

Cardiac Molecular Remodeling by Anticancer Drugs: Doxorubicin Affects More Metabolism While Mitoxantrone Impacts More Autophagy in Adult CD-1 Male Mice
 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Experimental Design
2.4. Blood Collection and Serum Analysis
2.5. Heart Collection and Homogenization
2.6. Histological Analysis
2.7. Western Blotting Analysis
2.8. Acylcarnitines and Amino Acids Analysis
2.9. Determination of Citrate Synthase (CS) Activity
2.10. Statistical Analysis
3. Results
3.1. DOX Had a Greater Impact on Whole-Body Weight and Serum Biomarkers of Heart Damage Than MTX
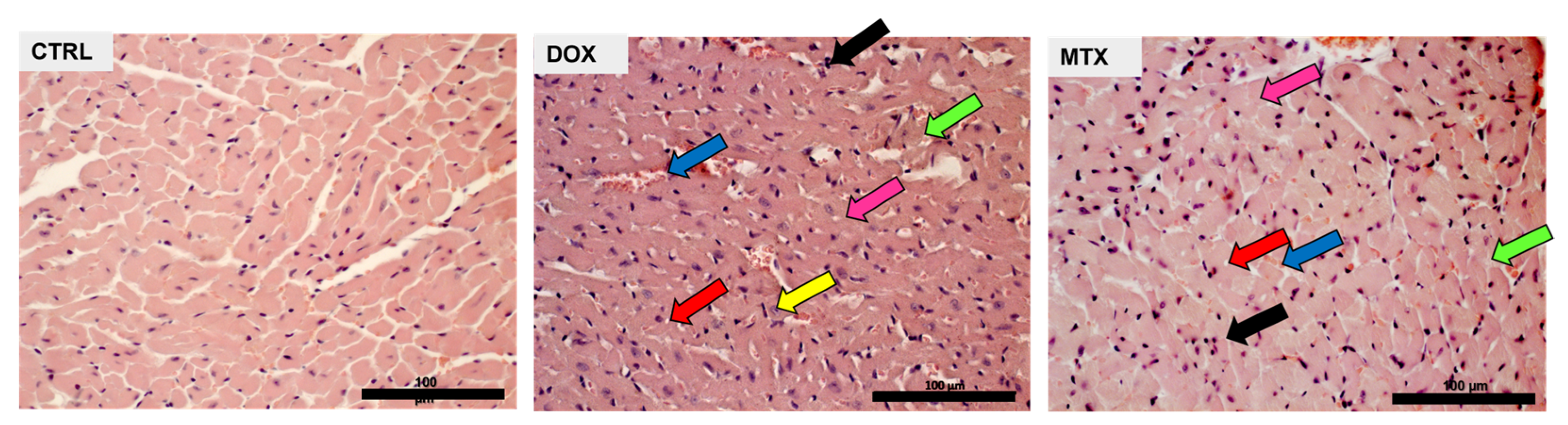
3.2. DOX Had a Higher Effect on Cardiac Structure Than MTX
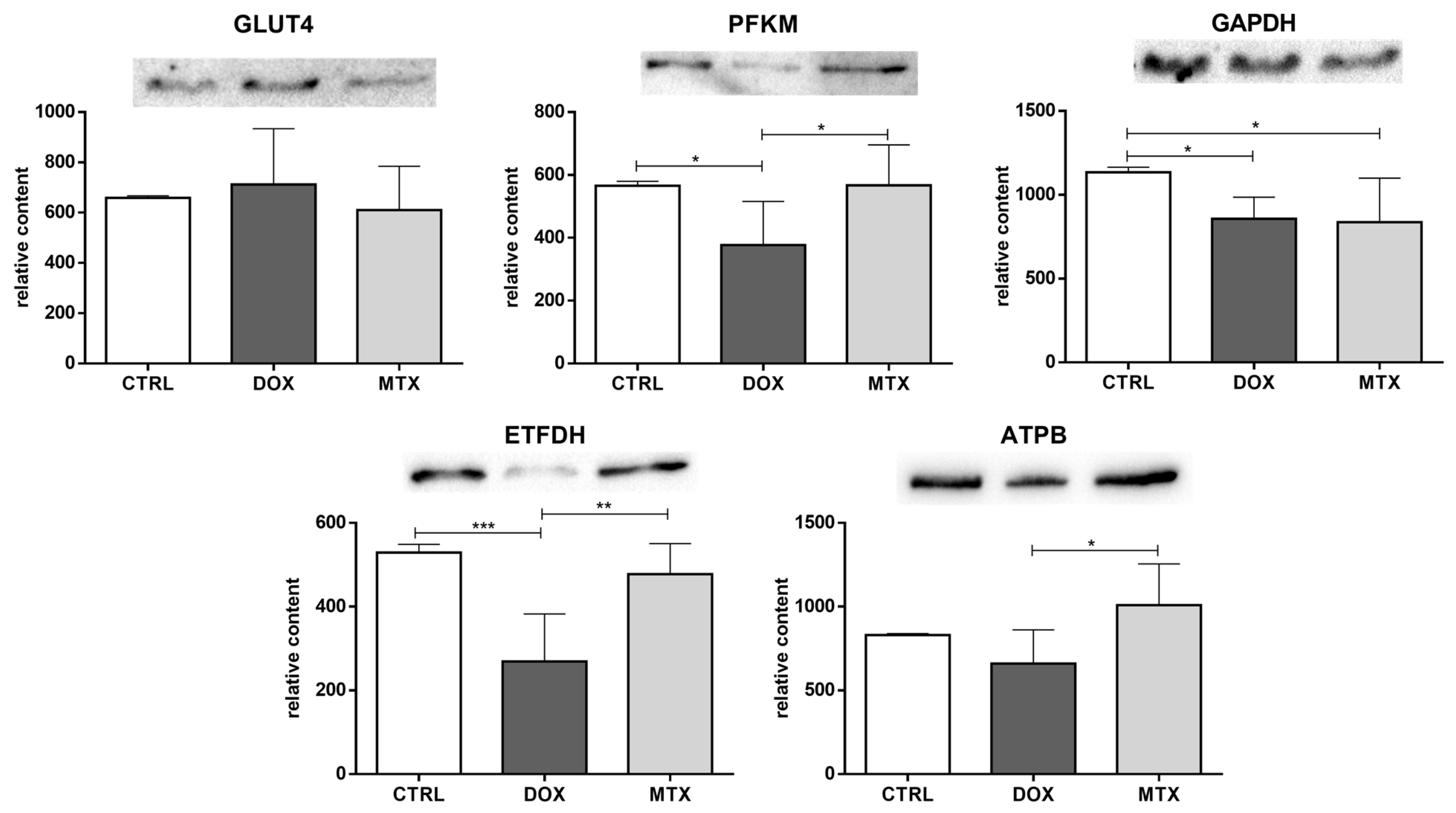
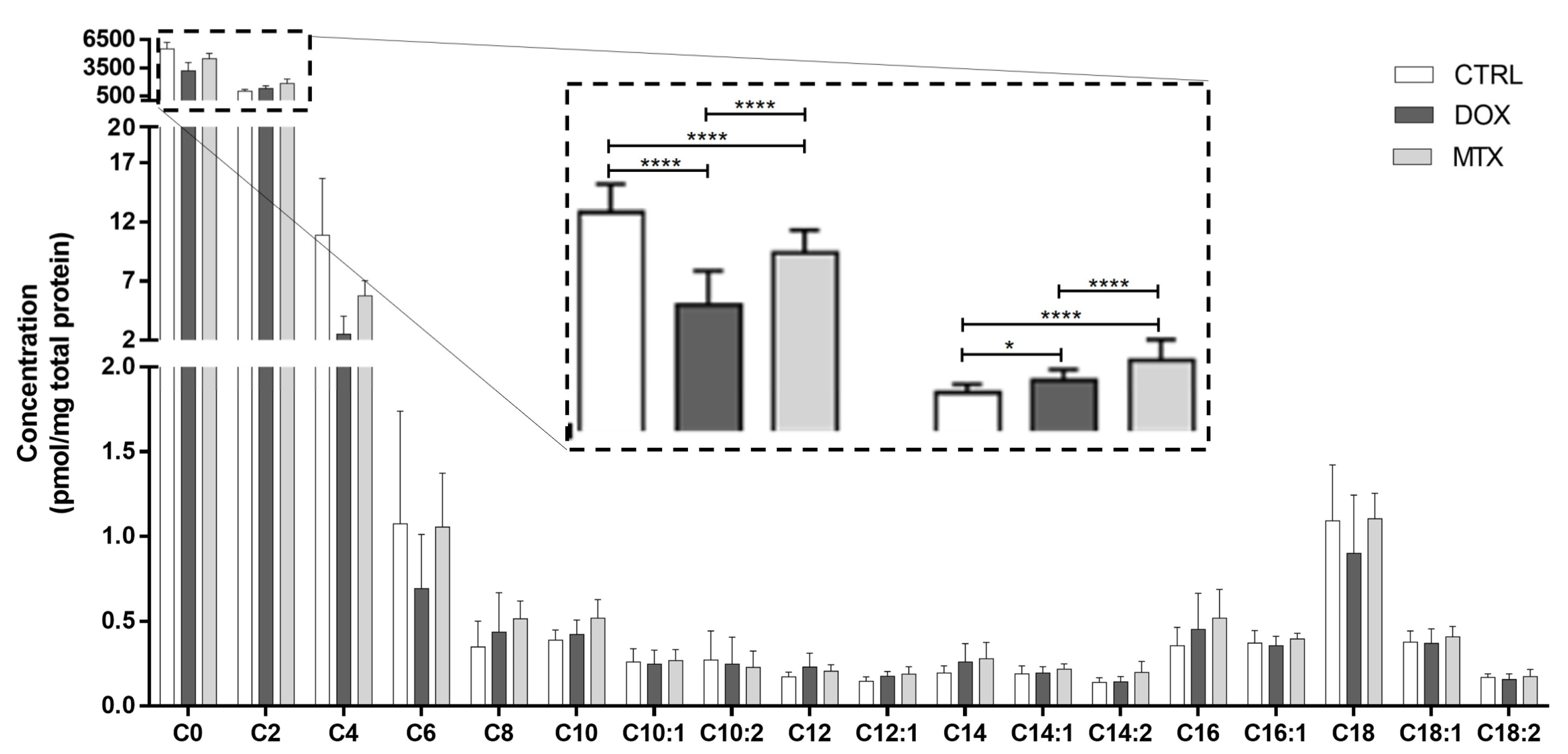
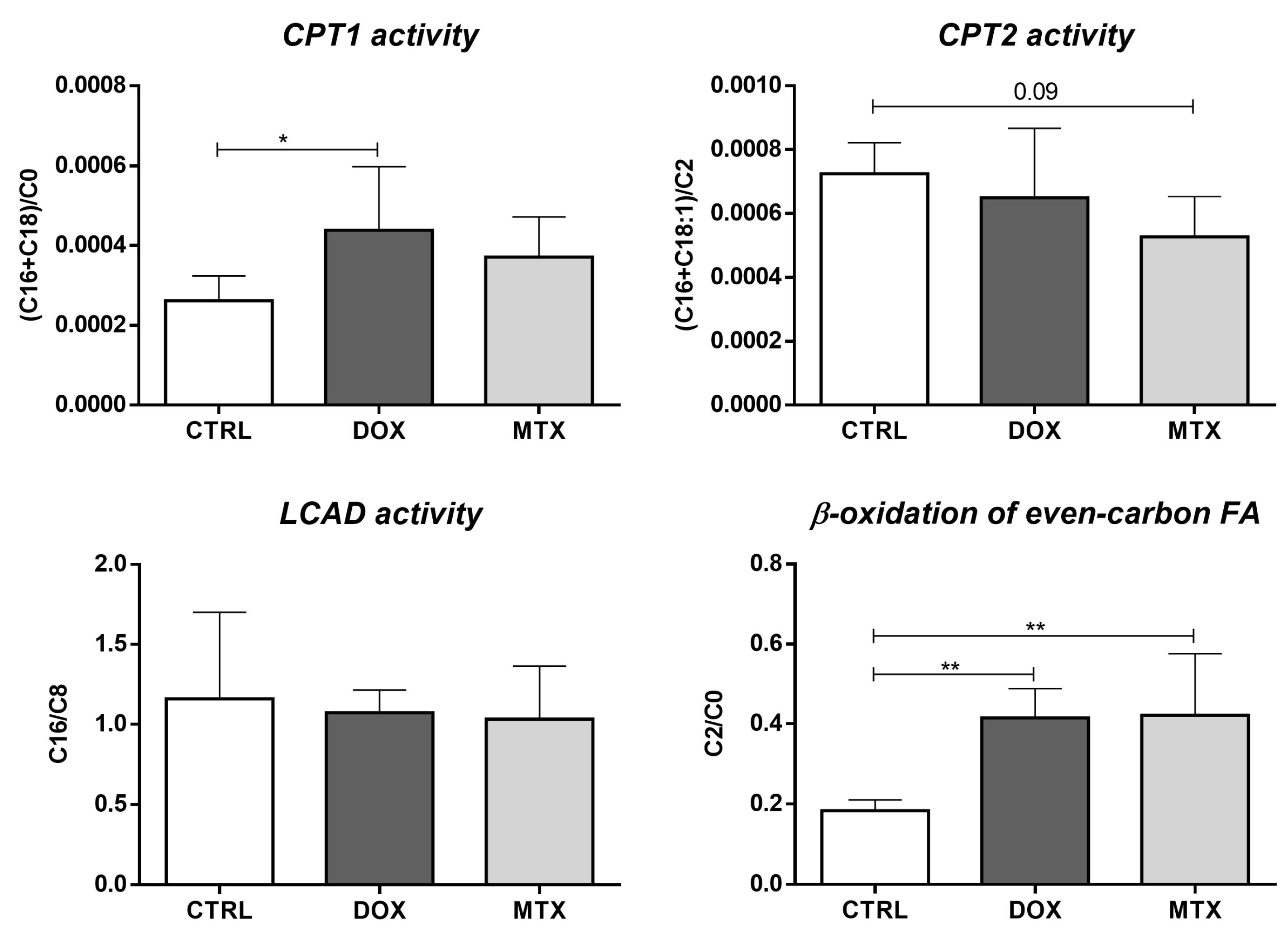
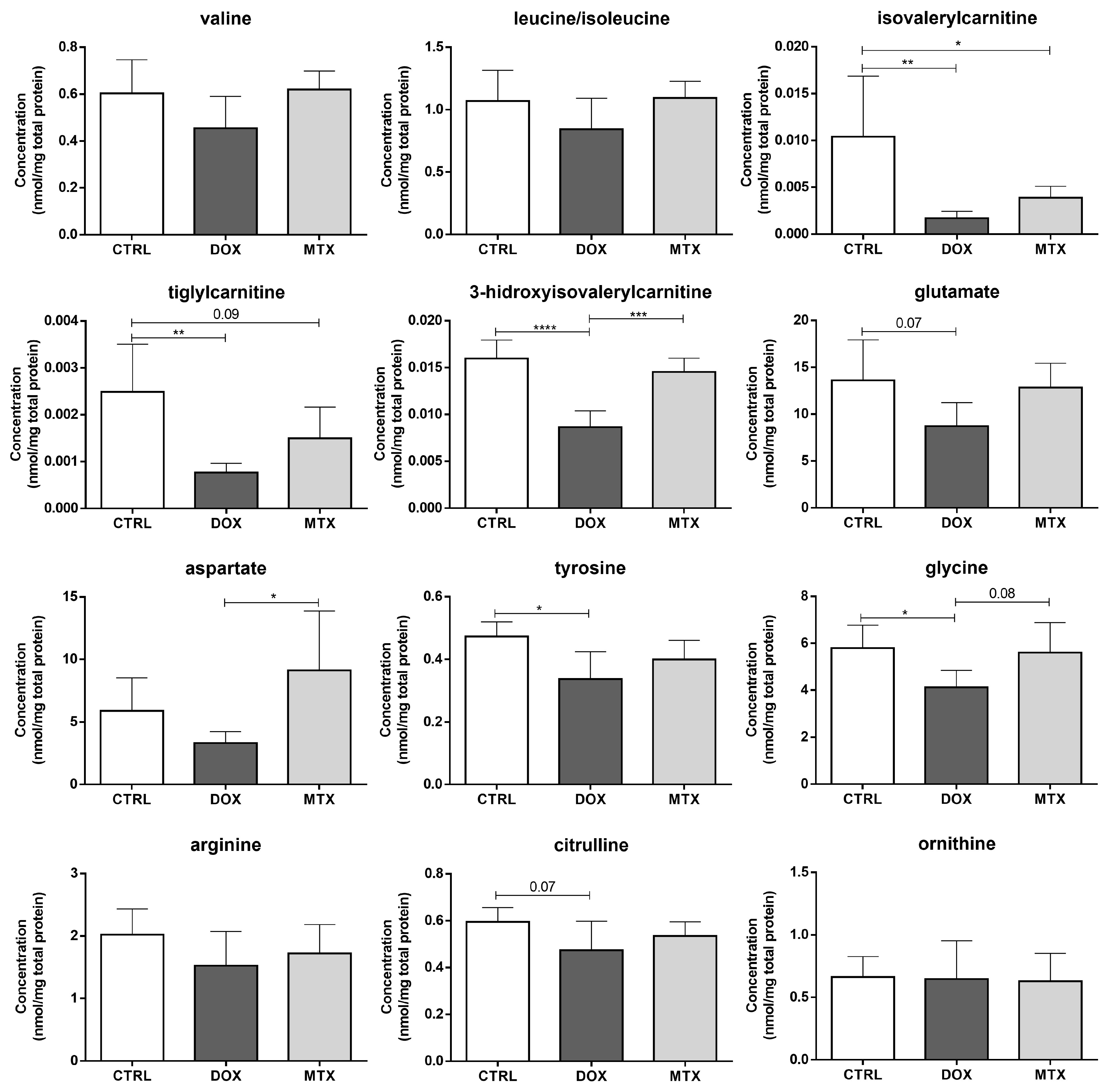
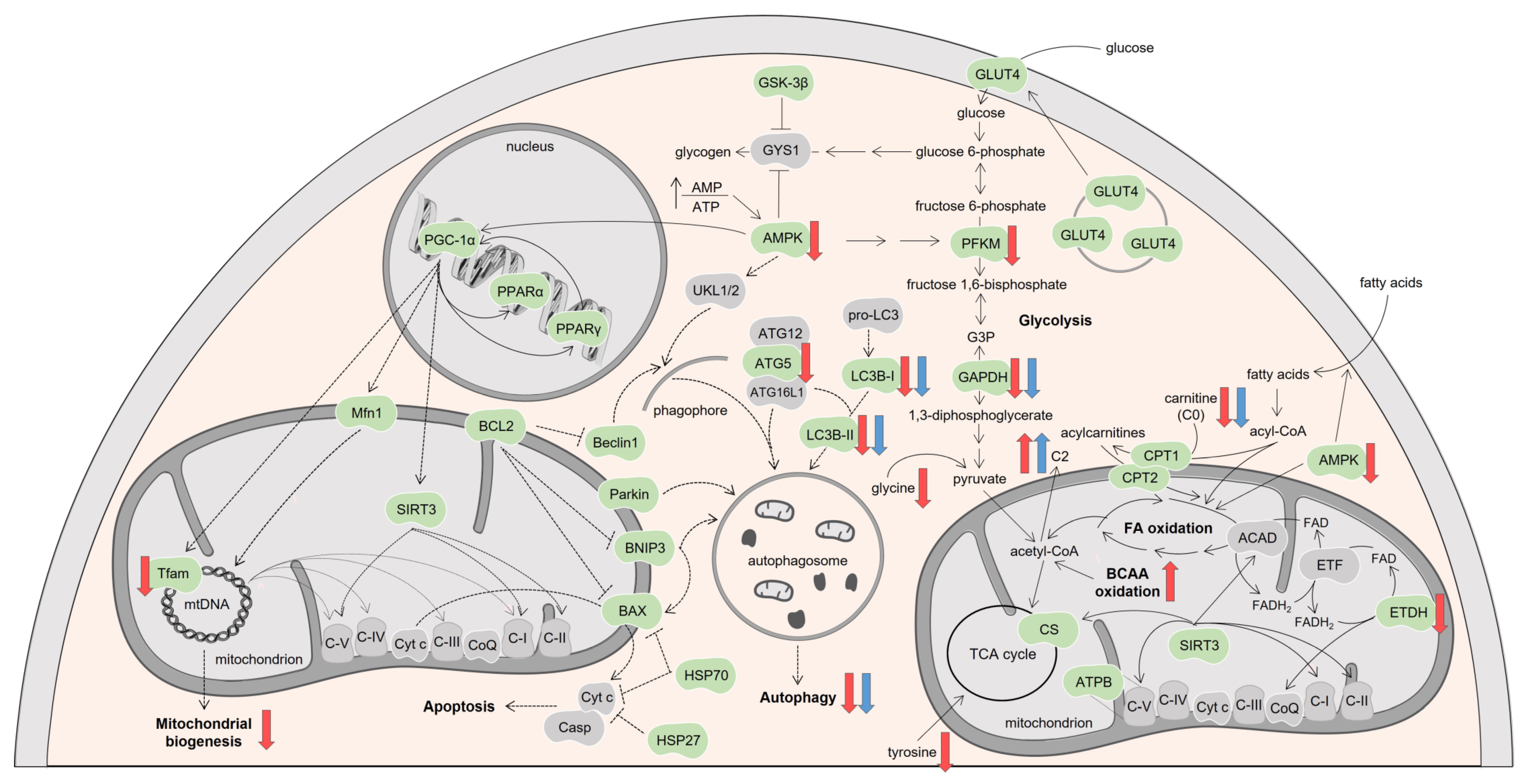
3.3. DOX Had a Higher Effect on Cardiac Oxidative Metabolism Than MTX
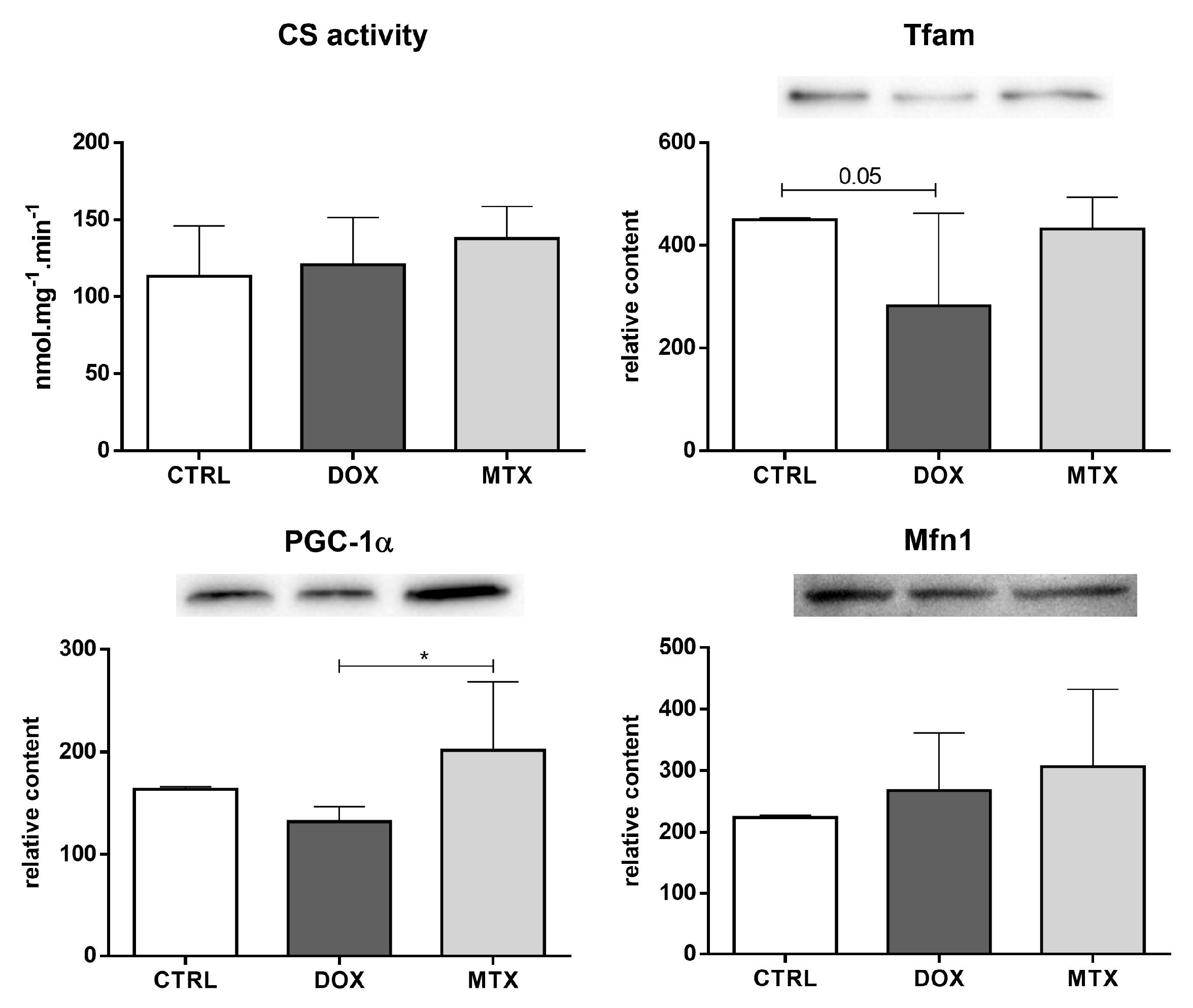
3.4. DOX Affected Cardiac Mitochondrial Biogenesis Differently from MTX
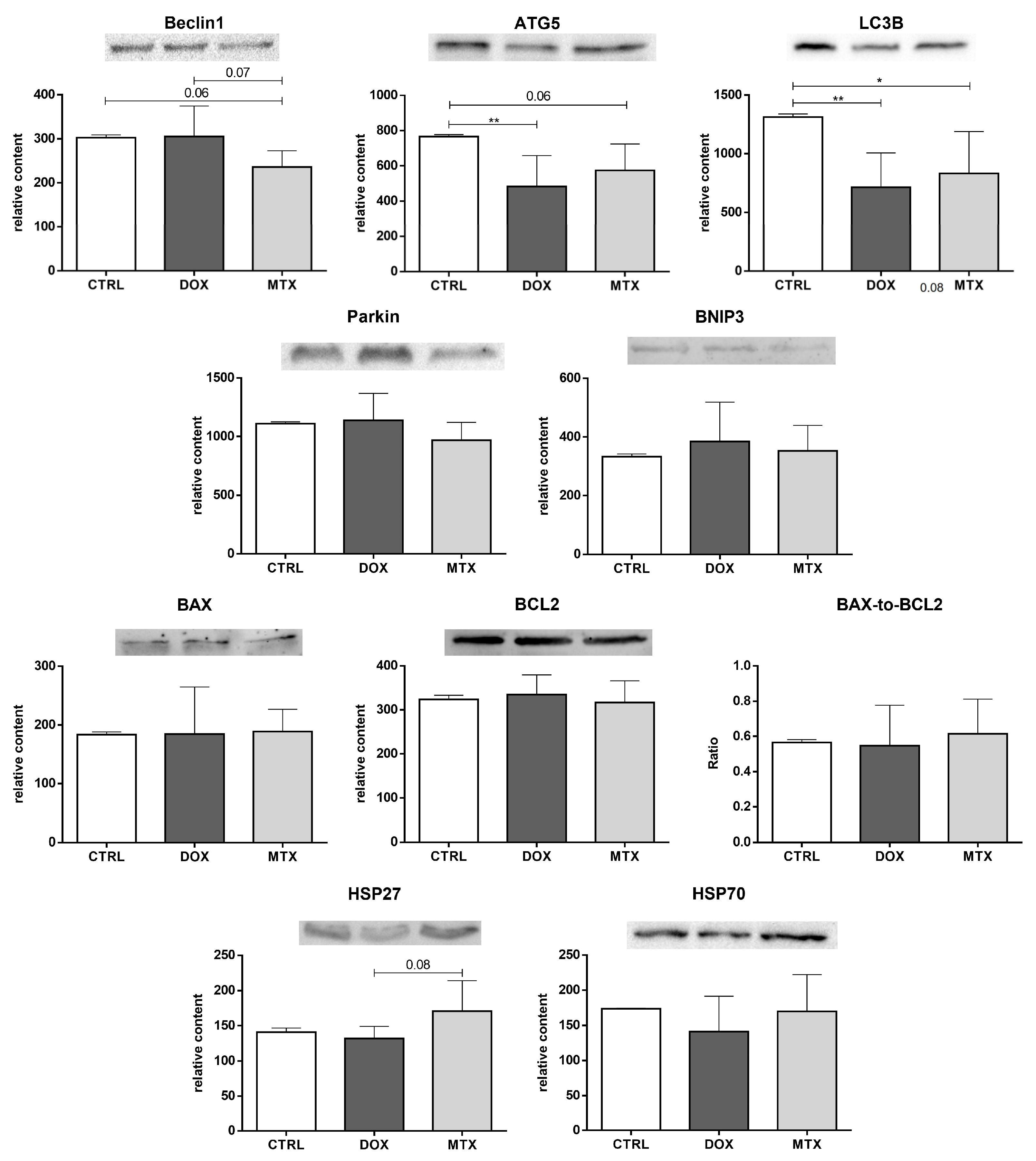
3.5. MTX Had a Bigger Impact on Cardiac Autophagy Than DOX
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Citron, M.L.; Berry, D.A.; Cirrincione, C.; Hudis, C.; Winer, E.P.; Gradishar, W.J.; Davidson, N.E.; Martino, S.; Livingston, R.; Ingle, J.N.; et al. Randomized Trial of Dose-Dense Versus Conventionally Scheduled and Sequential Versus Concurrent Combination Chemotherapy as Postoperative Adjuvant Treatment of Node-Positive Primary Breast Cancer: First Report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J. Clin. Oncol. 2003, 21, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, B.; Suciu, S.; Archimbaud, E.; Haak, H.; Stryckmans, P.; de Cataldo, R.; Dekker, A.W.; Berneman, Z.N.; Thyss, A.; van der Lelie, J.; et al. Mitoxantrone versus Daunorubicin in Induction-Consolidation Chemotherapy—The Value of Low-Dose Cytarabine for Maintenance of Remission, and an Assessment of Prognostic Factors in Acute Myeloid Leukemia in the Elderly: Final Report. European Organization for the Research and Treatment of Cancer and the Dutch-Belgian Hemato-Oncology Cooperative Hovon Group. J. Clin. Oncol. 1998, 16, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Seiter, K. Toxicity of the Topoisomerase II Inhibitors. Expert Opin. Drug Saf. 2005, 4, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Feijen, E.A.M.; Leisenring, W.M.; Stratton, K.L.; Ness, K.K.; van der Pal, H.J.H.; Caron, H.N.; Armstrong, G.T.; Green, D.M.; Hudson, M.M.; Oeffinger, K.C.; et al. Equivalence Ratio for Daunorubicin to Doxorubicin in Relation to Late Heart Failure in Survivors of Childhood Cancer. J. Clin. Oncol. 2015, 33, 3774–3780. [Google Scholar] [CrossRef] [PubMed]
- Feijen, E.A.M.; Leisenring, W.M.; Stratton, K.L.; Ness, K.K.; van der Pal, H.J.H.; van Dalen, E.C.; Armstrong, G.T.; Aune, G.J.; Green, D.M.; Hudson, M.M.; et al. Derivation of Anthracycline and Anthraquinone Equivalence Ratios to Doxorubicin for Late-Onset Cardiotoxicity. JAMA Oncol. 2019, 5, 864. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Aronow, W. Cardiotoxicity of Cancer Chemotherapy in Clinical Practice. Hosp. Pract. 2019, 47, 6–15. [Google Scholar] [CrossRef]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of Anticancer Treatments: Epidemiology, Detection, and Management: Cardiotoxicity of Anticancer Treatments. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef]
- Masood, I.; Kiani, M.H.; Ahmad, M.; Masood, M.I.; Sadaquat, H. Major Contributions towards Finding a Cure for Cancer through Chemotherapy: A Historical Review. Tumori J. 2016, 102, 6–17. [Google Scholar] [CrossRef]
- Hu, X.; Li, B.; Li, L.; Li, B.; Luo, J.; Shen, B. Asiatic Acid Protects against Doxorubicin-Induced Cardiotoxicity in Mice. Oxid. Med. Cell. Longev. 2020, 2020, 5347204. [Google Scholar] [CrossRef]
- Sabatino, J.; De Rosa, S.; Tammè, L.; Iaconetti, C.; Sorrentino, S.; Polimeni, A.; Mignogna, C.; Amorosi, A.; Spaccarotella, C.; Yasuda, M.; et al. Empagliflozin Prevents Doxorubicin-Induced Myocardial Dysfunction. Cardiovasc. Diabetol. 2020, 19, 66. [Google Scholar] [CrossRef]
- Stamm, P.; Kirmes, I.; Palmer, A.; Molitor, M.; Kvandova, M.; Kalinovic, S.; Mihalikova, D.; Reid, G.; Wenzel, P.; Münzel, T.; et al. Doxorubicin Induces Wide-Spread Transcriptional Changes in the Myocardium of Hearts Distinguishing between Mice with Preserved and Impaired Cardiac Function. Life Sci. 2021, 284, 119879. [Google Scholar] [CrossRef] [PubMed]
- Najafian, J.; Nasri, A.; Etemadifar, M.; Salehzadeh, F. Late Cardiotoxicity in MS Patients Treated with Mitoxantrone. Int. J. Prev. Med. 2019, 10, 211. [Google Scholar] [CrossRef]
- Shaikh, A.Y.; Suryadevara, S.; Tripathi, A.; Ahmed, M.; Kane, J.L.; Escobar, J.; Cerny, J.; Nath, R.; McManus, D.D.; Shih, J.; et al. Mitoxantrone-Induced Cardiotoxicity in Acute Myeloid Leukemia-A Velocity Vector Imaging Analysis. Echocardiography 2016, 33, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of Anticancer Treatments: What the Cardiologist Needs to Know. Nat. Rev. Cardiol. 2010, 7, 564–575. [Google Scholar] [CrossRef]
- Reis-Mendes, A.; Sousa, E.; de Lourdes Bastos, M.; Marisa Costa, V. The Role of the Metabolism of Anticancer Drugs in Their Induced-Cardiotoxicity. Curr. Drug Metab. 2015, 17, 75–90. [Google Scholar] [CrossRef]
- Anjos, M.; Fontes-Oliveira, M.; Costa, V.M.; Santos, M.; Ferreira, R. An Update of the Molecular Mechanisms Underlying Doxorubicin plus Trastuzumab Induced Cardiotoxicity. Life Sci. 2021, 280, 119760. [Google Scholar] [CrossRef]
- Ashley, N.; Poulton, J. Mitochondrial DNA Is a Direct Target of Anti-Cancer Anthracycline Drugs. Biochem. Biophys. Res. Commun. 2009, 378, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Brandão, S.R.; Carvalho, F.; Amado, F.; Ferreira, R.; Costa, V.M. Insights on the Molecular Targets of Cardiotoxicity Induced by Anticancer Drugs: A Systematic Review Based on Proteomic Findings. Metabolism 2022, 134, 155250. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Prasad, S.V.N.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of Doxorubicin Is Mediated through Mitochondrial Iron Accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Myers, C.; McGuire, W.; Liss, R.; Ifrim, I.; Grotzinger, K.; Young, R. Adriamycin: The Role of Lipid Peroxidation in Cardiac Toxicity and Tumor Response. Science 1977, 197, 165–167. [Google Scholar] [CrossRef]
- Pereira, G.C.; Pereira, S.P.; Tavares, L.C.; Carvalho, F.S.; Magalhães-Novais, S.; Barbosa, I.A.; Santos, M.S.; Bjork, J.; Moreno, A.J.; Wallace, K.B.; et al. Cardiac Cytochrome c and Cardiolipin Depletion during Anthracycline-Induced Chronic Depression of Mitochondrial Function. Mitochondrion 2016, 30, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Abilash, V.G.; Pichiah, P.B.T.; Arunachalam, S. Molecular Mechanism of Doxorubicin-Induced Cardiomyopathy—An Update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the Molecular Basis of Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.M.; Capela, J.P.; de Bastos, M.L.; Duarte, J.A.; Remião, F.; Carvalho, F. Pharmacological Concentrations of Mitoxantrone Are Able to Transiently Activate Caspases and Dually Modify Glutathione Pathways in HL-1 Cells. Toxicol. Lett. 2013, 221, S237. [Google Scholar] [CrossRef]
- Rossato, L.G.; Costa, V.M.; Dallegrave, E.; Arbo, M.; Silva, R.; Ferreira, R.; Amado, F.; Dinis-Oliveira, R.J.; Duarte, J.A.; de Lourdes Bastos, M.; et al. Mitochondrial Cumulative Damage Induced by Mitoxantrone: Late Onset Cardiac Energetic Impairment. Cardiovasc. Toxicol. 2014, 14, 30–40. [Google Scholar] [CrossRef]
- Rossato, L.G.; Costa, V.M.; Vilas-Boas, V.; de Lourdes Bastos, M.; Rolo, A.; Palmeira, C.; Remião, F. Therapeutic Concentrations of Mitoxantrone Elicit Energetic Imbalance in H9c2 Cells as an Earlier Event. Cardiovasc. Toxicol. 2013, 13, 413–425. [Google Scholar] [CrossRef]
- Barrett, K.E.; Boitano, S.; Barman, S.M.; Brooks, H.L.; Ganong, W.F. (Eds.) Ganong’s Review of Medical Physiology, 25th ed.; A Lange Medical Book; McGraw-Hill: New York, NY, USA, 2015; ISBN 978-0-07-182510-8. [Google Scholar]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure: Implications Beyond ATP Production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Moe, G.W.; Marín-García, J. Role of Cell Death in the Progression of Heart Failure. Heart Fail. Rev. 2016, 21, 157–167. [Google Scholar] [CrossRef]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro Phosphorylation Sites Regulate Parkin Recruitment and Mitochondrial Motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef]
- Kitmitto, A.; Baudoin, F.; Cartwright, E.J. Cardiomyocyte Damage Control in Heart Failure and the Role of the Sarcolemma. J. Muscle Res. Cell Motil. 2019, 40, 319–333. [Google Scholar] [CrossRef]
- Brandão, S.R.; Reis-Mendes, A.; Domingues, P.; Duarte, J.A.; Bastos, M.L.; Carvalho, F.; Ferreira, R.; Costa, V.M. Exploring the Aging Effect of the Anticancer Drugs Doxorubicin and Mitoxantrone on Cardiac Mitochondrial Proteome Using a Murine Model. Toxicology 2021, 459, 152852. [Google Scholar] [CrossRef] [PubMed]
- Dores-Sousa, J.L.; Duarte, J.A.; Seabra, V.; de Bastos, M.L.; Carvalho, F.; Costa, V.M. The Age Factor for Mitoxantrone’s Cardiotoxicity: Multiple Doses Render the Adult Mouse Heart More Susceptible to Injury. Toxicology 2015, 329, 106–119. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose Translation from Animal to Human Studies Revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.; Padrão, A.I.; Duarte, J.A.; Gonçalves-Monteiro, S.; Duarte-Araújo, M.; Remião, F.; Carvalho, F.; Sousa, E.; Bastos, M.L.; Costa, V.M. Role of Inflammation and Redox Status on Doxorubicin-Induced Cardiotoxicity in Infant and Adult CD-1 Male Mice. Biomolecules 2021, 11, 1725. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.; Dores-Sousa, J.L.; Padrão, A.I.; Duarte-Araújo, M.; Duarte, J.A.; Seabra, V.; Gonçalves-Monteiro, S.; Remião, F.; Carvalho, F.; Sousa, E.; et al. Inflammation as a Possible Trigger for Mitoxantrone-Induced Cardiotoxicity: An In Vivo Study in Adult and Infant Mice. Pharmaceuticals 2021, 14, 510. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Hu, R.-Y.; Chou, H.-C. Quercetin-Induced Cardioprotection against Doxorubicin Cytotoxicity. J. Biomed. Sci. 2013, 20, 11. [Google Scholar] [CrossRef]
- Cui, Y.; Piao, C.-S.; Ha, K.-C.; Kim, D.-S.; Lee, G.-H.; Kim, H.-K.; Chae, S.-W.; Lee, Y.-C.; Park, S.-J.; Yoo, W.-H.; et al. Measuring Adriamycin-Induced Cardiac Hemodynamic Dysfunction with a Proteomics Approach. Immunopharmacol. Immunotoxicol. 2010, 32, 376–386. [Google Scholar] [CrossRef]
- Holmgren, G.; Sartipy, P.; Andersson, C.X.; Lindahl, A.; Synnergren, J. Expression Profiling of Human Pluripotent Stem Cell-Derived Cardiomyocytes Exposed to Doxorubicin—Integration and Visualization of Multi-Omics Data. Toxicol. Sci. 2018, 163, 182–195. [Google Scholar] [CrossRef]
- Kumar, S.N.; Konorev, E.A.; Aggarwal, D.; Kalyanaraman, B. Analysis of Proteome Changes in Doxorubicin-Treated Adult Rat Cardiomyocyte. J. Proteomics 2011, 74, 683–697. [Google Scholar] [CrossRef]
- Yoon, C.; Kim, H.; Mishchenko, N.; Vasileva, E.; Fedoreyev, S.; Stonik, V.; Han, J. Spinochrome D Attenuates Doxorubicin-Induced Cardiomyocyte Death via Improving Glutathione Metabolism and Attenuating Oxidative Stress. Mar. Drugs 2018, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Petucci, C.; Rojas-Betancourt, S.; Gardell, S.J. Comparison of Tissue Harvest Protocols for the Quantitation of Acylcarnitines in Mouse Heart and Liver by Mass Spectrometry. Metabolomics 2012, 8, 784–792. [Google Scholar] [CrossRef]
- Coore, H.G.; Denton, R.M.; Martin, B.R.; Randle, P.J. Regulation of Adipose Tissue Pyruvate Dehydrogenase by Insulin and Other Hormones. Biochem. J. 1971, 125, 115–127. [Google Scholar] [CrossRef]
- Li, S.; Gao, D.; Jiang, Y. Function, Detection and Alteration of Acylcarnitine Metabolism in Hepatocellular Carcinoma. Metabolites 2019, 9, 36. [Google Scholar] [CrossRef]
- Manoli, I.; Venditti, C.P. Disorders of Branched Chain Amino Acid Metabolism. Transl. Sci. Rare Dis. 2016, 1, 91–110. [Google Scholar] [CrossRef]
- Arad, M.; Seidman, C.E.; Seidman, J.G. AMP-Activated Protein Kinase in the Heart: Role During Health and Disease. Circ. Res. 2007, 100, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of Mitochondrial Content in Skeletal Muscle of Healthy Young Human Subjects: Biomarkers of Mitochondrial Content. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Al-Hadi, H.A.; Fox, K.A. Cardiac Markers in the Early Diagnosis and Management of Patients with Acute Coronary Syndrome. Sultan Qaboos Univ. Med. J. 2009, 9, 231–246. [Google Scholar]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Comprehensive Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen Metabolism in Humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef]
- Krause, N.; Wegner, A. Fructose Metabolism in Cancer. Cells 2020, 9, 2635. [Google Scholar] [CrossRef]
- Carvalho, R.A.; Sousa, R.P.B.; Cadete, V.J.J.; Lopaschuk, G.D.; Palmeira, C.M.M.; Bjork, J.A.; Wallace, K.B. Metabolic Remodeling Associated with Subchronic Doxorubicin Cardiomyopathy. Toxicology 2010, 270, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Gratia, S.; Kay, L.; Michelland, S.; Sève, M.; Schlattner, U.; Tokarska-Schlattner, M. Cardiac Phosphoproteome Reveals Cell Signaling Events Involved in Doxorubicin Cardiotoxicity. J. Proteomics 2012, 75, 4705–4716. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Souza, T.; Verheijen, M.C.T.; Gmuender, H.; Selevsek, N.; Schlapbach, R.; Kleinjans, J.; Jennen, D. Translational Proteomics Analysis of Anthracycline-Induced Cardiotoxicity from Cardiac Microtissues to Human Heart Biopsies. Front. Genet. 2021, 12, 695625. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Pan, Y.; Bao, L.; Li, Y.; Cheng, C.; Liu, L.; Xiang, J.; Cheng, J.; Zhang, J.; Chu, W.; et al. Impact of Short-Term Starvation and Refeeding on the Expression of KLF15 and Regulatory Mechanism of Branched-Chain Amino Acids Metabolism in Muscle of Chinese Soft-Shelled Turtle (Pelodiscus Sinensis). Gene 2020, 752, 144782. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, H.; Tang, M.; Ryu, Y.-C.; Wang, X. A Therapeutic Dose of Doxorubicin Activates Ubiquitin-Proteasome System-Mediated Proteolysis by Acting on Both the Ubiquitination Apparatus and Proteasome. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2541–H2550. [Google Scholar] [CrossRef]
- Drake, K.J.; Shotwell, M.S.; Wikswo, J.P.; Sidorov, V.Y. Glutamine and Glutamate Limit the Shortening of Action Potential Duration in Anoxia-Challenged Rabbit Hearts. Physiol. Rep. 2015, 3, e12535. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino Acids in Cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive Quantification of Fuel Use by the Failing and Nonfailing Human Heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Zou, Z.; Hu, X.; Luo, T.; Ming, Z.; Chen, X.; Xia, L.; Luo, W.; Li, J.; Xu, N.; Chen, L.; et al. Naturally-Occurring Spinosyn A and Its Derivatives Function as Argininosuccinate Synthase Activator and Tumor Inhibitor. Nat. Commun. 2021, 12, 2263. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gong, B.; Duan, W.; Fan, C.; Zhang, J.; Li, Z.; Xue, X.; Xu, Y.; Meng, D.; Li, B.; et al. Melatonin Ameliorates Myocardial Ischemia/Reperfusion Injury in Type 1 Diabetic Rats by Preserving Mitochondrial Function: Role of AMPK-PGC-1α-SIRT3 Signaling. Sci. Rep. 2017, 7, 41337. [Google Scholar] [CrossRef]
- Hamstra, S.I.; Whitley, K.C.; Baranowski, R.W.; Kurgan, N.; Braun, J.L.; Messner, H.N.; Fajardo, V.A. The Role of Phospholamban and GSK3 in Regulating Rodent Cardiac SERCA Function. Am. J. Physiol. Cell Physiol. 2020, 319, C694–C699. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F. Roles of Glycogen Synthase Kinase-3 (GSK-3) in Cardiac Development and Heart Disease. J. UOEH 2018, 40, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Murugasamy, K.; Munjal, A.; Sundaresan, N.R. Emerging Roles of SIRT3 in Cardiac Metabolism. Front. Cardiovasc. Med. 2022, 9, 850340. [Google Scholar] [CrossRef]
- Ding, Q.; Qi, Y.; Tsang, S.-Y. Mitochondrial Biogenesis, Mitochondrial Dynamics, and Mitophagy in the Maturation of Cardiomyocytes. Cells 2021, 10, 2463. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.H.; Chaturvedi, P.; Tyagi, S.C. Mitochondrial Pathways to Cardiac Recovery: TFAM. Heart Fail. Rev. 2016, 21, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Kalkhoran, S.B.; Hernández-Reséndiz, S.; Samangouei, P.; Ong, S.-G.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-W.; Ducroux, A.; Jeang, K.-T.; Neuveut, C. Impact of Cellular Autophagy on Viruses: Insights from Hepatitis B Virus and Human Retroviruses. J. Biomed. Sci. 2012, 19, 92. [Google Scholar] [CrossRef]
- Li, M.; Russo, M.; Pirozzi, F.; Tocchetti, C.G.; Ghigo, A. Autophagy and Cancer Therapy Cardiotoxicity: From Molecular Mechanisms to Therapeutic Opportunities. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118493. [Google Scholar] [CrossRef]
- Reis-Mendes, A.; Carvalho, F.; Remião, F.; Sousa, E.; de Lourdes Bastos, M.; Costa, V.M. Autophagy (but Not Metabolism) Is a Key Event in Mitoxantrone-Induced Cytotoxicity in Differentiated AC16 Cardiac Cells. Arch. Toxicol. 2023, 97, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y. LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed]
- Sishi, B.J.N.; Loos, B.; van Rooyen, J.; Engelbrecht, A.-M. Autophagy Upregulation Promotes Survival and Attenuates Doxorubicin-Induced Cardiotoxicity. Biochem. Pharmacol. 2013, 85, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-M.; Li, C.-B.; Liu, Q.-L.; Li, P.; Yang, H. Ginsenoside Rg1 Prevents Doxorubicin-Induced Cardiotoxicity through the Inhibition of Autophagy and Endoplasmic Reticulum Stress in Mice. Int. J. Mol. Sci. 2018, 19, 3658. [Google Scholar] [CrossRef]
- Dorn, G.W. Central Parkin: The Evolving Role of Parkin in the Heart. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1857, 1307–1312. [Google Scholar] [CrossRef]
- Dorn, G.W. Mitochondrial Pruning by Nix and BNip3: An Essential Function for Cardiac-Expressed Death Factors. J. Cardiovasc. Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef]
- Pan, X.-C.; Xiong, Y.-L.; Hong, J.-H.; Liu, Y.; Cen, Y.-Y.; Liu, T.; Yang, Q.-F.; Tao, H.; Li, Y.-N.; Zhang, H.-G. Cardiomyocytic FoxP3 Is Involved in Parkin-Mediated Mitophagy during Cardiac Remodeling and the Regulatory Role of Triptolide. Theranostics 2022, 12, 2483–2501. [Google Scholar] [CrossRef]
- Ghayour-Mobarhan, M.; Saber, H.; Ferns, G.A.A. The Potential Role of Heat Shock Protein 27 in Cardiovascular Disease. Clin. Chim. Acta 2012, 413, 15–24. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, X.; Qian, B.; Min, X.; Gao, X.; Li, C.; Cheng, Y.; Huang, J. Over-Expression of Heat Shock Protein 27 Attenuates Doxorubicin-Induced Cardiac Dysfunction in Mice. Eur. J. Heart Fail. 2007, 9, 762–769. [Google Scholar] [CrossRef]
- Radons, J. The Human HSP70 Family of Chaperones: Where Do We Stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.Z.; Radicioni, S.M.; Mosser, D.D. Hsp70 Inhibits Heat-Induced Apoptosis Upstream of Mitochondria by Preventing Bax Translocation. J. Biol. Chem. 2005, 280, 38729–38739. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.K.; Vezyraki, P.; Kalaitzakis, A.; Zerikiotis, S.; Michalis, L.; Angelidis, C. Hsp70 Regulates the Doxorubicin-Mediated Heart Failure in Hsp70-Transgenic Mice. Cell Stress Chaperones 2014, 19, 853–864. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphometric Parameter | CTRL | DOX | MTX |
|---|---|---|---|
| Whole-body weight (g) | 42.671 ± 3.450 | 37.727 ± 6.163 | 41.264 ± 2.906 |
| Heart weight (g) | 0.228 ± 0.048 | 0.229 ± 0.037 | 0.227 ± 0.031 |
| Tibial length (cm) | 1.90 ± 0.08 | 1.84 ± 0.05 | 1.84 ± 0.07 |
| Heart weight-to-whole-body weight (mg/g) | 5.30 ± 0.81 | 6.35 ± 2.20 | 5.49 ± 0.63 |
| Heart weight-to-tibial length (g/cm) | 0.120 ± 0.025 | 0.125 ± 0.021 | 0.123 ± 0.016 |
| Serum Biochemical Parameter | CTRL | DOX | MTX |
|---|---|---|---|
| Total protein (g/L) | 47.84 ± 3.42 | 42.72 ± 3.34 | 43.76 ± 6.26 |
| Glucose (mg/dL) | 359.2 ± 57.47 | 269.7 ± 115.0 | 333.2 ± 102.0 |
| Albumin (g/L) | 38.41 ± 2.15 | 36.28 ± 2.22 | 36.31 ± 4.29 |
| ALAT (U/L) | 48.41 ± 19.61 | 70.52 ± 30.02 | 40.06 ± 23.89 |
| ASAT (U/L) | 26.31 ± 7.42 | 41.00 ± 16.64 | 31.09 ± 13.67 |
| ASAT-to-ALAT (U/L) | 0.62 ± 0.24 | 0.63 ± 0.22 | 0.89 ± 0.27 |
| CK-MB (U/L) | 66.40 ± 20.36 | 92.44 ± 26.60 | 76.50 ± 16.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandão, S.R.; Reis-Mendes, A.; Duarte-Araújo, M.; Neuparth, M.J.; Rocha, H.; Carvalho, F.; Ferreira, R.; Costa, V.M. Cardiac Molecular Remodeling by Anticancer Drugs: Doxorubicin Affects More Metabolism While Mitoxantrone Impacts More Autophagy in Adult CD-1 Male Mice. Biomolecules 2023, 13, 921. https://doi.org/10.3390/biom13060921
Brandão SR, Reis-Mendes A, Duarte-Araújo M, Neuparth MJ, Rocha H, Carvalho F, Ferreira R, Costa VM. Cardiac Molecular Remodeling by Anticancer Drugs: Doxorubicin Affects More Metabolism While Mitoxantrone Impacts More Autophagy in Adult CD-1 Male Mice. Biomolecules. 2023; 13(6):921. https://doi.org/10.3390/biom13060921
Chicago/Turabian StyleBrandão, Sofia Reis, Ana Reis-Mendes, Margarida Duarte-Araújo, Maria João Neuparth, Hugo Rocha, Félix Carvalho, Rita Ferreira, and Vera Marisa Costa. 2023. "Cardiac Molecular Remodeling by Anticancer Drugs: Doxorubicin Affects More Metabolism While Mitoxantrone Impacts More Autophagy in Adult CD-1 Male Mice" Biomolecules 13, no. 6: 921. https://doi.org/10.3390/biom13060921
APA StyleBrandão, S. R., Reis-Mendes, A., Duarte-Araújo, M., Neuparth, M. J., Rocha, H., Carvalho, F., Ferreira, R., & Costa, V. M. (2023). Cardiac Molecular Remodeling by Anticancer Drugs: Doxorubicin Affects More Metabolism While Mitoxantrone Impacts More Autophagy in Adult CD-1 Male Mice. Biomolecules, 13(6), 921. https://doi.org/10.3390/biom13060921