
Homogeneous, Synthetic, Non-Saccharide Glycosaminoglycan Mimetics as Potent Inhibitors of Human Cathepsin G

 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
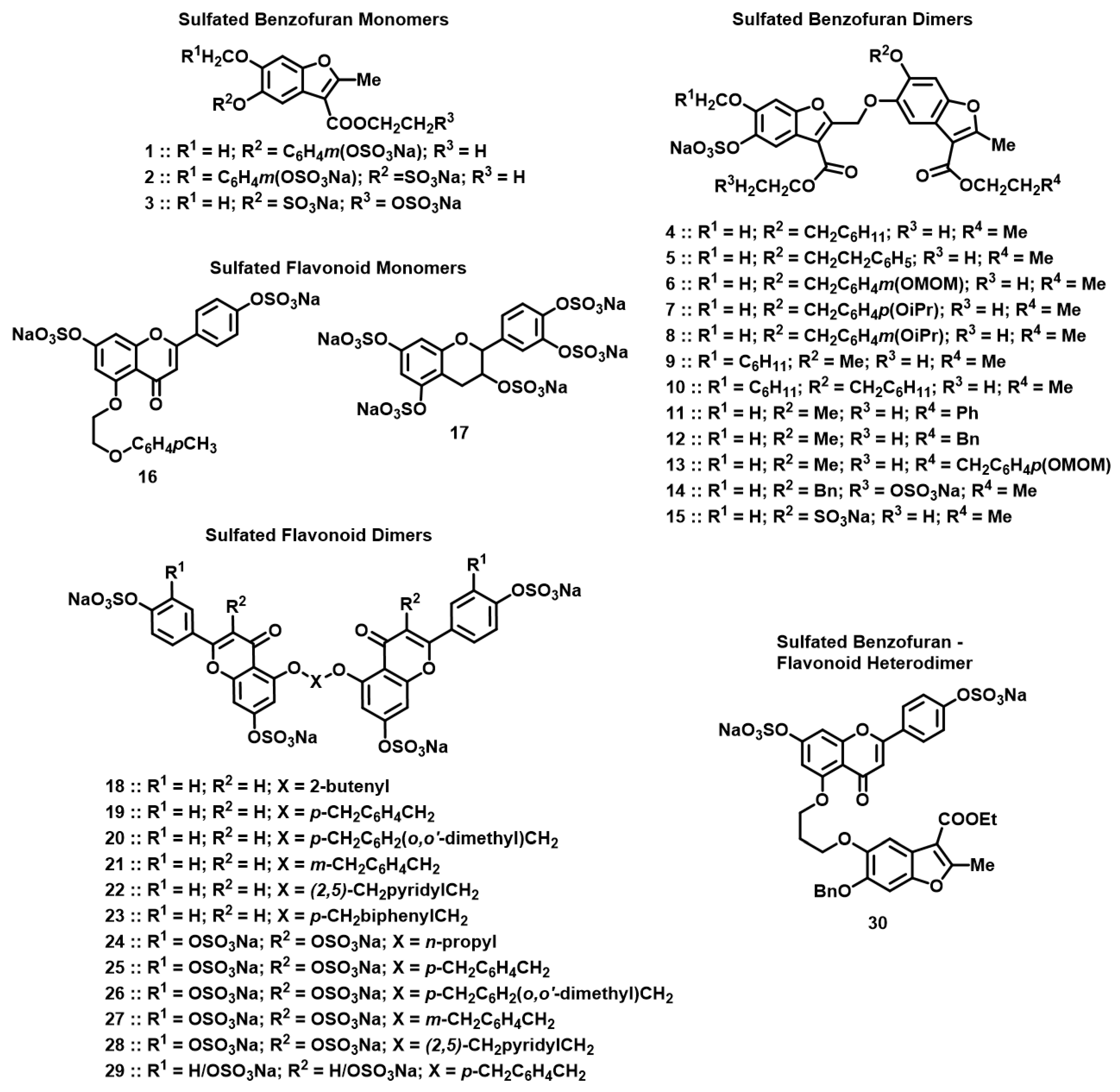
2.2. Chemistry
2.3. Inhibitor Screen
2.4. IC50 Determination
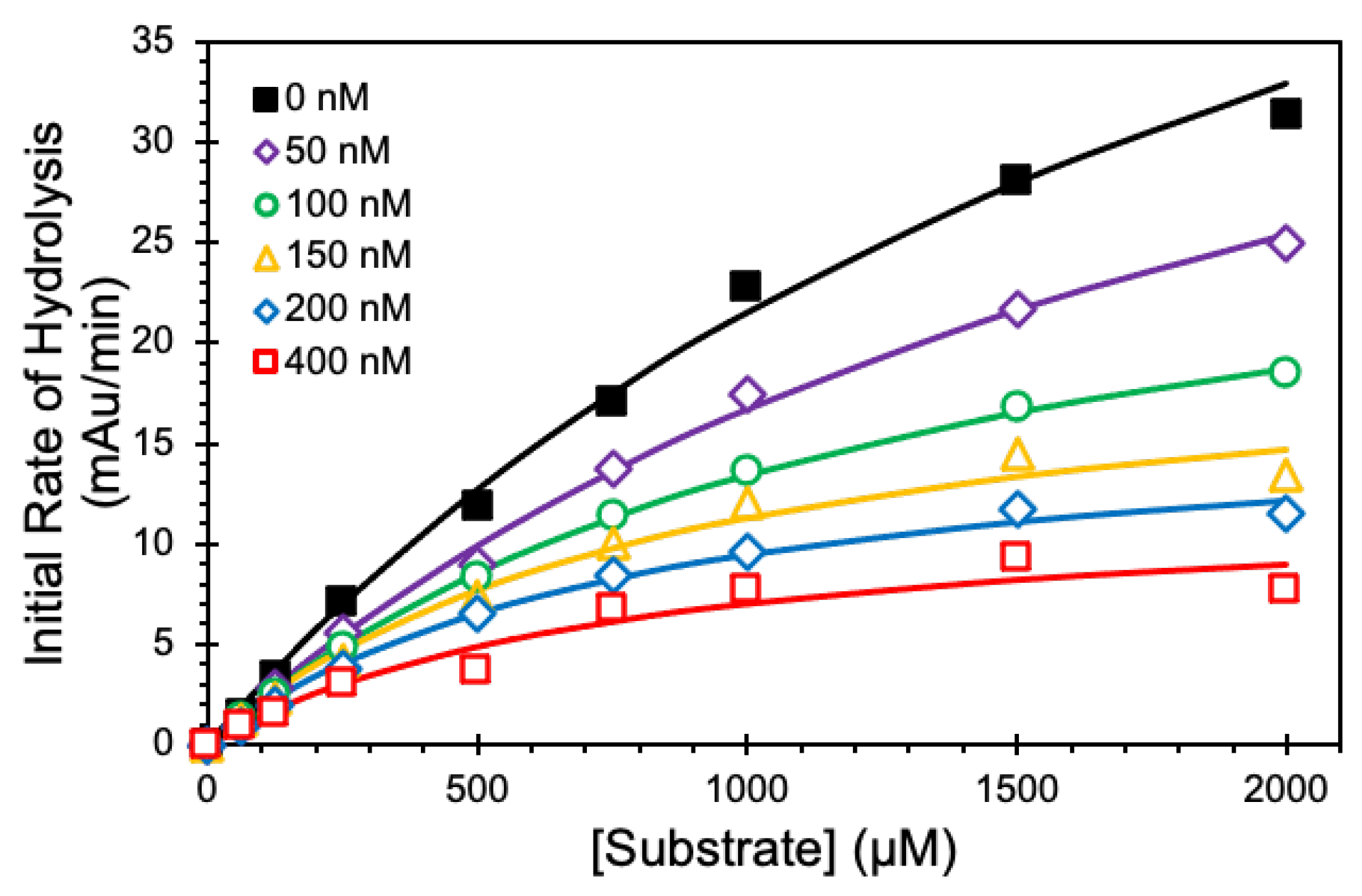
2.5. Michaelis–Menten Kinetics
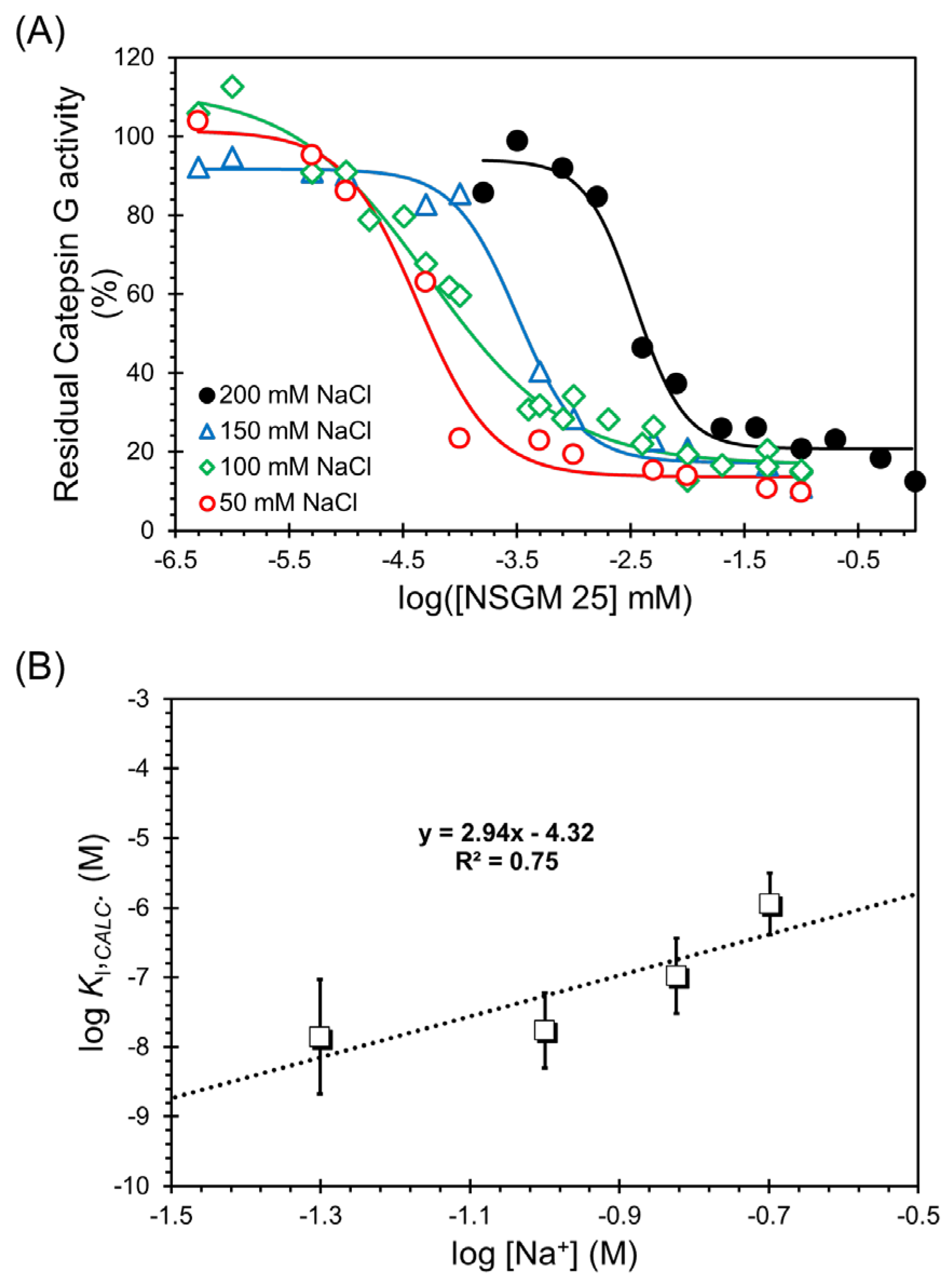
2.6. Salt Dependence of NSGM 25 Inhibition of CatG
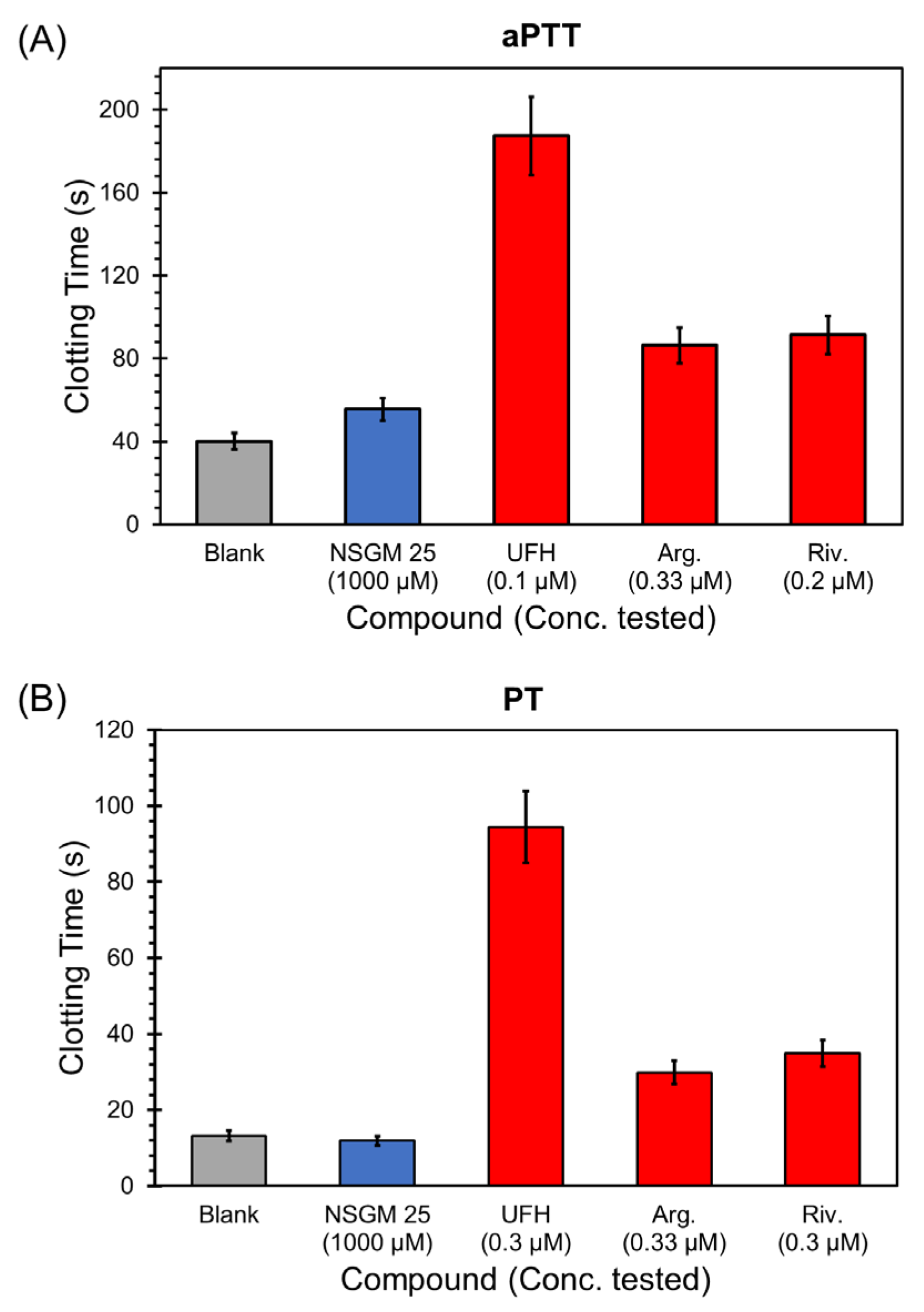
2.7. Impact of NSGM 25 on Clotting Assays
3. Results
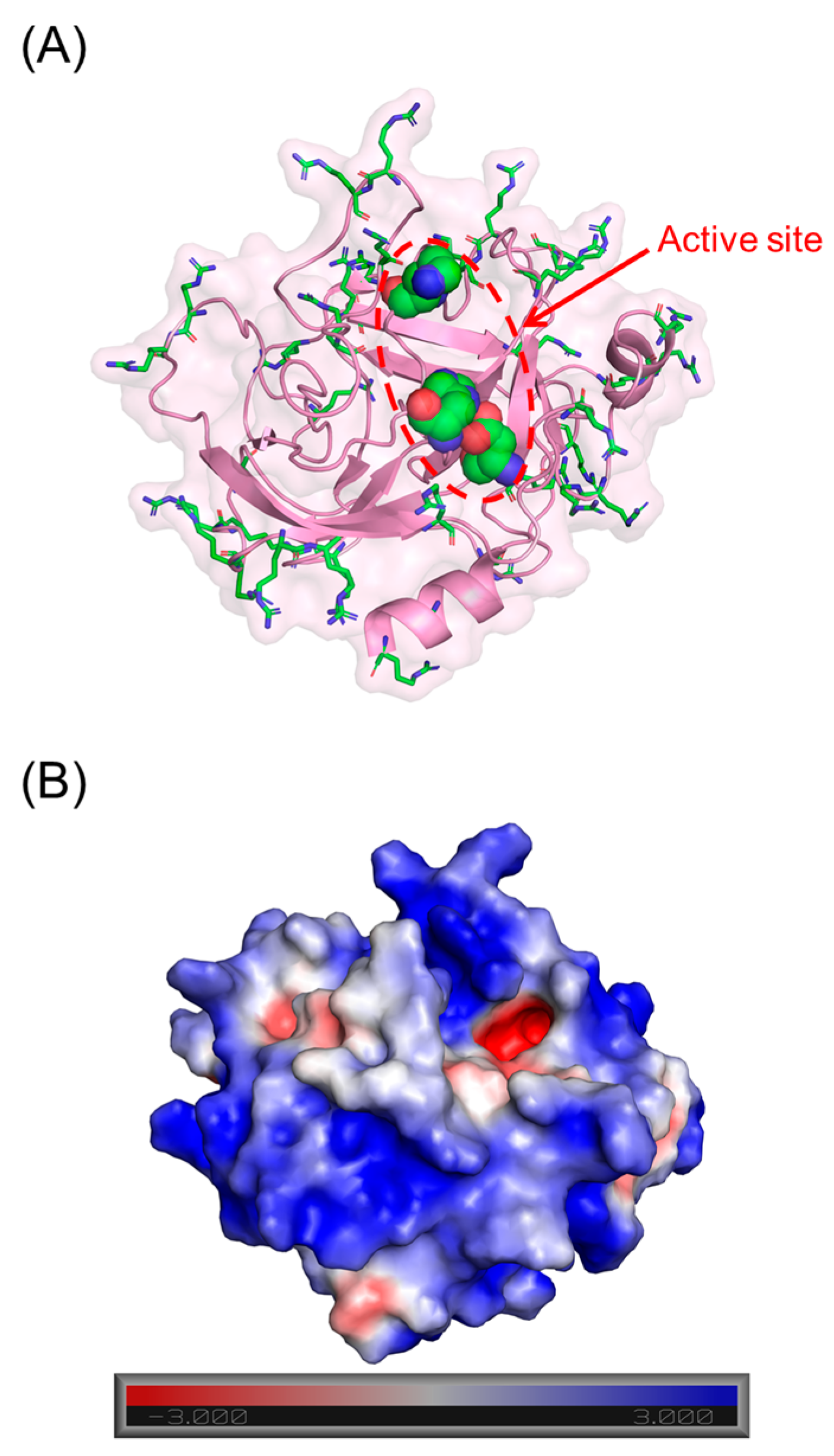
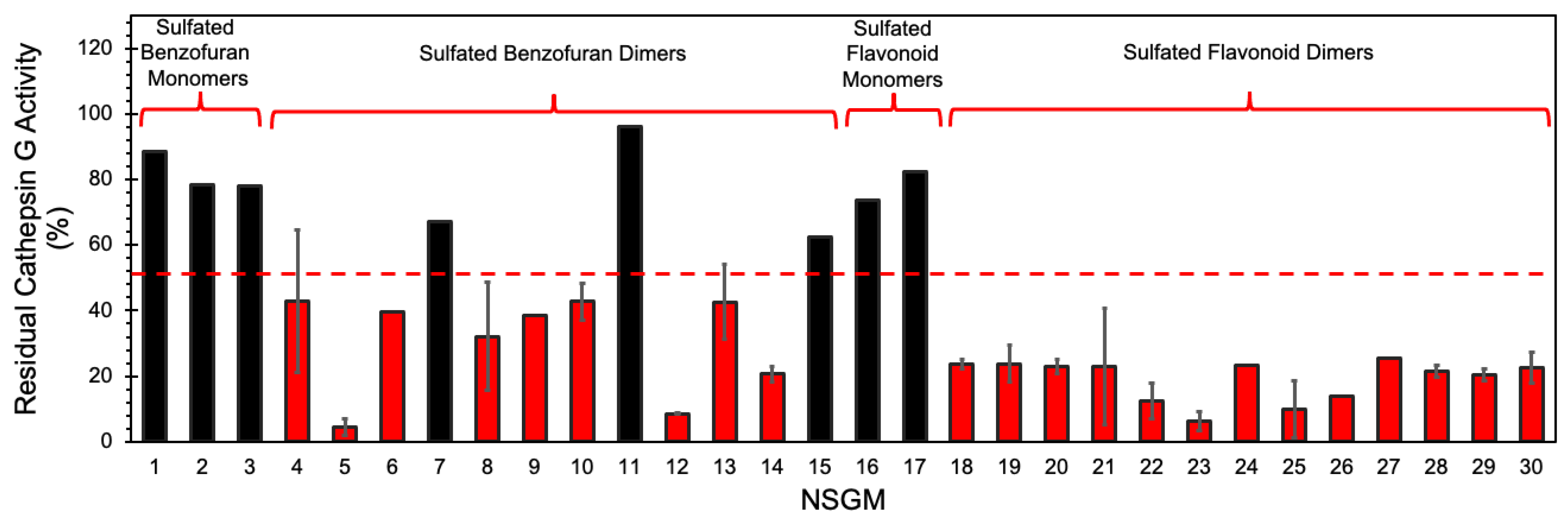
3.1. Screening for Cathepsin G Inhibition
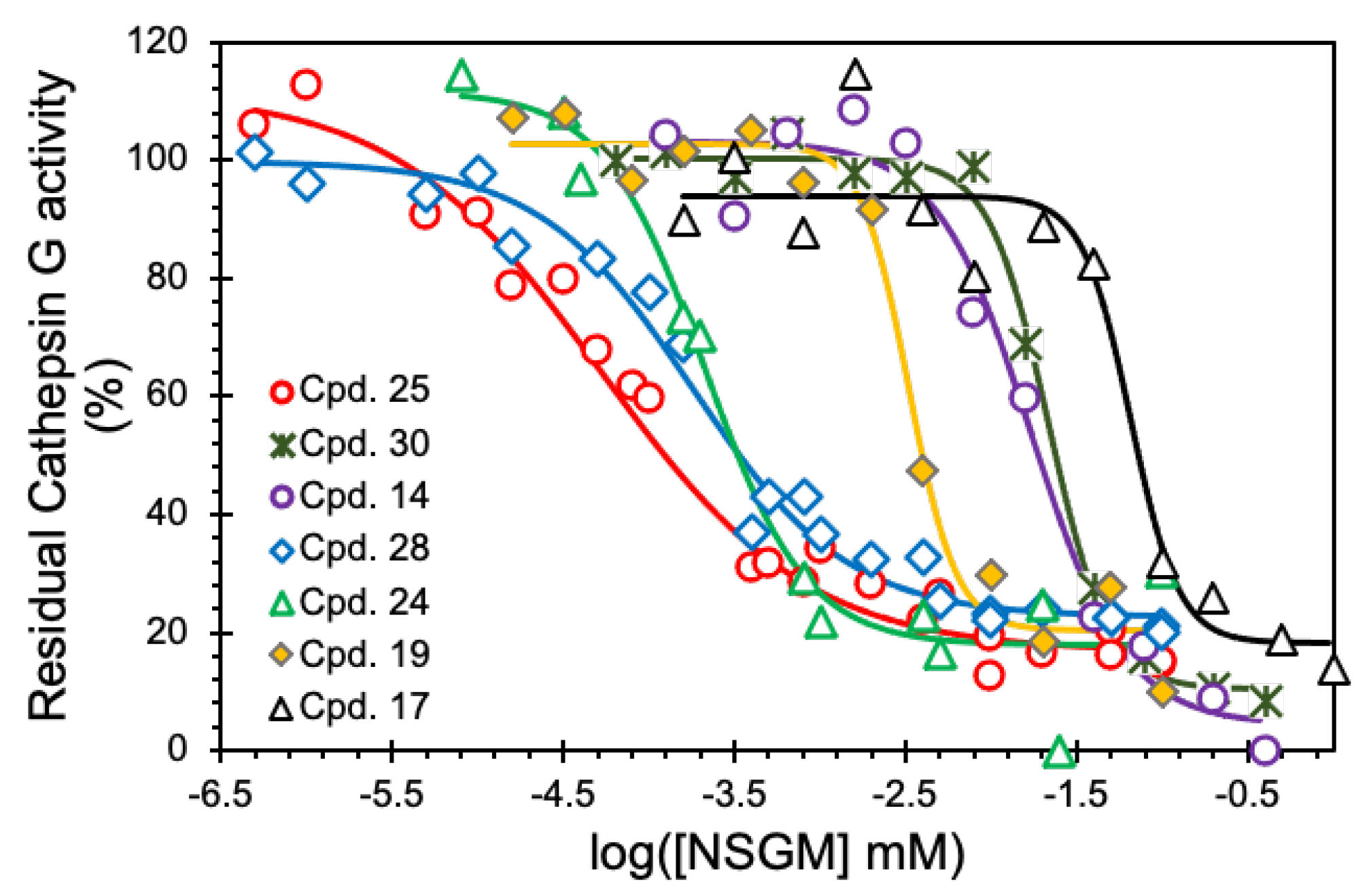
3.2. Structure–Activity Relationship (SAR)
3.3. Mechanism of CatG Inhibition by NSGM 25
3.4. Salt-Dependence of CatG Inhibition in the Presence of NSGM 25
3.5. Impact of NSGM 25 on Human Plasma Clotting
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stapels, D.A.C.; Geisbrecht, B.V.; Rooijakkers, S.H.M. Neutrophil Serine Proteases in Antibacterial Defense. Curr. Opin. Microbiol. 2015, 23, 42–48. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin Gas Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Hof, P.; Mayr, I.; Huber, R.; Korzus, E.; Potempa, J.; Travis, J.; Powers, J.C.; Bode, W. The 1.8 Å Crystal Structure of Human Cathepsin G in Complex with Suc-Val-Pro-Phe(P)-(OPh)2: A Janus-Faced Proteinase with Two Opposite Specificities. EMBO J. 1996, 15, 5481–5491. [Google Scholar] [CrossRef]
- Helske, S.; Syväranta, S.; Kupari, M.; Lappalainen, J.; Laine, M.; Lommi, J.; Turto, H.; Mäyränpää, M.; Werkkala, K.; Kovanen, P.T.; et al. Possible Role for Mast Cell-Derived Cathepsin G in the Adverse Remodelling of Stenotic Aortic Valves. Eur. Heart J. 2006, 27, 1495–1504. [Google Scholar] [CrossRef]
- Tani, K.; Murphy, W.J.; Chertov, O.; Oppenheim, J.J.; Wang, J.M. The Neutrophil Granule Protein Cathepsin G Activates Murine T Lymphocytes and Upregulates Antigen-Specific Ig Production in Mice. Biochem. Biophys. Res. Commun. 2001, 282, 971–976. [Google Scholar] [CrossRef]
- LaRosa, C.A.; Rohrer, M.J.; Benoit, S.E.; Rodino, L.J.; Barnard, M.R.; Michelson, A.D. Human Neutrophil Cathepsin G Is a Potent Platelet Activator. J. Vasc. Surg. 1994, 19, 306–318. [Google Scholar] [CrossRef]
- Rykl, J.; Thiemann, J.; Kurzawski, S.; Pohl, T.; Gobom, J.; Zidek, W.; Schlüter, H. Renal Cathepsin G and Angiotensin II Generation. J. Hypertens. 2006, 24, 1797–1807. [Google Scholar] [CrossRef]
- Shamamian, P.; Schwartz, J.D.; Pocock, B.J.Z.; Monea, S.; Whiting, D.; Marcus, S.G.; Mignatti, P. Activation of Progelatinase A (MMP-2) by Neutrophil Elastase, Cathepsin G, and Proteinase-3: A Role for Inflammatory Cells in Tumor Invasion and Angiogenesis. J. Cell. Physiol. 2001, 189, 197–206. [Google Scholar] [CrossRef]
- Wilson, T.J.; Nannuru, K.C.; Singh, R.K. Cathepsin G-Mediated Activation of pro-Matrix Metalloproteinase 9 at the Tumor-Bone Interface Promotes Transforming Growth Factor-β Signaling and Bone Destruction. Mol. Cancer Res. 2009, 7, 1224–1233. [Google Scholar] [CrossRef]
- Son, E.D.; Kim, H.; Choi, H.; Lee, S.H.; Lee, J.Y.; Kim, S.; Closs, B.; Lee, S.; Chung, J.H.; Hwang, J.S. Cathepsin G Increases MMP Expression in Normal Human Fibroblasts through Fibronectin Fragmentation, and Induces the Conversion of ProMMP-1 to Active MMP-1. J. Dermatol. Sci. 2009, 53, 150–152. [Google Scholar] [CrossRef]
- Jun, H.K.; Jung, Y.J.; Ji, S.; An, S.J.; Choi, B.K. Caspase-4 Activation by a Bacterial Surface Protein Is Mediated by Cathepsin G in Human Gingival Fibroblasts. Cell Death Differ. 2018, 25, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Thorpe, M.; Alemayehu, R.; Roy, A.; Kervinen, J.; de Garavilla, L.; Åbrink, M.; Hellman, L. Highly Selective Cleavage of Cytokines and Chemokines by the Human Mast Cell Chymase and Neutrophil Cathepsin G. J. Immunol. 2017, 198, 1474–1483. [Google Scholar] [CrossRef]
- Kosikowska, P.; Lesner, A. Inhibitors of Cathepsin G: A Patent Review (2005 to Present). Expert Opin. Ther. Pat. 2013, 23, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Wiedow, O.; Meyer-Hoffert, U. Neutrophil Serine Proteases: Potential Key Regulators of Cell Signalling during Inflammation. J. Intern. Med. 2005, 257, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T.N. Neutrophil Serine Proteases Fine-Tune the Inflammatory Response. Int. J. Biochem. Cell Biol. 2008, 40, 1317–1333. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T.N. Neutrophil Serine Proteases: Specific Regulators of Inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Meyer-Hoffert, U.; Wiedow, O. Neutrophil Serine Proteases: Mediators of Innate Immune Responses. Curr. Opin. Hematol. 2011, 18, 19–24. [Google Scholar] [CrossRef]
- Miyata, J.; Tani, K.; Sato, K.; Otsuka, S.; Urata, T.; Lkhagvaa, B.; Furukawa, C.; Sano, N.; Sone, S. Cathepsin G: The Significance in Rheumatoid Arthritis as a Monocyte Chemoattractant. Rheumatol. Int. 2007, 27, 375–382. [Google Scholar] [CrossRef]
- Krasavin, M.Y.; Gureev, M.A.; Garabadzhiu, A.V.; Pashkin, A.Y.; Zhukov, A.S.; Khairutdinov, V.R.; Samtsov, A.V.; Shvets, V.I. Inhibition of Neutrophil Elastase and Cathepsin G As a New Approach to the Treatment of Psoriasis: From Fundamental Biology to Development of New Target-Specific Drugs. Dokl. Biochem. Biophys. 2019, 487, 272–276. [Google Scholar] [CrossRef]
- Guo, J.; Tu, J.; Hu, Y.; Song, G.; Yin, Z. Cathepsin G Cleaves and Activates IL-36γ and Promotes the Inflammation of Psoriasis. Drug Des. Dev. Ther. 2019, 13, 581–588. [Google Scholar] [CrossRef]
- Gudmann, N.S.; Manon-Jensen, T.; Sand, J.M.B.; Diefenbach, C.; Sun, S.; Danielsen, A.; Karsdal, M.A.; Leeming, D.J. Lung Tissue Destruction by Proteinase 3 and Cathepsin G Mediated Elastin Degradation Is Elevated in Chronic Obstructive Pulmonary Disease. Biochem. Biophys. Res. Commun. 2018, 503, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.C.; De, S.; Mishra, P.K. Role of Proteases in Chronic Obstructive Pulmonary Disease. Front. Pharmacol. 2017, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Guyot, N.; Wartelle, J.; Malleret, L.; Todorov, A.A.; Devouassoux, G.; Pacheco, Y.; Jenne, D.E.; Belaaouaj, A. Unopposed Cathepsin G, Neutrophil Elastase, and Proteinase 3 Cause Severe Lung Damage and Emphysema. Am. J. Pathol. 2014, 184, 2197–2210. [Google Scholar] [CrossRef]
- Suter, S.; Schaad, U.B.; Morgenthaler, J.J.; Chevallier, I.; Schnebli, H.P. Fibronectin-Cleaving Activity in Bronchial Secretions of Patients with Cystic Fibrosis. J. Infect. Dis. 1988, 158, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.; Frey, D.; Hagner, M.; Dittrich, S.; Paulsen, M.; Mall, M.A.; Schultz, C. Cathepsin G Activity as a New Marker for Detecting Airway Inflammation by Microscopy and Flow Cytometry. ACS Cent. Sci. 2019, 5, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Li, C.Y.; de Veer, S.J.; Wang, C.K.; Craik, D.J. Design of Potent and Selective Cathepsin G Inhibitors Based on the Sunflower Trypsin Inhibitor-1 Scaffold. J. Med. Chem. 2017, 60, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Craciun, I.; Fenner, A.M.; Kerns, R.J. N-Arylacyl O-Sulfonated Aminoglycosides as Novel Inhibitors of Human Neutrophil Elastase, Cathepsin G and Proteinase 3. Glycobiology 2016, 26, 701–709. [Google Scholar] [CrossRef]
- Sieńczyk, M.; Lesner, A.; Wysocka, M.; Łegowska, A.; Pietrusewicz, E.; Rolka, K.; Oleksyszyn, J. New Potent Cathepsin G Phosphonate Inhibitors. Bioorg. Med. Chem. 2008, 16, 8863–8867. [Google Scholar] [CrossRef]
- Sissi, C.; Lucatello, L.; Naggi, A.; Torri, G.; Palumbo, M. Interactions of Low-Molecular-Weight Semi-Synthetic Sulfated Heparins with Human Leukocyte Elastase and Human Cathepsin G. Biochem. Pharmacol. 2006, 71, 287–293. [Google Scholar] [CrossRef]
- Ledoux, D.; Merciris, D.; Barritault, D.; Caruelle, J.P. Heparin-like Dextran Derivatives as Well as Glycosaminoglycans Inhibit the Enzymatic Activity of Human Cathepsin G. FEBS Lett. 2003, 537, 23–29. [Google Scholar] [CrossRef]
- Burster, T.; Mustafa, Z.; Myrzakhmetova, D.; Zhanapiya, A.; Zimecki, M. Hindrance of the proteolytic activity of neutrophil-derived serine proteases by serine protease inhibitors as a management of cardiovascular diseases and chronic inflammation. Front. Chem. 2021, 9, 784003. [Google Scholar] [CrossRef] [PubMed]
- Morla, S. Glycosaminoglycans and Glycosaminoglycan Mimetics in Cancer and Inflammation. Int. J. Mol. Sci. 2019, 20, 1963. [Google Scholar] [CrossRef]
- Voynow, J.A.; Zheng, S.; Kummarapurugu, A.B. Glycosaminoglycans as Multifunctional Anti-Elastase and Anti-Inflammatory Drugs in Cystic Fibrosis Lung Disease. Front. Pharmacol. 2020, 11, 1011. [Google Scholar] [CrossRef] [PubMed]
- Kummarapurugu, A.B.; Afosah, D.K.; Sankaranarayanan, N.V.; Gangji, R.N.; Zheng, S.; Kennedy, T.; Rubin, B.K.; Voynow, J.A.; Desai, U.R. Molecular Principles for Heparin Oligosaccharide-Based Inhibition of Neutrophil Elastase in Cystic Fibrosis. J. Biol. Chem. 2018, 293, 12480–12490. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Naggi, A.; Torri, G. Heparin-Derived Heparan Sulfate Mimics to Modulate Heparan Sulfate-Protein Interaction in Inflammation and Cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef]
- Griffin, K.L.; Fischer, B.M.; Kummarapurugu, A.B.; Zheng, S.; Kennedy, T.P.; Rao, N.V.; Foster, W.M.; Voynow, J.A. 2-O, 3-O-Desulfated Heparin Inhibits Neutrophil Elastase-Induced HMGB-1 Secretion and Airway Inflammation. Am. J. Respir. Cell Mol. Biol. 2014, 50, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Afosah, D.K.; Kar, S.; Aliter, K.F.; Mottamal, M. Sulfated Penta-Galloyl Glucopyranoside (SPGG) Is Glycosaminoglycan Mimetic Allosteric Inhibitor of Cathepsin G. RPS Pharm. Pharmacol. Rep. 2023, 2, rqad001. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Desai, U.R. Designing Allosteric Inhibitors of Factor XIa. Lessons from the Interactions of Sulfated Pentagalloylglucopyranosides. J. Med. Chem. 2014, 57, 4805–4818. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Ponnusamy, P.; Mehta, A.Y.; Gailani, D.; Desai, U.R. Sulfated Pentagalloylglucoside Is a Potent, Allosteric, and Selective Inhibitor of Factor XIa. J. Med. Chem. 2013, 56, 867–878. [Google Scholar] [CrossRef]
- Afosah, D.K.; Verespy, S.; Al-Horani, R.A.; Boothello, R.S.; Karuturi, R.; Desai, U.R. A Small Group of Sulfated Benzofurans Induces Steady-State Submaximal Inhibition of Thrombin. Bioorg. Med. Chem. Lett. 2018, 28, 1101–1105. [Google Scholar] [CrossRef]
- Sidhu, P.S.; Liang, A.; Mehta, A.Y.; Abdel Aziz, M.H.; Zhou, Q.; Desai, U.R. Rational Design of Potent, Small, Synthetic Allosteric Inhibitors of Thrombin. J. Med. Chem. 2011, 54, 5522–5531. [Google Scholar] [CrossRef]
- Abdel Aziz, M.H.; Sidhu, P.S.; Liang, A.; Kim, J.Y.; Mosier, P.D.; Zhou, Q.; Farrell, D.H.; Desai, U.R. Designing Allosteric Regulators of Thrombin. Monosulfated Benzofuran Dimers Selectively Interact with Arg173 of Exosite 2 to Induce Inhibition. J. Med. Chem. 2012, 55, 6888–6897. [Google Scholar] [CrossRef] [PubMed]
- Gangji, R.N.; Sankaranarayanan, N.V.; Elste, J.; Al-Horani, R.A.; Afosah, D.K.; Joshi, R.; Tiwari, V.; Desai, U.R. Inhibition of Herpes Simplex Virus-1 Entry into Human Cells by Nonsaccharide Glycosaminoglycan Mimetics. ACS Med. Chem. Lett. 2018, 9, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Afosah, D.K.; Al-Horani, R.A.; Sankaranarayanan, N.V.; Desai, U.R. Potent, Selective, Allosteric Inhibition of Human Plasmin by Sulfa Non-Saccharide Glycosaminoglycan Mimetics. J. Med. Chem. 2017, 60, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Karuturi, R.; White, D.T.; Desai, U.R. Plasmin Regulation through Allosteric, Sulfated, Small Molecules. Molecules 2015, 20, 608–624. [Google Scholar] [CrossRef]
- Salvesen, G.; Cathespin, G. Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: Oxford, UK, 2013; Volume 1–3, pp. 2661–2666. [Google Scholar] [CrossRef]
- Hileman, R.E.; Jennings, R.N.; Linhardt, R.J. Thermodynamic Analysis of the Heparin Interaction with a Basic Cyclic Peptide Using Isothermal Titration Calorimetry. Biochemistry 1998, 37, 15231–15237. [Google Scholar] [CrossRef]
- Henry, B.L.; Connell, J.; Liang, A.; Krishnasamy, C.; Desai, U.R. Interaction of Antithrombin with Sulfated, Low Molecular Weight Lignins. Opportunities for Potent, Selective Modulation of Antithrombin Function. J. Biol. Chem. 2009, 284, 20897–20908. [Google Scholar] [CrossRef]
- Abdelfadiel, E.I.; Gunta, R.; Villuri, B.K.; Afosah, D.K.; Sankaranarayanan, N.V.; Desai, U.R. Designing Smaller, Synthetic, Functional Mimetics of Sulfated Glycosaminoglycans as Allosteric Modulators of Coagulation Factors. J. Med. Chem. 2023, 6, 4503–4531. [Google Scholar] [CrossRef]
- Afosah, D.K.; Al-Horani, R.A. Sulfated Non-Saccharide Glycosaminoglycan Mimetics as Novel Drug Discovery Platform for Various Pathologies. Curr. Med. Chem. 2018, 27, 3412–3447. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and Their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Olson, S.T.; Bjork, I. Predominant Contribution of Surface Approximation to the Mechanism of Heparin Acceleration of the Antithrombin-Thrombin Reaction: Elucidation from Salt Concentration Effects. J. Biol. Chem. 1991, 266, 6353–6364. [Google Scholar] [CrossRef] [PubMed]
- Jairajpuri, M.A.; Lu, A.; Desai, U.; Olson, S.T.; Bjork, I.; Bock, S.C. Antithrombin III Phenylalanines 122 and 121 Contribute to Its High Affinity for Heparin and Its Conformational Activation. J. Biol. Chem. 2003, 278, 15941–15950. [Google Scholar] [CrossRef] [PubMed]
- Desai, U.R.; Petitou, M.; Björk, I.; Olson, S.T. Mechanism of Heparin Activation of Antithrombin. Role of Individual Residues of the Pentasaccharide Activating Sequence in the Recognition of Native and Activated States of Antithrombin. J. Biol. Chem. 1998, 273, 7478–7487. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Lippi, G.; Koutts, J. Laboratory Testing of Anticoagulants: The Present and the Future. Pathology 2011, 43, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Fleddermann, J.; Pichert, A.; Arnhold, J. Interaction of Serine Proteases from Polymorphonuclear Leucocytes with the Cell Surface and Heparin. Inflammation 2012, 35, 81–88. [Google Scholar] [CrossRef]
- Belorgey, D.; Bieth, J.G. DNA Binds Neutrophil Elastase and Mucus Proteinase Inhibitor and Impairs Their Functional Activity. FEBS Lett. 1995, 361, 265–268. [Google Scholar] [CrossRef]
- Lin, S.J.; Dong, K.C.; Eigenbrot, C.; Van Lookeren Campagne, M.; Kirchhofer, D. Structures of Neutrophil Serine Protease 4 Reveal an Unusual Mechanism of Substrate Recognition by a Trypsin-Fold Protease. Structure 2014, 22, 1333–1340. [Google Scholar] [CrossRef]
- Morla, S.; Sankaranarayanan, N.V.; Afosah, D.K.; Kumar, M.; Kummarapurugu, A.B.; Voynow, J.A.; Desai, U.R. On the Process of Discovering Leads That Target the Heparin-Binding Site of Neutrophil Elastase in the Sputum of Cystic Fibrosis Patients. J. Med. Chem. 2019, 62, 5501–5511. [Google Scholar] [CrossRef]
- Motta, J.P.; Rolland, C.; Edir, A.; Florence, A.C.; Sagnat, D.; Bonnart, C.; Rousset, P.; Guiraud, L.; Quaranta-Nicaise, M.; Mas, E.; et al. Epithelial Production of Elastase Is Increased in Inflammatory Bowel Disease and Causes Mucosal Inflammation. Mucosal. Immunol. 2021, 14, 667–678. [Google Scholar] [CrossRef]
- Schuliga, M.; Westall, G.; Xia, Y.; Stewart, A.G. The Plasminogen Activation System: New Targets in Lung Inflammation and Remodeling. Curr. Opin. Pharmacol. 2013, 13, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ke, F.; Zhang, W.; Shen, X.; Xu, Q.; Wang, H.; Yu, X.Z.; Leng, Q.; Wang, H. Plasmin Plays an Essential Role in Amplification of Psoriasiform Skin Inflammation in Mice. PLoS ONE 2011, 6, e16483. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.K.; Strickland, S. A Critical Role for Plasminogen in Inflammation. J. Exp. Med. 2020, 217, e20191865. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Abdelfadiel, E.I.; Afosah, D.K.; Morla, S.; Sistla, J.C.; Mohammed, B.; Martin, E.J.; Sakagami, M.; Brophy, D.F.; Desai, U.R. A Synthetic Heparin Mimetic That Allosterically Inhibits Factor XIa and Reduces Thrombosis in Vivo without Enhanced Risk of Bleeding. J. Thromb. Haemost. 2019, 17, 2110–2122. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NSGM | IC50 (µM) a | ΔY (%) b | HS c |
|---|---|---|---|
| 1 | >50 | ND | ND |
| 2 | >50 | ND | ND |
| 3 | >50 | ND | ND |
| 4 | 48.3 ± 6.8 d | 96 ± 6 | 1.3 ± 0.2 |
| 5 | 5.5 ± 0.8 | 93 ± 5 | 5.3 ± 1.9 |
| 6 | ~50 | ND | ND |
| 7 | >50 | ND | ND |
| 8 | 27.0 ± 3.1 | 101 ± 5 | 1.7 ± 0.3 |
| 9 | 45.5 ± 3.3 | 99 ± 4 | 2.5 ± 0.4 |
| 10 | 29.0 ± 6.5 | 97 ± 10 | 1.5 ± 0.5 |
| 11 | >50 | ND | ND |
| 12 | 19 ± 12 | 87 ± 4 | 8.6 ± 5.1 |
| 13 | 42.1 ± 4.2 | 90 ± 5 | 2.3 ± 0.5 |
| 14 | 17.3 ± 2.8 | 96 ± 7 | 1.6 ± 0.4 |
| 15 | >50 | ND | ND |
| 16 | 126 ± 11 | 115 ± 4 | 1.9 ± 0.3 |
| 17 | 65 ± 13 | 76 ± 7 | 3.1 ± 1.3 |
| NSGM | IC50 (µM) a | Δ Y (%) b | HS c |
|---|---|---|---|
| 18 | 4.7 ± 0.7 d | 81 ± 7 | 1.5 ± 0.3 |
| 19 | 3.3 ± 0.3 | 82 ± 4 | 3.1 ± 1.0 |
| 20 | 1.5 ± 0.2 | 89 ± 4 | 1.4 ± 0.2 |
| 21 | 10.3 ± 4.8 | 81 ± 7 | 1.3 ± 0.8 |
| 22 | 1.3 ± 0.2 | 105 ± 7 | 1.2 ± 0.2 |
| 23 | 3.4 ± 0.2 | 103 ± 4 | 4.1 ± 1.0 |
| 24 | 0.21 ± 0.04 | 94 ± 9 | 1.5 ± 0.5 |
| 25 | 0.05 ± 0.01 | 95 ± 7 | 0.7 ± 0.1 |
| 26 | 0.07 ± 0.01 | 101 ± 5 | 1.2 ± 0.2 |
| 27 | 0.15 ± 0.06 | 77 ± 10 | 0.9 ± 0.4 |
| 28 | 0.18 ± 0.04 | 77 ± 5 | 0.9 ± 0.2 |
| 29 | 0.14 ± 05 | 110 ± 16 | 1.1 ± 0.1 |
| 30 | 18.9 ± 2.2 | 87 ± 5 | 2.1 ± 0.5 |
| [NSGM 25] (nM) | KM (mM) | VMAX (mAu/min) |
|---|---|---|
| 0 | 2.26 ± 0.28 b | 70.0 ± 5.5 |
| 50 | 2.18 ± 0.27 | 52.9 ± 4.1 |
| 100 | 1.31 ± 0.06 | 31.0 ± 0.8 |
| 150 | 0.87 ± 0.2 | 21.1 ± 2.1 |
| 200 | 0.80 ± 0.11 | 17.0 ± 1.0 |
| 400 | 0.77 ± 0.30 | 12.4 ± 2.1 |
| [NaCl] | IC50 (nM) a | ΔY (%) b | HS c | KI (nM) d |
|---|---|---|---|---|
| 50 | 42.7 ± 10 | 87.7 ± 7.3 b | 1.3 ± 0.4 | 10.9 ± 3.9 |
| 100 | 52.6 ± 12.2 | 94.9 ± 17.9 | 0.7 ± 0.1 | 19.2 ± 5.3 |
| 150 | 318.6 ± 44.5 | 74.4 ± 2.9 | 1.6 ± 0.3 | 148 ± 54 |
| 200 | 3453 ± 517 | 73.2 ± 4.9 | 2.0 ± 0.5 | 1671 ± 480 |
| Slope | Intercept | KI,NON-IONIC (µM) | ΔGNON-IONIC (kcal/mol) | ΔGNON-IONIC (%) | ΔGIONIC (%) |
|---|---|---|---|---|---|
| 2.94 | −4.32 | 158.5 | 6.12 | 55.6 | 44.4 |
| Sample | aPTT (EC2X) (µM) | PT (EC2X) (µM) |
|---|---|---|
| NSGM 25 | >1000 | >1000 |
| UFH | 0.045 | 0.170 |
| Argatroban | 0.34 | 0.36 |
| Rivaroxaban | 0.12 | 0.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afosah, D.K.; Fayyad, R.M.; Puliafico, V.R.; Merrell, S.; Langmia, E.K.; Diagne, S.R.; Al-Horani, R.A.; Desai, U.R. Homogeneous, Synthetic, Non-Saccharide Glycosaminoglycan Mimetics as Potent Inhibitors of Human Cathepsin G. Biomolecules 2023, 13, 760. https://doi.org/10.3390/biom13050760
Afosah DK, Fayyad RM, Puliafico VR, Merrell S, Langmia EK, Diagne SR, Al-Horani RA, Desai UR. Homogeneous, Synthetic, Non-Saccharide Glycosaminoglycan Mimetics as Potent Inhibitors of Human Cathepsin G. Biomolecules. 2023; 13(5):760. https://doi.org/10.3390/biom13050760
Chicago/Turabian StyleAfosah, Daniel K., Rawan M. Fayyad, Valerie R. Puliafico, Spencer Merrell, Eltice K. Langmia, Sophie R. Diagne, Rami A. Al-Horani, and Umesh R. Desai. 2023. "Homogeneous, Synthetic, Non-Saccharide Glycosaminoglycan Mimetics as Potent Inhibitors of Human Cathepsin G" Biomolecules 13, no. 5: 760. https://doi.org/10.3390/biom13050760
APA StyleAfosah, D. K., Fayyad, R. M., Puliafico, V. R., Merrell, S., Langmia, E. K., Diagne, S. R., Al-Horani, R. A., & Desai, U. R. (2023). Homogeneous, Synthetic, Non-Saccharide Glycosaminoglycan Mimetics as Potent Inhibitors of Human Cathepsin G. Biomolecules, 13(5), 760. https://doi.org/10.3390/biom13050760