
The Crystal Structure of the Hsp90-LA1011 Complex and the Mechanism by Which LA1011 May Improve the Prognosis of Alzheimer’s Disease

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Methods and Materials
2.1. Protein Purification
2.2. Crystallization and Refinement
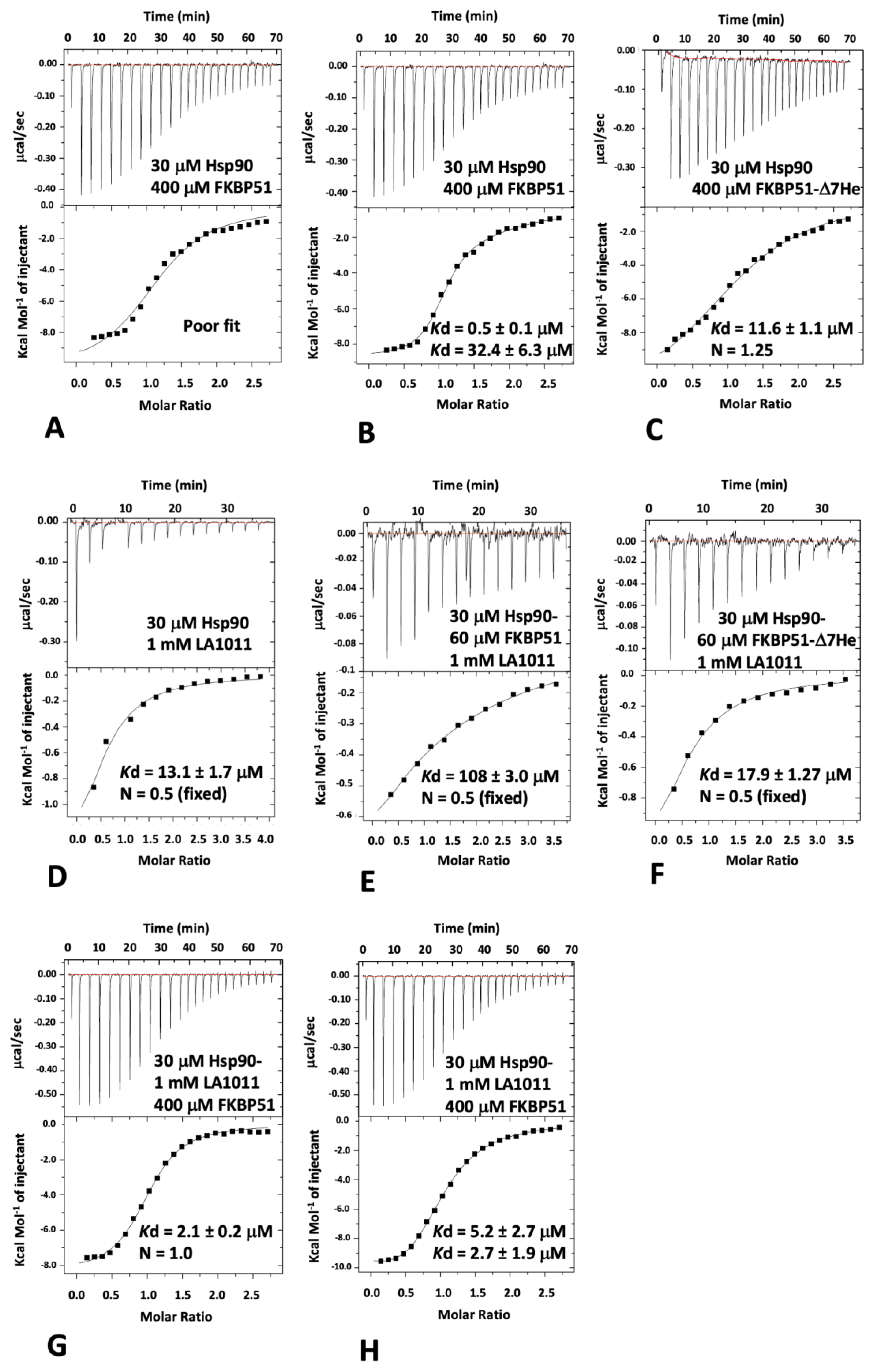
2.3. Isothermal Titration Calorimetry
3. Results and Conclusions
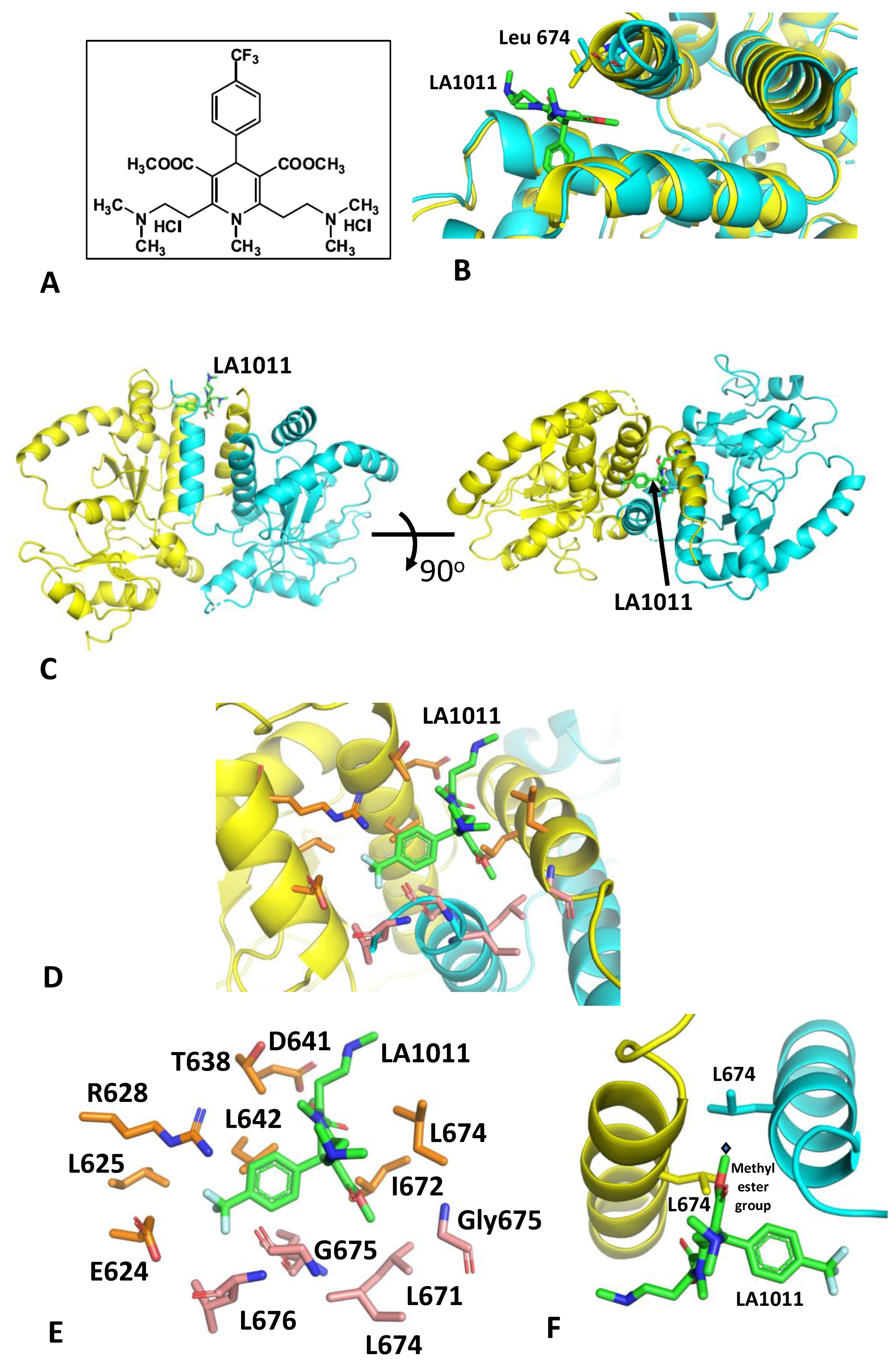
3.1. The Crystal Structure of Hsp90-LA1011
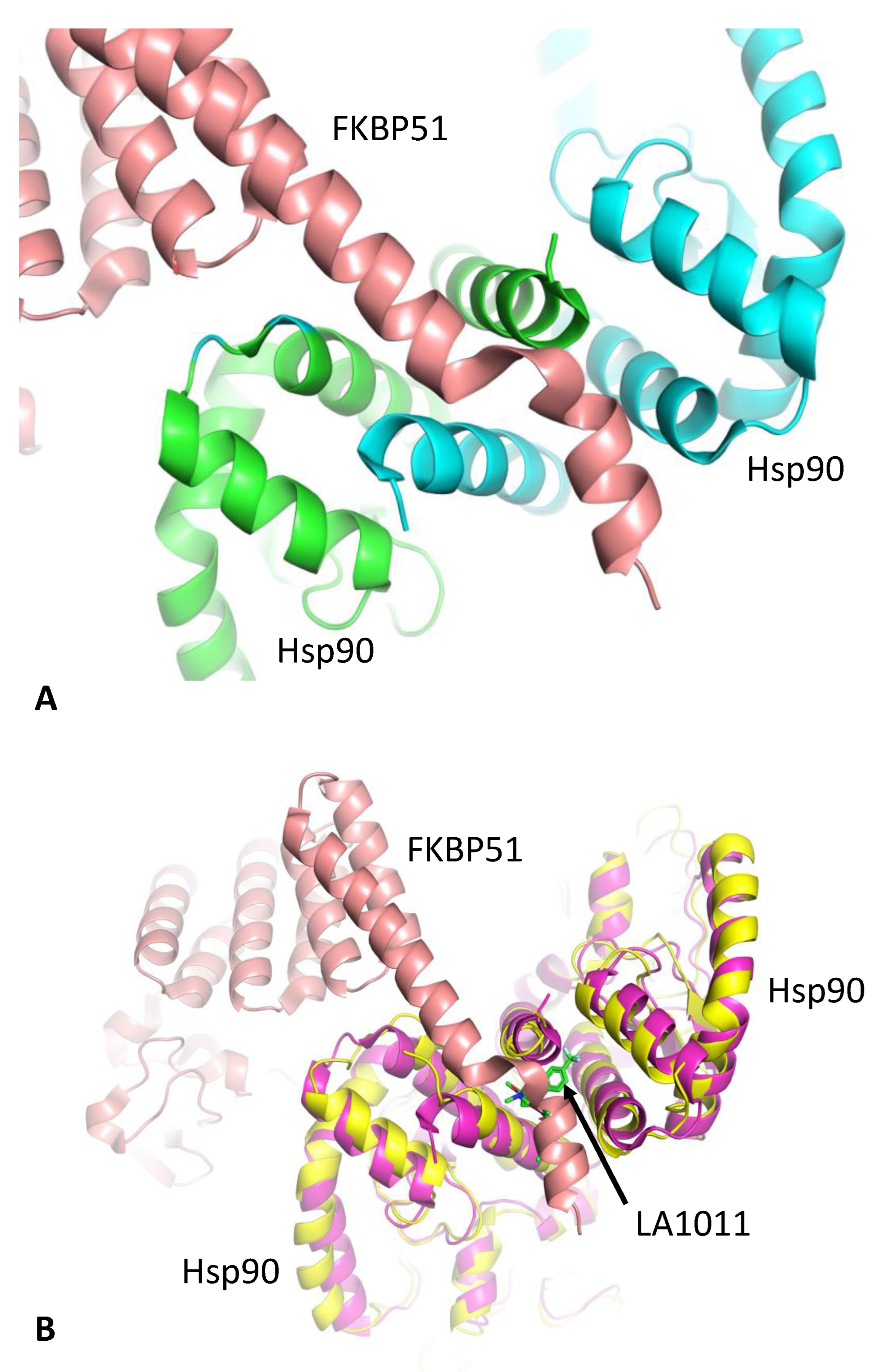
3.2. LA1011 and FKBP51 Competitively Bind to Hsp90
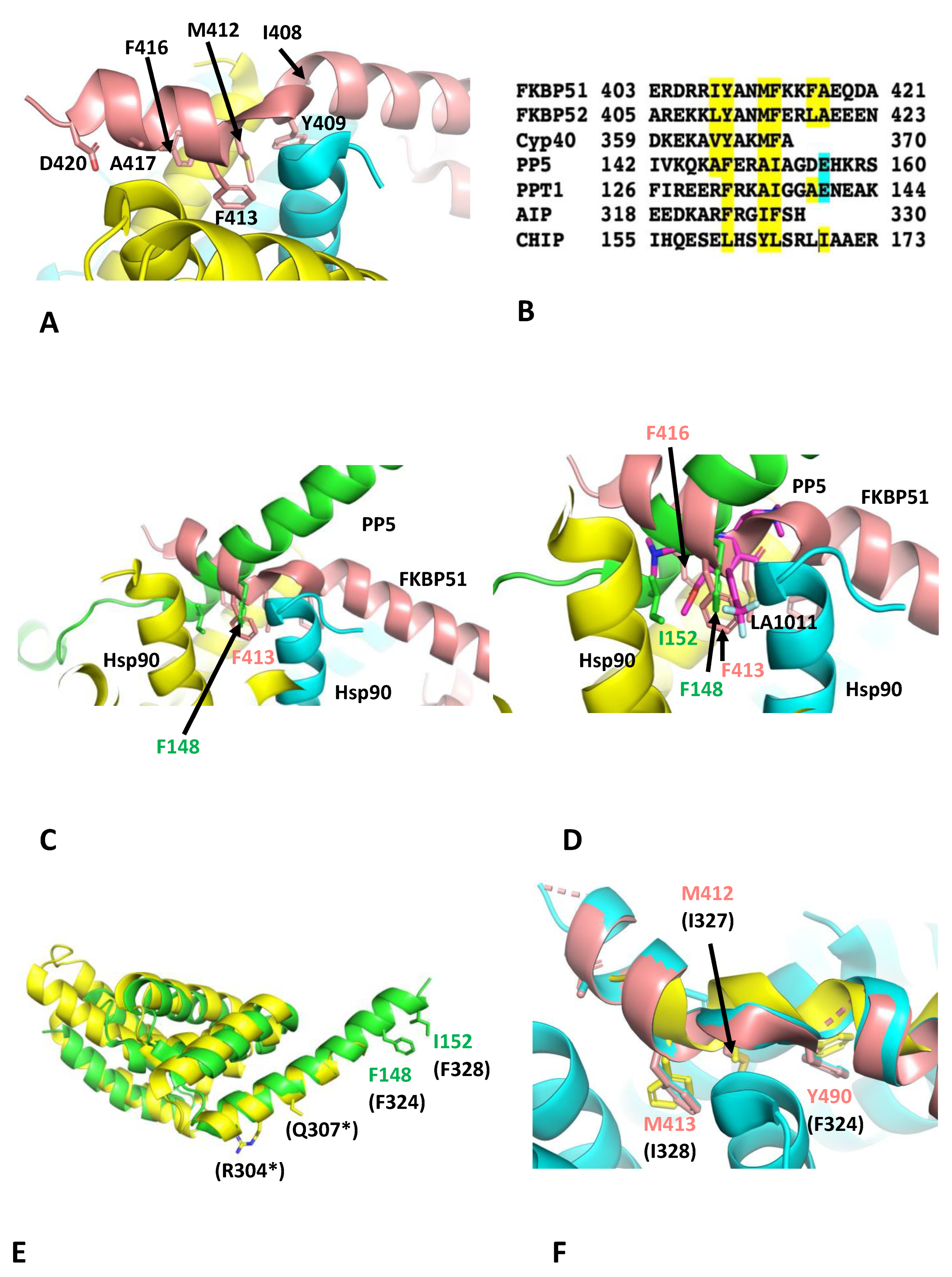
3.3. Effects of LA1011 on the Binding of Other TPR-Domain-Containing Co-Chaperones of Hsp90
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prodromou, C.; Bjorklund, D.M. Advances towards Understanding the Mechanism of Action of the Hsp90 Complex. Biomolecules 2022, 12, 600. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; D’avanzo, C.; Aronson, J.; Tanzi, R.E.; Kim, O.Y. Recapitulating amyloid β and tau pathology in human neural cell culture models: Clinical implications. US Neurol. 2015, 11, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.-R.; Tan, M.-S.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat Shock Protein 90 in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.B.; Rüdiger, S.G.D. Alzheimer Cells on Their Way to Derailment Show Selective Changes in Protein Quality Control Network. Front. Mol. Biosci. 2020, 7, 214. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Duarte, A.M.; Akoury, E.; Ippel, H.; Biernat, J.; Luengo, T.M.; Radli, M.; Didenko, T.; Nordhues, B.A.; Veprintsev, D.B.; et al. Hsp90-Tau Complex Reveals Molecular Basis for Specificity in Chaperone Action. Cell 2014, 156, 963–974. [Google Scholar] [CrossRef]
- Weickert, S.; Wawrzyniuk, M.; John, L.H.; Rüdiger, S.G.D.; Drescher, M. The mechanism of Hsp90-induced oligomerizaton of Tau. Sci. Adv. 2020, 6, eaax6999. [Google Scholar] [CrossRef]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Koren, J., 3rd; Dickey, C.A. Reconstructing the Hsp90/Tau Machine. Curr. Enzym. Inhib. 2013, 9, 41–45. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Trotter, J.H.; Abisambra, J.F.; Koren, J., 3rd; Lawson, L.Y.; Vestal, G.D.; O’Leary, J.C., 3rd; Johnson, A.G.; Jin, Y.; Jones, J.R.; et al. The Hsp90 Kinase Co-chaperone Cdc37 Regulates Tau Stability and Phosphorylation Dynamics. J. Biol. Chem. 2011, 286, 16976–16983. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Annaert, W.; De Strooper, B. Alzheimer’s Disease Neurons Fail the Acid Test. Cell 2010, 141, 1112–1114. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Munari, F.; Mollica, L.; Valente, C.; Parolini, F.; Kachoie, E.A.; Arrigoni, G.; D’Onofrio, M.; Capaldi, S.; Assfalg, M. Structural Basis for Chaperone-Independent Ubiquitination of Tau Protein by Its E3 Ligase CHIP. Angew. Chem. Int. Ed. Engl. 2022, 61, e202112374. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, Z.-W.; Mao, C.-Y.; Shi, C.-H.; Xu, Y.-M. CHIP as a therapeutic target for neurological diseases. Cell Death Dis. 2020, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Shelton, L.B.; Koren, J., 3rd; Blair, L.J. Imbalances in the Hsp90 Chaperone Machinery: Implications for Tauopathies. Front. Neurosci. 2017, 11, 724. [Google Scholar] [CrossRef]
- Baker, J.D.; Shelton, L.B.; Zheng, D.; Favretto, F.; Nordhues, B.A.; Darling, A.; Sullivan, L.E.; Sun, Z.; Solanki, P.K.; Martin, M.D.; et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017, 15, e2001336. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Koren, J., 3rd; Borysov, S.I.; Schmid, A.B.; Abisambra, J.F.; Blair, L.J.; Johnson, A.G.; Jones, J.R.; Shults, C.L.; O’Leary, J.C., 3rd; et al. The Hsp90 Cochaperone, FKBP51, Increases Tau Stability and Polymerizes Microtubules. J. Neurosci. 2010, 30, 591–599. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; O’Leary, J.C., 3rd; Blair, L.J.; Klengel, T.; Nordhues, B.A.; Fontaine, S.N.; Binder, E.B.; Dickey, C.A. Age-Associated Epigenetic Upregulation of the FKBP5 Gene Selectively Impairs Stress Resiliency. PLoS ONE 2014, 9, e107241. [Google Scholar] [CrossRef]
- Kuznetsova, I.L.; Ponomareva, N.V.; Alemastseva, E.A.; Manakhov, A.D.; Andreeva, T.V.; Gusev, F.E.; Rogaev, E.I. The Interactive Effect of Genetic and Epigenetic Variations in FKBP5 and ApoE Genes on Anxiety and Brain EEG Parameters. Genes 2022, 13, 164. [Google Scholar] [CrossRef]
- Justice, N.J. The relationship between stress and Alzheimer’s disease. Neurobiol. Stress 2018, 8, 127–133. [Google Scholar] [CrossRef]
- Lee, K.; Thwin, A.C.; Nadel, C.M.; Tse, E.; Gates, S.N.; Gestwicki, J.E.; Southworth, D.R. The structure of an Hsp90-immunophilin complex reveals cochaperone recognition of the client maturation state. Mol. Cell 2021, 81, 3496–3508.e3495. [Google Scholar] [CrossRef]
- Kamah, A.; Cantrelle, F.X.; Huvent, I.; Giustiniani, J.; Guillemeau, K.; Byrne, C.; Jacquot, Y.; Landrieu, I.; Baulieu, E.; Smet, C.; et al. Isomerization and Oligomerization of Truncated and Mutated Tau Forms by FKBP52 are Independent Processes. J. Mol. Biol. 2016, 428, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Giustiniani, J.; Guillemeau, K.; Dounane, O.; Sardin, E.; Huvent, I.; Schmitt, A.; Hamdane, M.; Buée, L.; Landrieu, I.; Lippens, G.; et al. The FK506-binding protein FKBP52 in vitro induces aggregation of truncated Tau forms with prion-like behavior. FASEB J. 2015, 29, 3171–3181. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.; Guillemeau, K.; Dounane, O.; Sazdovitch, V.; Duyckaerts, C.; Chambraud, B.; Baulieu, E.E.; Giustiniani, J. Caspase-cleaved Tau-D421 is colocalized with the immunophilin FKBP52 in the autophagy-endolysosomal system of Alzheimer’s disease neurons. Neurobiol. Aging 2016, 46, 124–137. [Google Scholar] [CrossRef]
- Conde, R.; Xavier, J.; McLoughlin, C.; Chinkers, M.; Ovsenek, N. Protein phosphatase 5 Is a negative modulator of heat shock factor 1. J. Biol. Chem. 2005, 280, 28989–28996. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Wu, G.; Rossie, S.; Wegiel, J.; Li, L.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of microtubule-associated protein tau by protein phosphatase 5. J. Neurochem. 2004, 88, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Hamos, J.E.; Oblas, B.; Pulaski-Salo, D.; Welch, W.J.; Bole, D.G.; Drachman, D.A. Expression of heat shock proteins in Alzheimer’s disease. Neurology 1991, 41, 345–350. [Google Scholar] [CrossRef]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.S.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef]
- Kasza, A.; Hunya, A.; Frank, Z.; Fülöp, F.; Török, Z.; Balogh, G.; Sántha, M.; Bálind, A.; Bernáth, S.; Blundell, K.L.; et al. Dihydropyridine Derivatives Modulate Heat Shock Responses and have a Neuroprotective Effect in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 53, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Roe, M.S.; Wahab, B.; Török, Z.; Horváth, I.; Vigh, L.; Prodromou, C. Dihydropyridines Allosterically Modulate Hsp90 Providing a Novel Mechanism for Heat Shock Protein Co-induction and Neuroprotection. Front. Mol. Biosci. 2018, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.M.; Hernández-Ramírez, L.C.; Trivellin, G.; Zhou, L.; Roe, S.M.; Korbonits, M.; Prodromou, C. Structure of the TPR Domain of AIP: Lack of Client Protein Interaction with the C-Terminal α-7 Helix of the TPR Domain of AIP Is Sufficient for Pituitary Adenoma Predisposition. PLoS ONE 2012, 7, e53339. [Google Scholar] [CrossRef] [PubMed]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase activity of Hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Struct. Biol. Crystallogr. 2004, D60, 2126–2132. [Google Scholar] [CrossRef]
- Oberoi, J.; Guiu, X.A.; Outwin, E.A.; Schellenberger, P.; Roumeliotis, T.I.; Choudhary, J.S.; Pearl, L.H. HSP90-CDC37-PP5 forms a structural platform for kinase dephosphorylation. Nat. Commun. 2022, 13, 7343. [Google Scholar] [CrossRef]
- Jaime-Garza, M.; Nowotny, C.A.; Coutandin, D.; Wang, F.; Tabios, M.; Agard, D.A. Hsp90 provides a platform for kinase dephosphorylation by PP5. Nat. Commun. 2023, 14, 2197. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set (Highest Shell in Parentheses) | AutoPROC | STARANISO |
|---|---|---|
| a (Å) | 97.71 | |
| b (Å) | 97.71 | |
| c (Å) | 275.57 | |
| α (°) | 90.0 | |
| β (°) | 90.0 | |
| γ (°) | 90.0 | |
| Space group | P 41 21 21 | |
| Wavelength (Å) | 0.96864 | |
| Resolution limit (Å) | 92.1–3.18 (3.23–3.18) | 92.1–2.94 (3.19–2.94) |
| Number of unique obs. | 22,841 (1103) | 23,054 (1153) |
| Completeness (%) | 97.4 (97.0) | 91.9 (58.1) |
| Multiplicity | 9.3 (5.9) | 9.2 (4.0) |
| Rmerge (I) % | 0.218 (1.867) | 0.217 (1.215) |
| Rpim (I) % | 0.073 (0.803) | 0.073 (0.666) |
| CC1/2 | 0.995 (0.408) | 0.995 (0.361) |
| I/σI | 9.0 (1.1) | 9.0 (1.4) |
| Refinement | ||
| Resolution range (Å) | 79.7–2.94 | |
| Rcryst | 0.2470 | |
| Rfree | 0.3062 | |
| Number of protein atoms | 6816 | |
| Number of ligand atoms | 0 | |
| Number of solvent atoms | 1 | |
| Mean B | 70.11 | |
| Rmsd bond lengths (Å) | 0.005 | |
| Rmsd bond angles (°) | 1.051 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roe, S.M.; Török, Z.; McGown, A.; Horváth, I.; Spencer, J.; Pázmány, T.; Vigh, L.; Prodromou, C. The Crystal Structure of the Hsp90-LA1011 Complex and the Mechanism by Which LA1011 May Improve the Prognosis of Alzheimer’s Disease. Biomolecules 2023, 13, 1051. https://doi.org/10.3390/biom13071051
Roe SM, Török Z, McGown A, Horváth I, Spencer J, Pázmány T, Vigh L, Prodromou C. The Crystal Structure of the Hsp90-LA1011 Complex and the Mechanism by Which LA1011 May Improve the Prognosis of Alzheimer’s Disease. Biomolecules. 2023; 13(7):1051. https://doi.org/10.3390/biom13071051
Chicago/Turabian StyleRoe, S. Mark, Zsolt Török, Andrew McGown, Ibolya Horváth, John Spencer, Tamás Pázmány, László Vigh, and Chrisostomos Prodromou. 2023. "The Crystal Structure of the Hsp90-LA1011 Complex and the Mechanism by Which LA1011 May Improve the Prognosis of Alzheimer’s Disease" Biomolecules 13, no. 7: 1051. https://doi.org/10.3390/biom13071051
APA StyleRoe, S. M., Török, Z., McGown, A., Horváth, I., Spencer, J., Pázmány, T., Vigh, L., & Prodromou, C. (2023). The Crystal Structure of the Hsp90-LA1011 Complex and the Mechanism by Which LA1011 May Improve the Prognosis of Alzheimer’s Disease. Biomolecules, 13(7), 1051. https://doi.org/10.3390/biom13071051