-cells, CD8+ T-cells and B-cells, compared to that in actual humans.
Briefly, the first set of experiments was performed for four groups (three formulations and one control group as a preliminary test). Briefly, 10 HIS mice (five male and five female) were split into four groups. In each group, mice were immunized with each NanoVac formulation two doses in 3-week intervals via intramuscular injection (IM) and 50 µg of immunogen per mouse was administrated. In the second set of experiments, 10 mice were used for the studies. The NanoVac formulation was administrated by either IM or IN routes for two doses at 3–week intervals. For IM administration, 50 µg of immunogen per mouse was injected into the thigh at a volume of 0.1 mL. For IN administration, 20 µL of the formulation (50 µg of immunogen) was reconstituted for intranasal instillation using a micropipette. Two–weeks after the last boost, sera were collected from immunized, as well as naïve mice, and the titers of mouse IgG against HIV glycoprotein were determined via ELISA. Briefly, the ELISA microplates (Thermo Scientific, Durham, NC, USA) were coated with 75 µL per well of HIV V1V2 protein at 20 µg/mL, which was diluted in a coating buffer (Thermo Scientific). The plates were left overnight at room temperature (RT) with a sealer covered on top. On the next day, the plates were washed 3 times with PBS, blocked by adding a blocking buffer (Thermo Scientific, Durham, NC, USA) at 200 µL/well and left for at least 2 h at RT. Meanwhile, the serum samples were prepared with planned dilutions on a separate non-ELISA, non-coated 96-well round-bottom plate. The serum samples were diluted with the blocking buffer. The duplicates were prepared for each sample and dilution. After blocking, the plates were washed twice with PBS. Then, 100 µL/well of samples in appropriate serial dilutions were added to plates and they were left for at least 90 min at RT. The plates were washed 4 times with PBS-0.05% Tween, 100 µL/well of a 1:5000 (diluted-in-PBS-Tween) secondary antibody (HRP conjugated goat anti-human IgG Fc fragment; Bethyl Lab Cat#: A80-104P) was added and the plates were left for at least 1 h at RT. The plates were washed 4 times with PBS-0.05% Tween and 3 times with PBS, 75 µL/well of TMB High Sensitivity Substrate at RT (BioLegend Cat#: 421501, San Diego, CA, USA) was added and the plates were left for 10 to 15 min in the dark. Briefly, 75 µL/well of 2.00 Normal sulfuric acid was then added to the plates at RT to stop the reaction. Finally, the plates were placed on a spectrophotometer plate reader and the optical density (OD) was measured at 450 nm.
The spleens were harvested and splenocytes isolated, followed by IFN-γ ELISpot assay to determine the relative number of HIV glycoprotein-specific human T-cell response. The relative numbers of splenic HIV V1V2–specific, IFN-γ-secreting human CD8+ T-cells of Luna Labs’ vaccines-immunized HIS-A2/DR4 mice were determined by an ELISpot assay, using a human IFN-γ ELISpotPRO kit (Mabtech AB, Stockholm, Sweden) and a HIV V1V2 protein. A single cell suspension of splenocytes was isolated from the spleens harvested from the HIS-A2/DR4 mice 12 days after the last immunization with vaccines. Then, 5 × 105 splenocytes in 200 µL/well of culture medium (RPMI-1640 with 10% fetal bovine serum and antibiotics and 5 × 10−5 M 2-mercaptoethanol) were placed on each well of the 96-well ELISpot plates pre-coated with anti-human IFN-γ monoclonal antibody (1-D1K) and incubated with or without 20 μg/mL of HIV-V1V2 protein for 24 h at 37 °C in a CO2 incubator. On the next day, the ELISpot plates were washed 5 times with PBS, and 100 µL/well of horseradish peroxidase-conjugated anti-human IFN-γ detection monoclonal antibody (7-B6-HRP) (diluted 1:200 in filtered PBS containing 0.5% fetal calf serum according to the company’s instruction) was added and the plates were left for at least 2 h at RT. The ELISpot plates were then washed 5 times with PBS, 100 µL/well of the ready-to-use TMB substrate solution was added and the plates were left until distinct spots were developed (5–10 min at RT in the dark). To identify the number of IFN-γ-secreting CD8+ T-cells in each well, the mean number of spots (for duplicates) counted in the wells incubated with splenocytes in the presence of the peptide was subtracted by the mean number of spots (for duplicates) counted in the wells that were incubated with splenocytes only.
2.12. Safety Studies for SCNT Vaccine Candidates
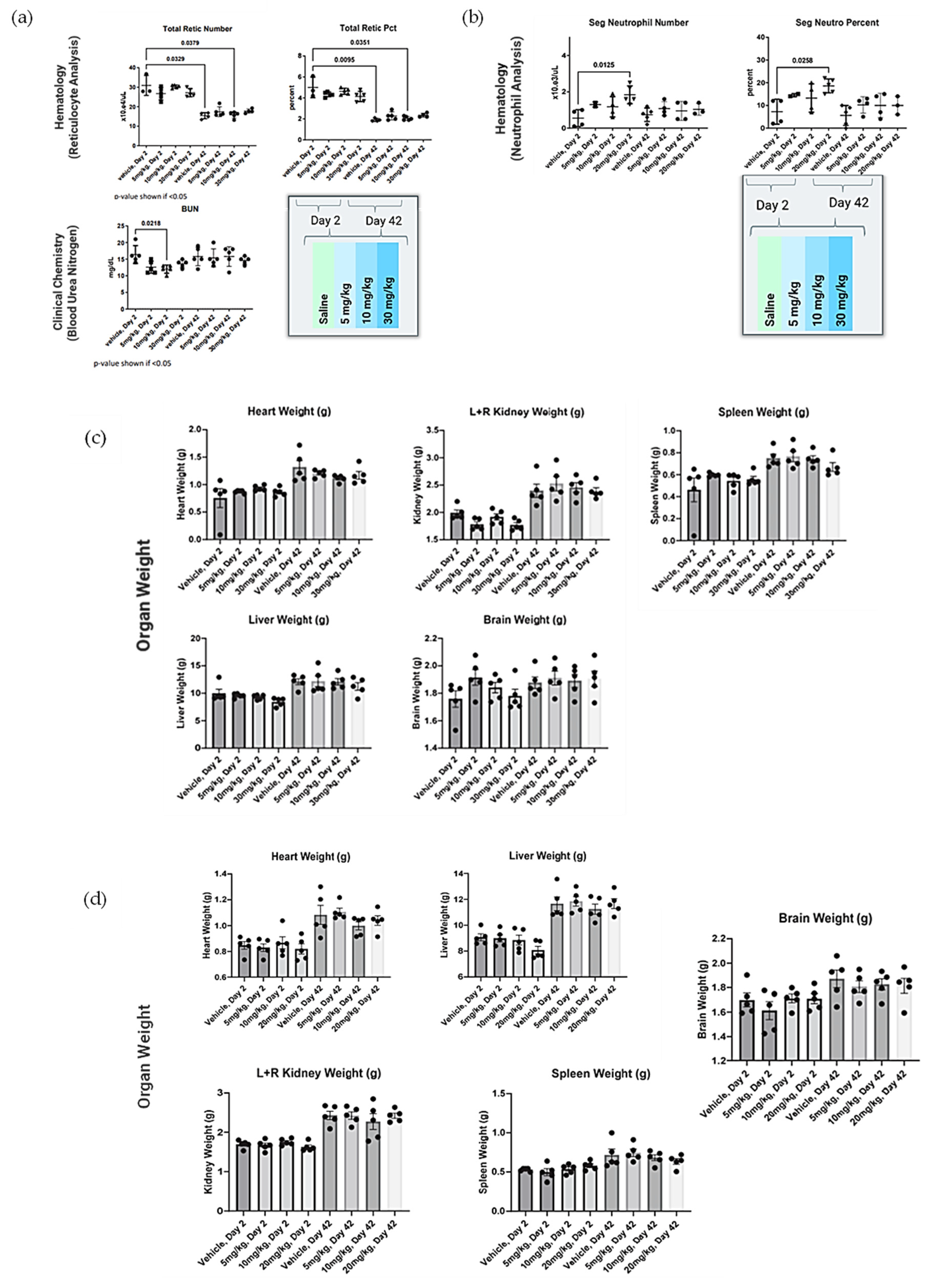
Forty (40) male Sprague Dawley rats (8–10 weeks old, Charles River Laboratories, Wilmington, DE, USA) were housed individually in solid-bottom cages with aspen bedding for the duration of the study. The study was conducted following a minimum 72 h acclimation, body weights were recorded (224–249 g) and animals were assigned to 8 groups of 5 rats each, achieving equal mean group body weights to the extent possible. One or three doses of NanoVac at a dose level of 0–30 mg/kg were intramuscularly or intranasally administrated at a dose level of 0–20 mg/kg for rats on Day 1, 14 and 28. Clinical observations were conducted daily. Body weight was collected approximately weekly and at the time of necropsy. Groups 1–4 were euthanized with isoflurane on Day 2, approximately 24 h post-dose. Groups 5–8 were euthanized on Day 42. Terminal blood was collected and processed for hematology and clinical chemistry as described below. All blood samples contained K3EDTA as an anticoagulant and were analyzed. Heart, lungs, kidneys, liver and spleen were collected, weighed, and fixed in 10% neutral-buffered formalin. The brain was removed, weighed and discarded. Slides of fixed tissue were prepared, stained with standard hematoxylin and eosin and reviewed by a qualified veterinary pathologist. Data were collected on a Microsoft 365 Excel spreadsheet. Graphic interpretation of the data was performed using GraphPad Prism 9.0.0 (GraphPad Software, Boston, MA, USA). Statistical comparisons of body weight, organ weight, hematology, and biochemistry parameters to vehicle control were made using Kruskal–Wallis one-way analysis of variance followed by post hoc testing using Dunn’s multiple comparisons test. In all cases, p < 0.05 was considered statistically significant.
2.13. Efficacy Studies Using HIS Mice
NSG-B2m KO mice were first transduced with human genes encoding HLA-A2/DR4 and human cytokines (IL-3, Il-4, IL-6, IL-7, IL-15 and GM-CSF) by AAV9 vectors, followed by being sub-lethally irradiated and engrafted with HLA-matched HSCs, as described above. Fifteen weeks later, more than 60% of PBMCs were found to be human CD45+ leukocytes with the composition of human B-cells and CD4+ T-cells, and the CD4+ T-cell subsets were similar to those observed in humans. Two groups of vaccine candidates were used for efficacy studies. One group was the SCNT-mRNA final formulation, and the second group was MC3 lipid nanoparticle (LNP)–encapsulated mRNA prepared according to published methods [
10], serving as a benchmark control. The LNP mRNA contained the same amount of immunogen (50 µg) for delivery as the NanoVac candidate did. LNP was synthesized using a microfluidic device [
10]. A third group was the naïve group, with no sample vaccine formulation delivered. Groups of 6 HIS mice (3 male and 3 female) were immunized intramuscularly with each mRNA vaccine two times in 3-week intervals. Then, 3 weeks after the last immunization, groups of immunized as well as naïve HIS mice were challenged via the intravenous administration of 2 × 10
5 infectious units of a CCR5-tropic HIV-1 variant that is based on the LAI strain but carries part of the env gene from the CCR5-tropic Bal strain [
17]. To confirm whether or not the HIS mice were susceptible to HIV-1 infection, we monitored the viral load in the plasma and CD4+ T-cell count in the blood after infecting the animals [
17].
4. Discussion
The dimension of the as-produced nanotubes typically varies widely. They also have the tendency to form bundles and aggregates with varying shapes and sizes. To overcome this problem, Luna Labs first selected multiple-walled carbon nanotubes (MWCNTs) which have initial diameters of 60–100 nm and lengths of 1–2 µm as the starting materials. We then used concentrated H
2SO
4/HNO
3 mixtures to cut the highly entangled long ropes of CNTs into narrow-size-distribution, short, open-ended non-entangled tubes. In this effort to specifically mimic the HIV-1 virus for subsequent vaccination, we refined the dimension of SCNTs as close to that of the HIV-1 virus as possible (120 nm in diameter). During the modification process, the MWCNTs become more biocompatible, biodegradable and stable in the aqueous solution [
20] which is different than the case of unmodified CNTs as discussed previously [
21]. We have demonstrated the gram-scale production of SCNT and achieved a narrow size distribution of high-purity short carbon nanotubes (SCNT) as the delivery vehicle. The purity of SCNT can reach greater than 99.64% as monitored using inductively coupled plasma optical emission spectroscopy (ICP-OES), as reported [
13]. The purity of CNT is very important for potential product development, quality control and regulatory compliance for clinical use. We then successfully prepared NanoVac formulations as the vaccine candidates for mRNA delivery known as NanoVac–mRNA; antigen/protein delivery known as NanoVac–V1V2; or the codelivery of mRNA and V1V2 protein. Surface chemistries of the short multiwalled carbon nanotubes were also modified to guide antigen/mRNA density and presentation at the surface to better mimic the HIV-1 virion. This is a critical step for the use of this technology in vaccination efforts, where mimicking the virus and surface epitope presentation is critical to enhanced immunization [
22]. As a result, the density of the antigen on the surface is a key factor. Luna Labs has demonstrated an optimized density for V1V2 protein conjugation and the control of the epitope orientation using small lipid-PEG as reported previously [
13]. Recent technological advances have demonstrated multiple mRNA vaccine platforms that use the lipid nanoparticle (LNP) delivery of mRNA against infectious diseases. However, it cannot be modified for epitope presentation or the delivery of an mRNA-encoded protein with a controlled morphology. NanoVac shares its advantages with those of carbon-based materials which allows the better control of antigen presentation on the surfaces.
Two potential administration routes were explored using NanoVac as the vehicle, since the formulations were specifically designed/engineered for compatibility with both intramuscular and intranasal administration. To deliver mRNA into cells, we further modified/functionalized the surface of SCNT to achieve high loading capacity and delivery efficiency. We also engineered surface chemistry and functional design to stabilize loaded mRNA from degradation. One key challenge in creating nanodelivery mRNA complexes is that their transfection efficiency (TE) is adversely affected by serum, both in vitro and in vivo [
23]. As a result, the design and formation of stable nanocomplexes of mRNA are critical, and the evaluation of their transfection efficiency for mRNA is required. Polyethyleneimine (PEI) has been demonstrated as an efficient gene delivery reagent both in vitro and in vivo [
24]. Many studies using different types of PEI have shown relatively high efficiency in gene expression compared with other polymer vectors [
25]. In our studies, after PEI was conjugated on the SCNT surface it enhanced cellular transfection efficiency. Additionally, the MW of PEI was found to be another key factor for transfection. We determined that using PEI at MWs of 25 K and 600 were the best conjugation candidates on the SCNT surface for transfection. Our data showed that the SCNT to PEI weight ratio of 1:1–1:5 was found to be the optimized condition of using the formulation. Additionally, to further block the surface negative charge of mRNA to form a protective layer, the optimized combinations of a multivalent lipid EPC and PEI on the SCNT surface stabilized mRNA and effectively achieved the maximum transfection delivery efficiency in vitro while also reducing vehicle cytotoxicity. The final formulation showed relatively high surface charges greater than 40 mV after centrifugation purification to remove the excess PEI and EPC polymer from the solution. Due to the positive charge density, it forms positively charged complexes with mRNA with high efficiency and provides efficient transfection and protection against nuclease-mediated degradation. Successful nonviral gene transfection currently requires compromises to achieve a useful level of transfection efficiency while minimizing toxicity. Thus, this purification step is critical to reduce the cytotoxicity concerns from these excess polymers observed in the TEER studies. The final formulation suspension was demonstrated to be very stable without any precipitation during injection/administration to animals. The size of the final formulation was 268.5 ± 117.0 nm with a surface charge of 29.5 ± 5.5 mV.
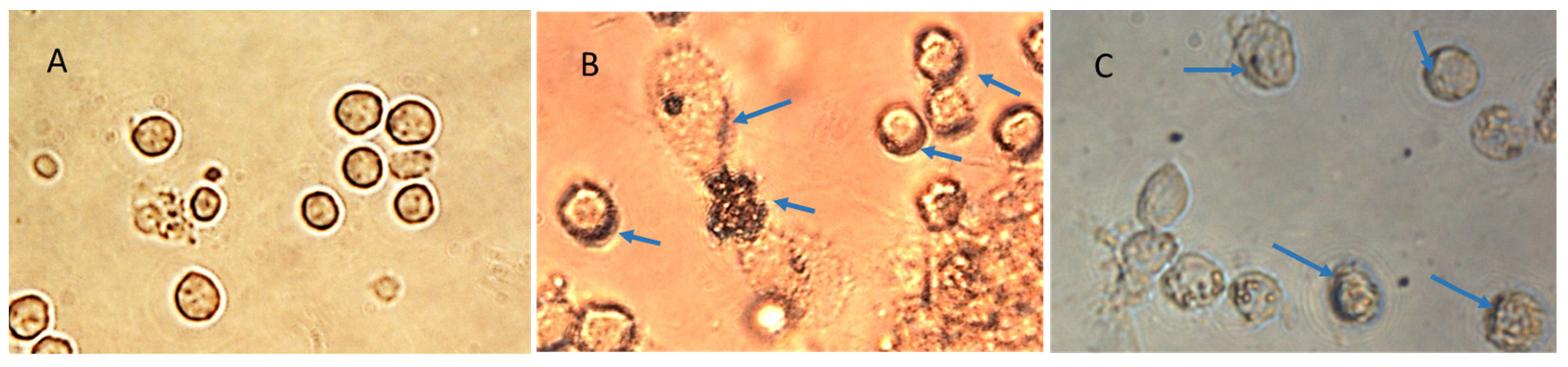
The specifically designed and functional NanoVac–mRNA promoted rapid uptake within 5 min of exposure to antigen-presenting cells (APCs), such as dendritic cells (DCs). The uptake pattern is punctate, presenting numerous vesicles (
Figure 13B) within the first 5 min. With a longer SCNT exposure time, the SCNTs began to be visible inside cells, suggesting that increasingly more SCNT–antigen conjugates were internalized (
Figure 13C). The rapid and vesicular uptake is consistent with micropinocytosis, which is known to be an important mechanism of macromolecular antigen uptake in dendritic cells. As shown in
Figure 13, we demonstrated that DCs internalized large amounts of NanoVac conjugates without compromising cellular integrity. Additionally, the chemistry of PEI plays a dual role to both bind nucleic acids and induce endosomal rupture via the so-called “proton-sponge” effect [
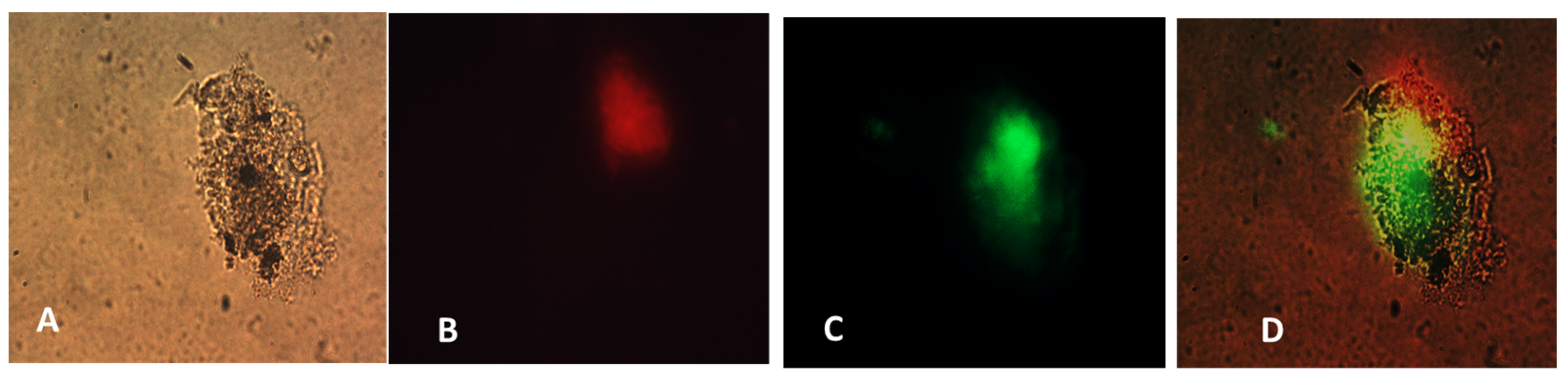
26]. We also determined that the NanoVac activated DCs to express high levels of CD83, the cell surface marker for mature human DCs. DCs showed much greater responses to NanoVac compared to the antigen alone, likely due to the NanoVac shape and its functionality. NanoVac was able to fully activate the DC cells to become mature, as demonstrated via the CD83 expression and morphology (
Figure 14), which indicated that the DCs matured and were able to promote MHC class II (major histocompatibility complex) and CD86 expression, critical to T-cell activation and the immune response. Due to the higher efficiency of NanoVac uptake into APCs compared to that of the antigen without a delivery vehicle, it will take significantly less time to complete the dosing, thus shortening the time to develop an effective antibody response.
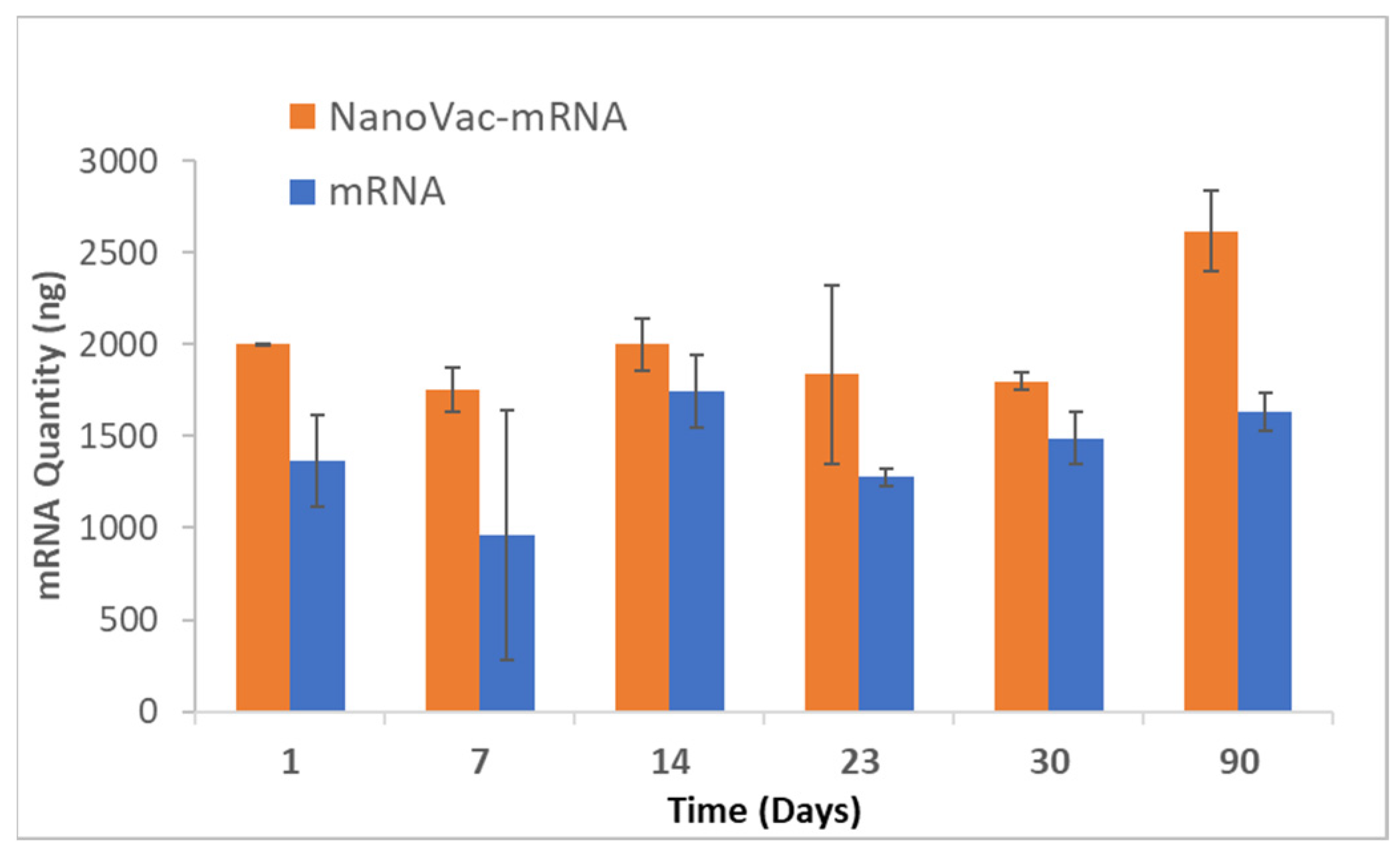
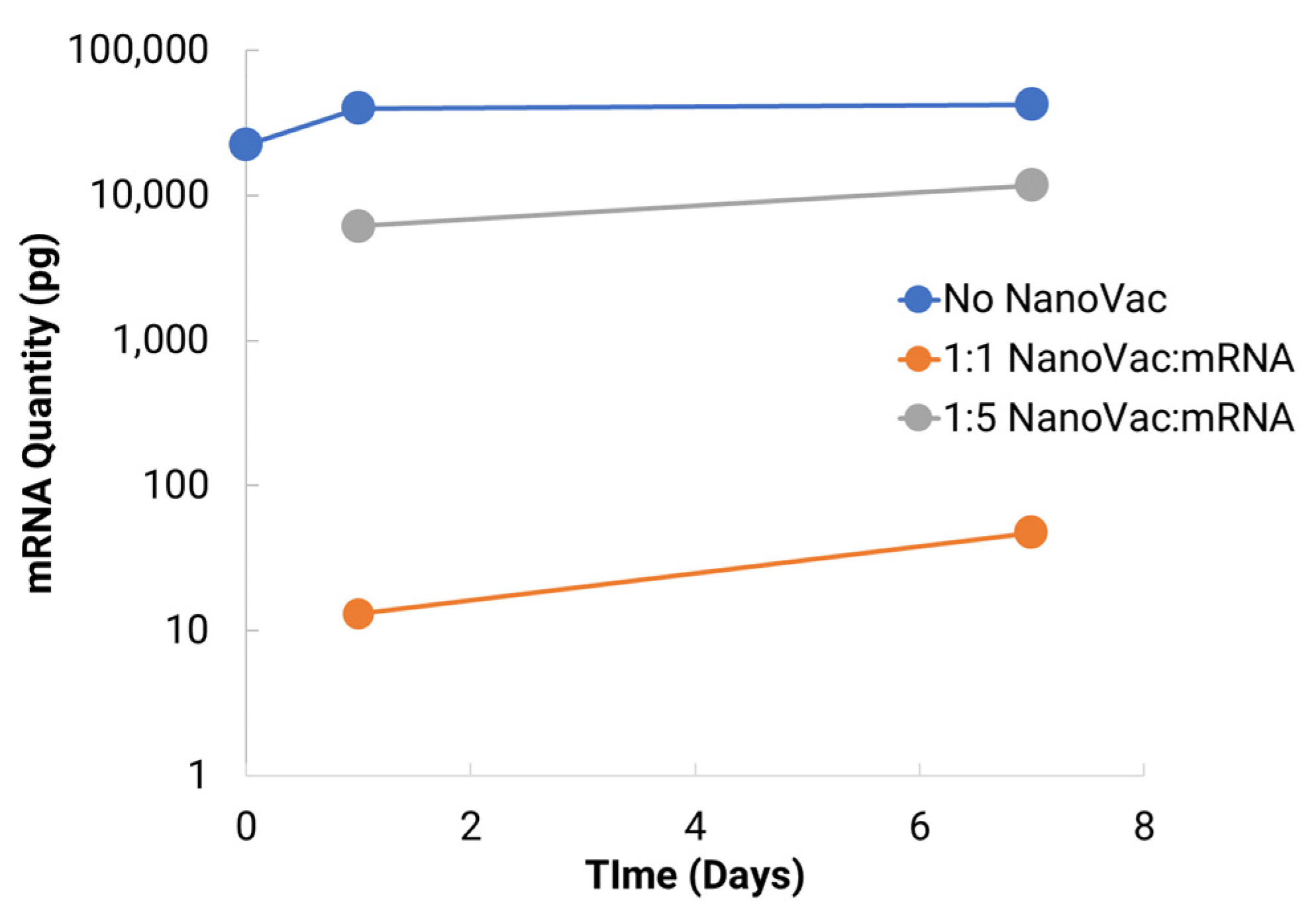
To increase translatability and RNA stability, along with in vivo delivery to activate innate immunity, we worked with TriLink to modify the mRNA encoding for V1V2. A 5′ cap, optimized 5′- and 3′-UTRs, and a coding sequence and poly(A)-tail modifications were introduced into the construction. We also modified nucleosides, using N1-methylpseudouridine (Ψ) to increase protein translation. Using NanoVac as the delivery vehicle, we further stabilized the mRNA from degradation during the storage under refrigerated conditions (4 °C) by tuning the chemistry on the SCNT surface. This was confirmed both by the qPCR results demonstrating recoverable mRNA, and an RNA gel at the 3-month time point that demonstrated that the mRNA bands were bright and solid with minimal apparent degradation. Interestingly, the quantity of mRNA recovered as monitored via qPCR appeared to increase slightly in quantity based on the qPCR results. It is inferred that over time, more of the mRNA bound to NanoVac was released and was able to be detected during the cDNA and qPCR reactions. This study looked at two different ratios—1:1 and 1:5—of NanoVac:mRNA, keeping the mRNA amount constant for both (2 μg) along with an additional sample containing no NanoVac. These results showed that significantly more mRNA was detected in the 1:5 ratio compared to that in the 1:1 ratio. We could potentially control the mRNA delivery via controlling the strength of binding and thus the releases from the surface.
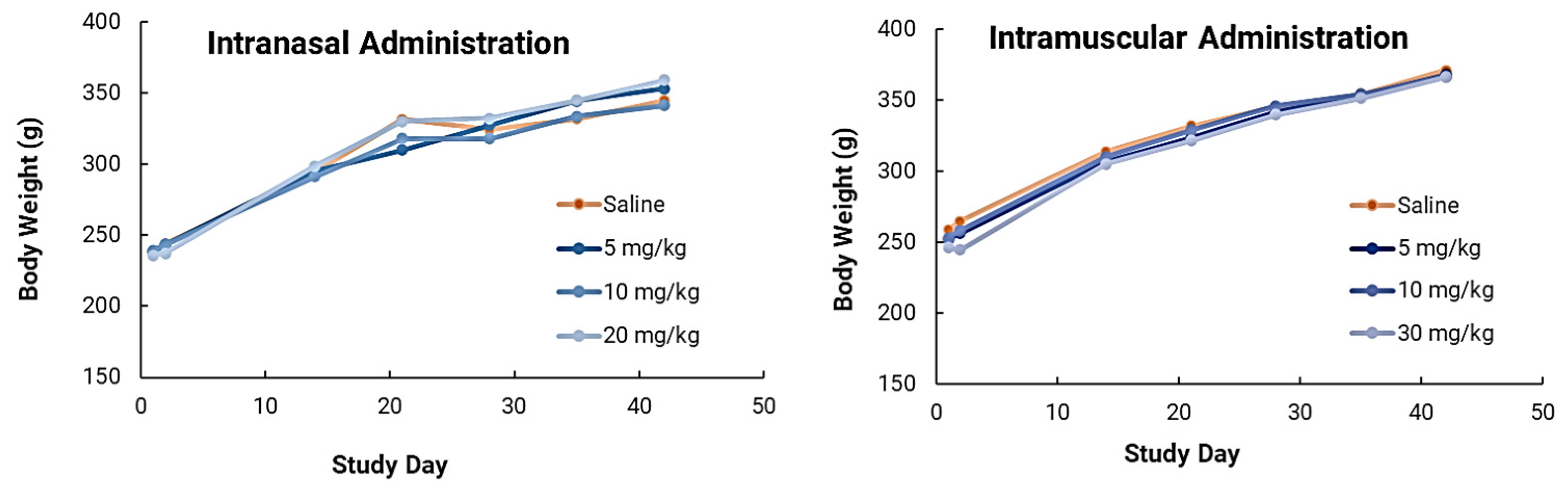
In safety studies for both intranasal and intramuscular administration, short carbon nanotubes were clinically well-tolerated to very high doses (20–30mg/kg) in male Sprague Dawley rats when evaluated after two weeks. There were no biologically relevant effects or clinical biochemistry parameter concerns. In ongoing research efforts, long-term safety studies will be performed because of the increased attention and safety concerns related to carbon nanotube use in human clinical trials. This issue is especially important because CNTs are extremely physically and chemically stable, and therefore may not be biodegradable. However, novel approaches for degrading CNTs have been developed and were implemented in this study. Luna Labs modified the SCNT surface through a strong oxidation reaction, which has previously been demonstrated to result in SCNT biodegradation in macrophages through a biological pathway [
20]. This is an encouraging result, and, as demonstrated in our safety studies, adverse effects were limited to inflammation at the site of SCNT administration and local sequestration of the black pigment, likely SCNTs, within macrophages and in lymphoid tissue. Clinically, there have been promising published results of using CNT nanoparticles as a safe lymph node-harvesting tool [
27], which indicates that SCNTs can be applied for human uses. Our results indicated that NanoVac is a safe platform that could be used for vaccine delivery. In the future, we will continue to expand toxicity studies, with specific focus on NanoVac intranasal formulations. We will use radiolabeled biodistribution studies to determine if, where and how much of the vaccine enters the nasal cavity, or even the brain, as this will help eliminate safety concerns with intranasal administration. This effort will support fully developed immunological profiles and a preclinical safety package before the performance of clinical studies.
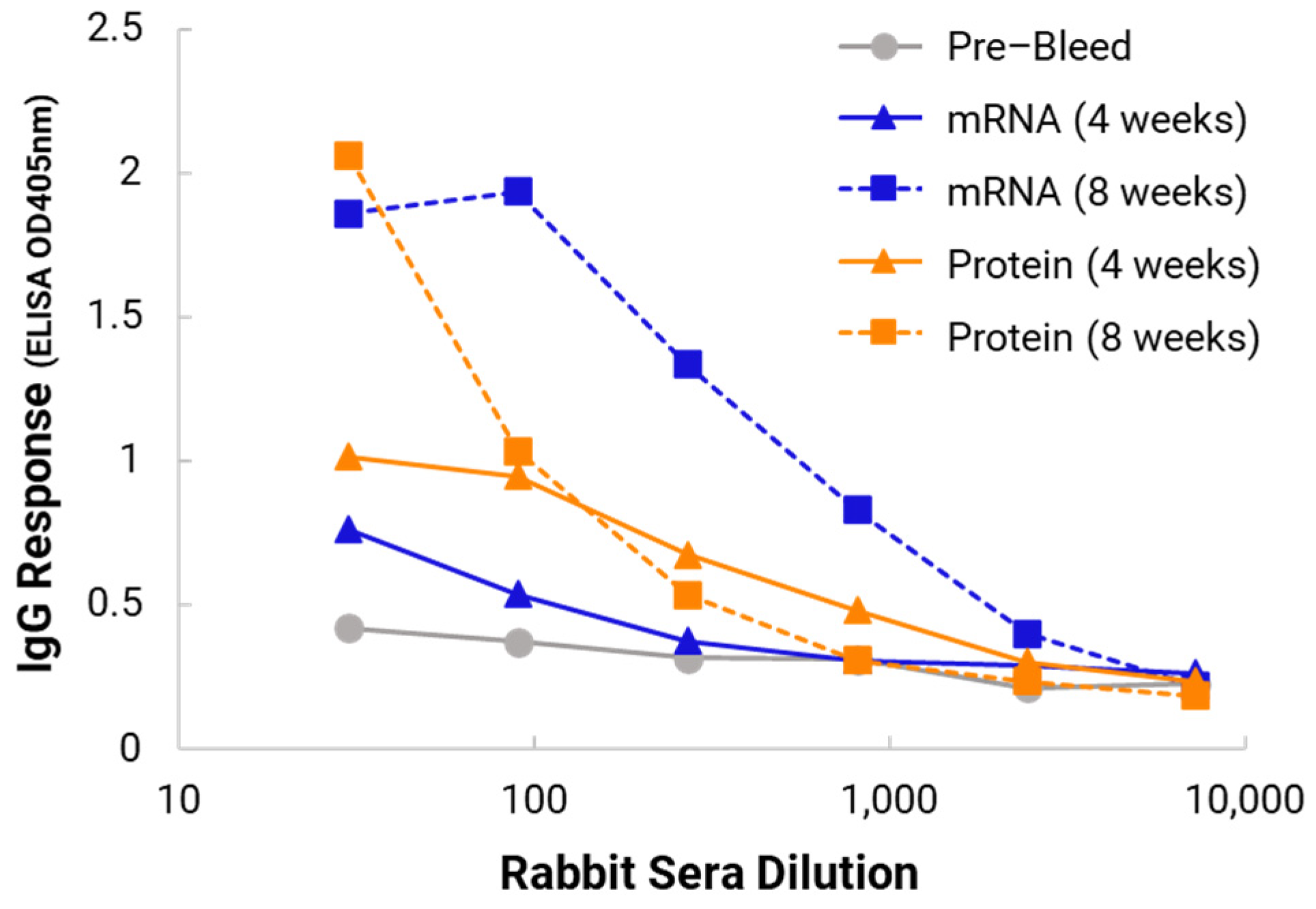
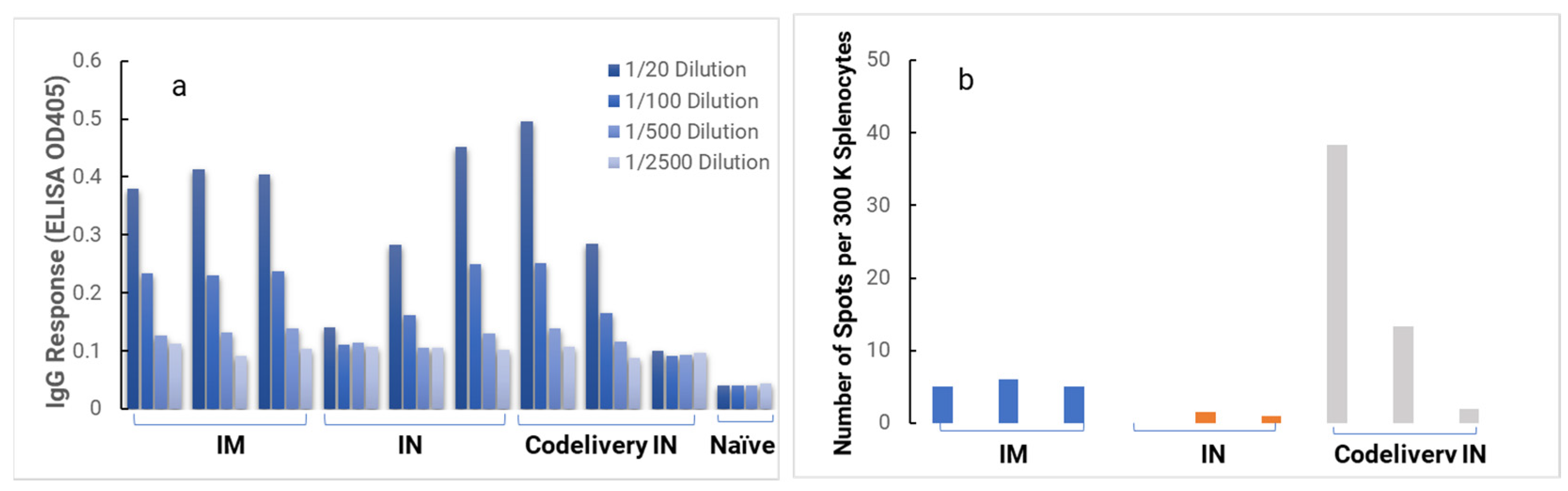
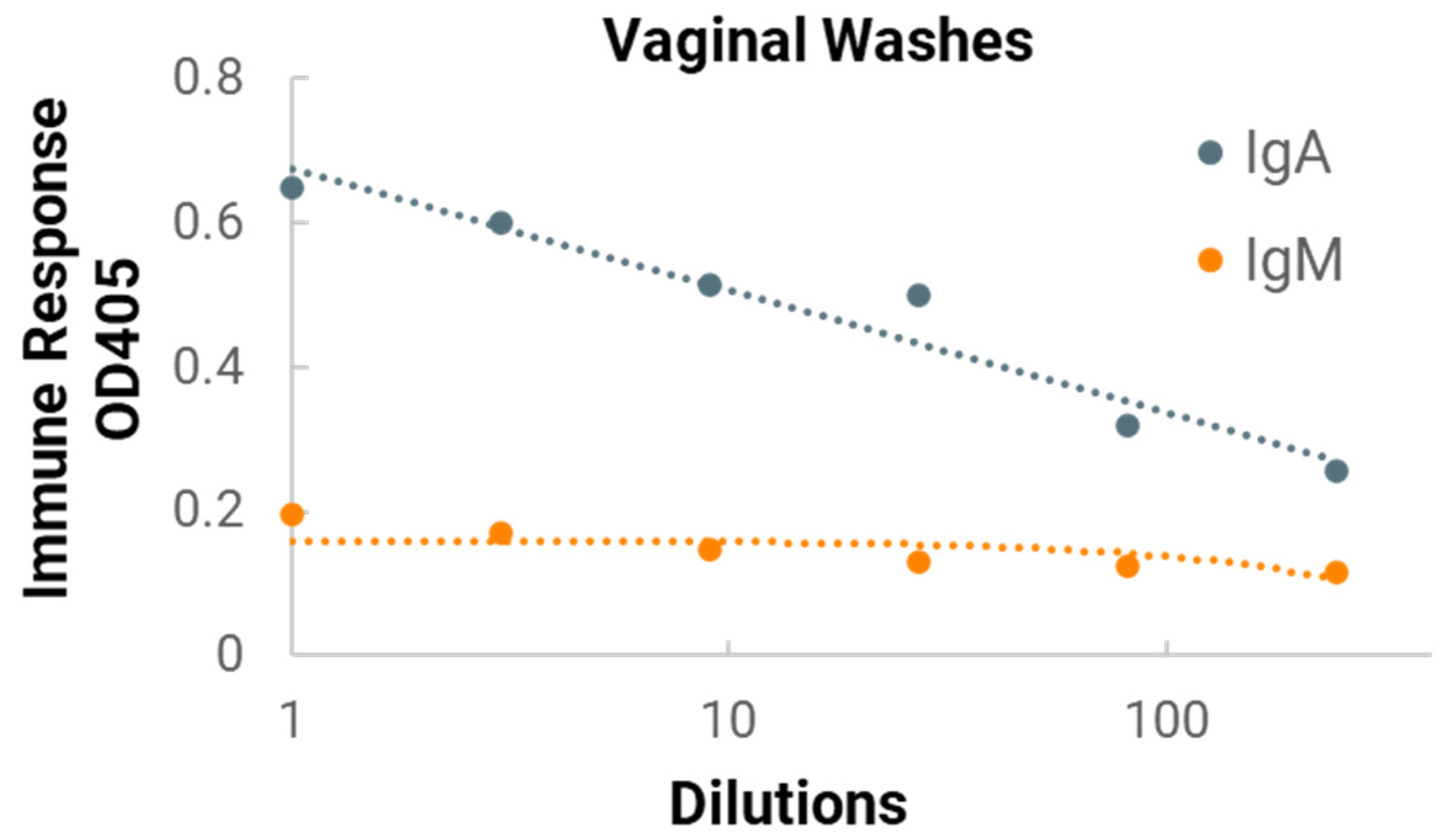
In the in vivo immunogenicity studies on rabbits, we compared the NanoVac–mRNA formulation and NanoVac–V1V2 formulation in eliciting immune responses. As shown in
Figure 7, we did not see a high enough immune response during the earlier time point at 4 weeks for NanoVac–mRNA. However, after 8 weeks and the third booster of the NanoVac–mRNA vaccine, we observed significantly higher immune responses against the HIV-1 V1V2 (ZM53)-2F5K target antigen in the animal group than what we obtained from the protein V1V2 group. Even at a titer of 5000, we observed significant higher responses for the NanoVac–mRNA formulation compared to those with the administration of NanoVac–V1V2 protein as the control. It elicited two-fold higher IgG responses compared to those of the protein groups. Additionally, it is interesting to see IgA responses which were observed in the rabbit vaginal wash samples as shown in
Figure A2 in
Appendix A.
Systemic immunization has generally been considered incapable of generating protective mucosal immune responses; however, cumulative data from recent studies suggest that some systemically administered vaccines are capable of eliciting mucosal immune responses, including the secretion of IgA antibodies [
28]. The main hypothesis is that the antigen first diffuses from the IM immunization site to the regional draining lymph nodes, and from there is taken up by local APCs (such as DCs, B-cells and macrophages). These APC cells then migrate to mucosa-associated lymphoid tissue (MALT), such as Peyer’s patches (PPs) and nasopharynx-associated lymphoid tissue (NALT), where they activate CD4
+ T-cells and B-cells [
29]. The mechanism of this induction remains poorly understood. Though evidence points towards a mucosal response being generated using the NanoVac delivery vehicle, more comprehensive studies will be designed and performed in the future, including those comparing IgA response following IM and IN administration. A summary of the immune responses that have been evaluated from rabbit or mice studies to date, including those reported in our previous publication [
13], is provided in
Table A2,
Appendix A. Combined, these results demonstrate that NanoVac is an effective adjuvant/delivery system for HIV-1 mRNA. Based on the rabbit results discussed above, we selected a HIS mouse model for further immunogenicity and efficacy studies to better interpret the results before moving on to NHP and human trials.
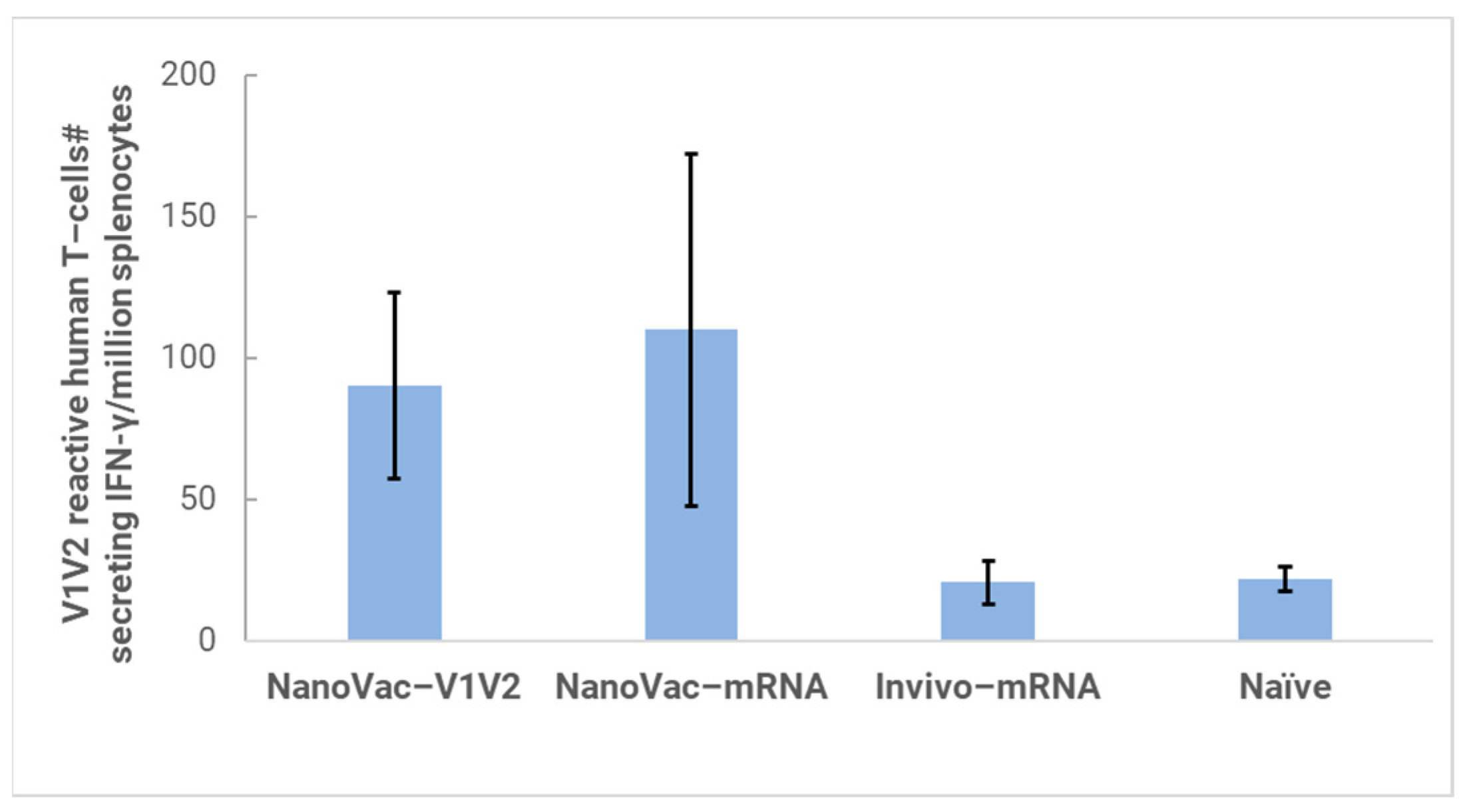
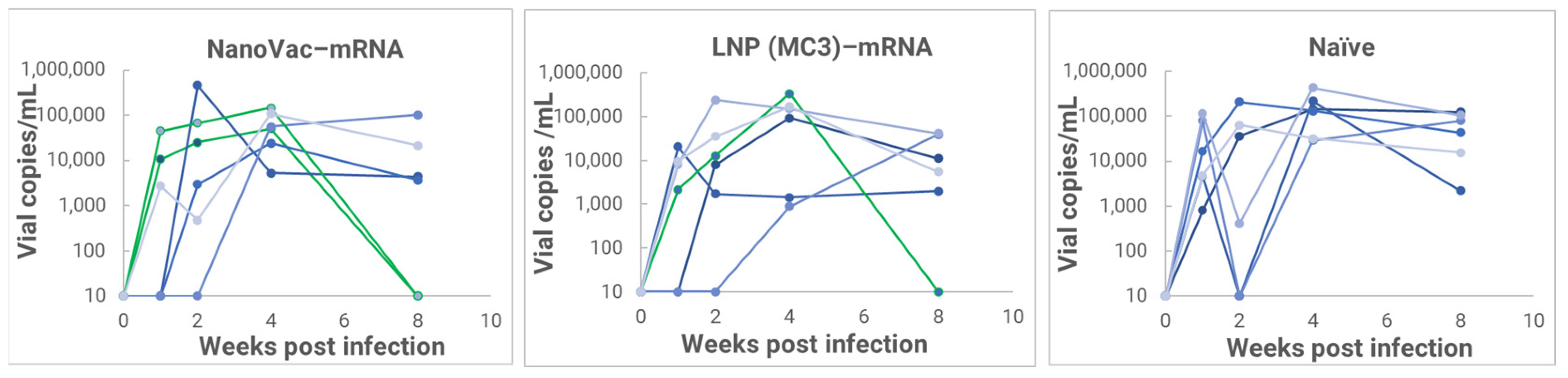
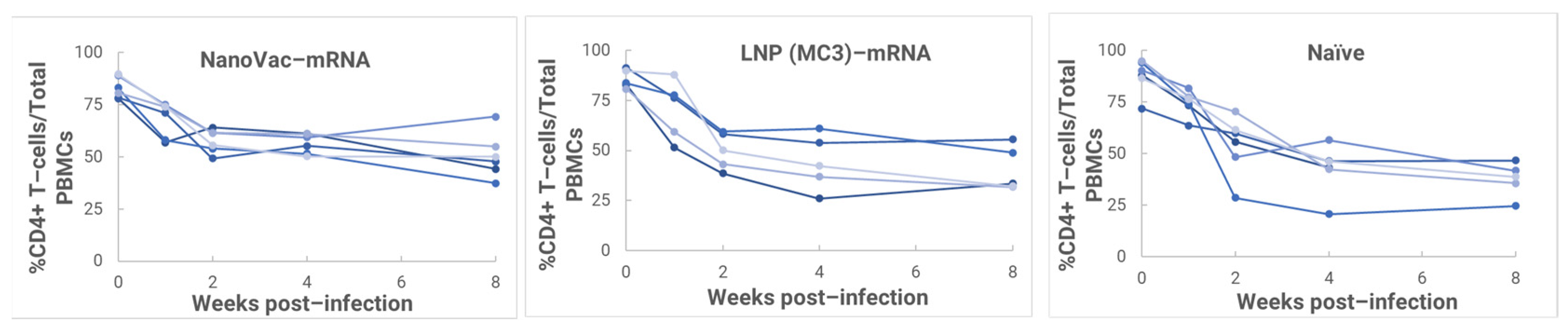
In the HIS model, the produced HIS-CD4/B mice could mount functional human CD4+ T-cell and B-cell responses and were used for immunogenicity and challenge studies. In the HIS mice, more than 60% of PBMCs were found to be human CD45+ leukocytes with the composition of human B-cells and CD4+ T-cells, and the CD4+ T-cell subsets were similar to those observed in humans. In the HIS mouse model, Luna Labs clearly demonstrated that NanoVac-V1V2 and NanoVac–mRNA vaccine candidates were able to induce both human-derived humoral and cellular responses against HIV V1V2 antigen in HIS-A2/DR4 mice. Both the IN and IM administration of NanoVac elicited a robust IgG response in HIS mice models. The codelivery of protein and mRNA via the IN administration of HIS mice stimulated much higher T-cells responses than other individually administrated ones did either via the IM or IN route. In the efficacy studies, we found that inoculation with HIV-1 resulted in productive and persistent infection in all our HIS-CD4/B mice infected with Lai-BaL with the viral load peaking at 3–4 weeks after the challenge and that the percentage of CD4+ T-cells in the blood began to decrease gradually at 8 weeks post-challenge compared to that in uninfected HIS-CD4/B mice, as a control. It is noteworthy that two of the HIV-1-infected mice previously vaccinated with the NanoVac–mRNA vaccine was cleared of virus infection by 8 weeks post-infection. Using a benchmark LNP–mRNA vaccine, only one mouse was cleared of infection. Further, the NanoVac delivery vehicle decreased the lipid content delivered by 10x, which could explain the negligible concerns arising from toxicity studies.



 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















