
Advanced Omics Techniques for Understanding Cochlear Genome, Epigenome, and Transcriptome in Health and Disease




Abstract
1. Introduction
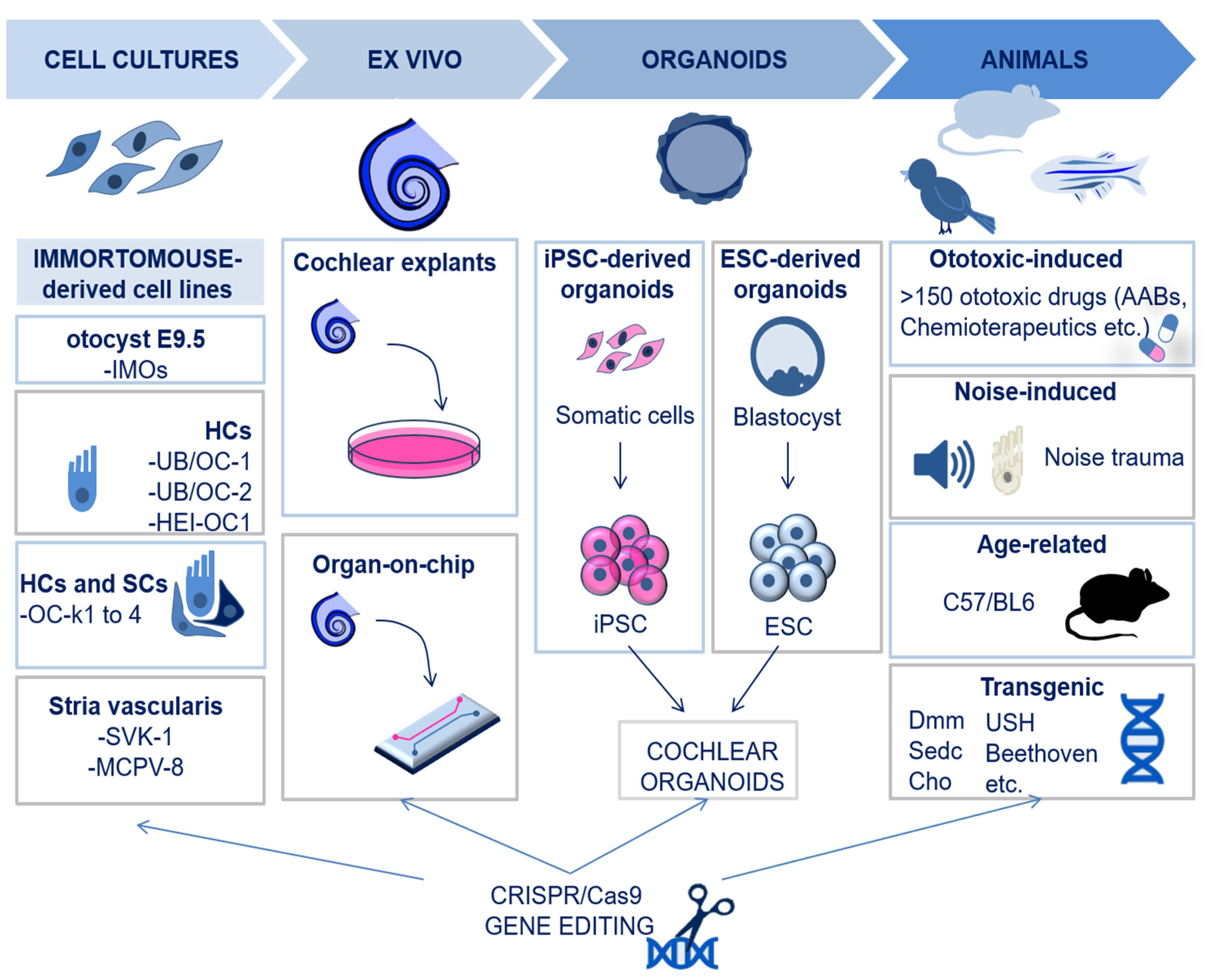
2. Experimental Models in Inner Ear Research
2.1. In Vitro and Ex Vivo Models: Cochlear Cell Lines, Organotypic Cultures, and Organoids
2.2. In Vivo Models
2.3. New Models Created by CRISPR/Cas9 Technology
3. Omics Techniques
3.1. Introduction to Omics: Principles and Advancements
3.2. Principles of Single-Cell Omics
3.3. Spatial Omics
4. The Role of Bioinformatics in Analyzing Omics Data
5. Genomics
5.1. Principles of Sequencing
5.2. Single-Cell and Spatial Genomics
5.3. Genomic Studies Have Delivered Unprecedented Knowledge on the Genetic Background and Early Diagnosis of Inherited Hearing Loss
6. Transcriptomics
6.1. Single-Cell Transcriptomics
6.2. Spatial Transcriptomics
6.2.1. Imaging-Based Technologies
6.2.2. Sequencing-Based Technologies
6.3. scRNA-seq Is a Key Tool for Deciphering the Complex Cellular Heterogeneity of the Cochlea
6.4. Spatial Transcriptomics Have Enabled Us to Understand the Cellular and Molecular Architecture of the Cochlea
7. Epigenomics
7.1. Principles of Epigenomics
7.2. Single-Cell Epigenomics
7.3. Spatial Epigenomics
7.4. Epigenetic Profiling of the Cochlea Has Provided New Insights into the Mechanisms Whereby Genes Responsible for Auditory Function Are Regulated
8. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omics Categories | Techniques | Applications in Hearing Research | Models Utilized | Reference |
|---|---|---|---|---|
| Genomics | WGS, WES | Identification of novel structural variants and rare mutations in genes associated with deafness | Humans (affected individuals with the CRDHL) | [65] |
| WES | Early detection of hearing loss for diagnostic purposes | Humans (individuals with diagnosis of hearing loss) | [83] | |
| Target exome panel | Improvement in the clinical diagnostic yield and thereby routine genetic screening | Humans (deaf patients suspected with underlying genetic causes of deafness) | [84] | |
| Humans (patients diagnosed with SNHL) | [82] | |||
| Transcriptomics | TruSeq | Identification of differential and preferential gene expression patterns and characterization of novel molecular pathways of the cochlea | Humans (patients with tumors of the skull base with normal hearing) | [118] |
| Engineered mouse models of genes related to circadian rhythm with noise damage | [121] | |||
| Comprehension of mechanisms involved in hair cell regeneration | Ototoxic (neomycin)-treated zebrafish | [184] | ||
| SMART-Seq v4 | Insights into the transcriptional changes in HCs during the process of ageing and damage | 1-, 9-, 18-, 22-, and 26-month-old CBA/J mice | [120] | |
| RNA-Seq V2 | Unraveling the genes specific to SGNs and their dynamicity in developmental processes | Mouse at different stages: E15.5, P1, P8, P14, and P30 | [185] | |
| Single-cell transcriptomics | SMART-Seq2 | Identification of novel subtypes of cochlear cells | Chicken | [123] |
| Identification of new markers of HCs | Mouse (C3HeB/FeJ) | [126] | ||
| 10x Genomics | Identification of gene regulatory networks involved in HC regeneration | Zebrafish (transgenic model for HC ablation) | [164] | |
| Identification of genes associated with Tmprss3-related hearing loss | Mouse (Tmprss3-KO organoids) | [127] | ||
| Delineation of key regulatory mechanisms in HC regeneration | Rats | [186] | ||
| Spatial transcriptomics | Single-molecule FISH (smFISH) | Annotating distinct transcriptome of SC populations in specific anatomic locations of the cochlea | Mouse | [131] |
| Whole-mount ISH | Spatiotemporal cadence of key signaling pathways in the context of developmental processes of the cochlea | Mouse organotypic cultures | [132] | |
| Genetically engineered mouse models of genes related to developmental processes | [129] | |||
| Uncovering quantitative differential transcriptional profile in pre-mature and mature HCs, revealing novel role of genes in the differentiation process | Mice (P4 and 3 weeks old) | [130] | ||
| LCM-NGS | Discovery of quantitative information of transcripts relevant in deafness in the organ of Corti, spiral ganglion, lateral wall, and spiral limbus | Mice (C57BL/6J) | [104] | |
| Epigenomics | ChIP-seq and ChIP-qPCR | Epigenetic modifications in the promoters of genes involved in SGN differentiation | In vitro immortalized multipotent otic progenitors (iMOP cells) | [165] |
| ATAC-seq | Identification of dynamics in chromatin accessibility of key transcriptional factors during the reprogramming of SCs into HCs | Mouse (Atoh1-nGFP, Sox2-GFP or Lgr5-GFP) and cochlear organoids | [167] | |
| Single-cell epigenomics | scATAC-seq | Regulation of chromatin accessibility during the process of regeneration and identification of genetically conserved regenerative response elements necessary for injury/regenerative responses | Zebrafish (transgenic model for HC ablation) | [164] |
| Identification of the epigenetic mechanisms responsible for the inability of SCs to trans-differentiate into HCs in the adult mammalian cochlea | Transgenic mouse models expressing transcription factors | [163] |
9. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Deafness and Hearing Loss. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 23 August 2023).
- Ma, Y.; Wise, A.K.; Shepherd, R.K.; Richardson, R.T. New Molecular Therapies for the Treatment of Hearing Loss. Pharmacol. Ther. 2019, 200, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.C.; Kujawa, S.G. Cochlear Synaptopathy in Acquired Sensorineural Hearing Loss: Manifestations and Mechanisms. Hear. Res. 2017, 349, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.K.; Pujol, R.; Landry, T.G.; Fallon, J.B.; Shepherd, R.K. Structural and Ultrastructural Changes to Type I Spiral Ganglion Neurons and Schwann Cells in the Deafened Guinea Pig Cochlea. J. Assoc. Res. Otolaryngol. 2017, 18, 751. [Google Scholar] [CrossRef] [PubMed]
- Smith-Cortinez, N.; Tan, A.K.; Stokroos, R.J.; Versnel, H.; Straatman, L.V. Regeneration of Hair Cells from Endogenous Otic Progenitors in the Adult Mammalian Cochlea: Understanding Its Origins and Future Directions. Int. J. Mol. Sci. 2023, 24, 7840. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cortez, R.L.P.; Yarza, T.K.L.; Bootpetch, T.C.; Tantoco, M.L.C.; Mohlke, K.L.; Cruz, T.L.G.; Perez, M.E.C.; Chan, A.L.; Lee, N.R.; Tobias-Grasso, C.A.M.; et al. Identification of Novel Candidate Genes and Variants for Hearing Loss and Temporal Bone Anomalies. Genes 2021, 12, 566. [Google Scholar] [CrossRef]
- Kelley, M.W. Cochlear Development; New Tools and Approaches. Front. Cell Dev. Biol. 2022, 10, 884240. [Google Scholar] [CrossRef]
- Wu, M.; Xia, M.; Li, W.; Li, H. Single-Cell Sequencing Applications in the Inner Ear. Front. Cell Dev. Biol. 2021, 9, 637779. [Google Scholar] [CrossRef]
- Bommakanti, K.; Iyer, J.S.; Stankovic, K.M. Cochlear Histopathology in Human Genetic Hearing Loss: State of the Science and Future Prospects. Hear. Res. 2019, 382, 107785. [Google Scholar] [CrossRef]
- Yamakami, I.; Ito, S.; Higuchi, Y. Retrosigmoid Removal of Small Acoustic Neuroma: Curative Tumor Removal with Preservation of Function: Clinical Article. J. Neurosurg. 2014, 121, 554–563. [Google Scholar] [CrossRef]
- Nicoli, T.K.; Atula, T.; Sinkkonen, S.T.; Korpi, J.; Vnencak, M.; Tarkkanen, J.; Mäkitie, A.A.; Jero, J. Ear Canal and Middle-Ear Tumors: A Single-Institution Series of 87 Patients. Acta Oto-Laryngol. 2022, 142, 132–139. [Google Scholar] [CrossRef]
- Gao, S.S.; Xia, A.; Applegate, B.E.; Shelton, R.L.; Yuan, T.; Raphael, P.D.; Oghalai, J.S. Quantitative Imaging of Cochlear Soft Tissues in Wild-Type and Hearing-Impaired Transgenic Mice by Spectral Domain Optical Coherence Tomography. Opt. Express 2011, 19, 15415–15428. [Google Scholar] [CrossRef]
- Rivolta, M.N.; Holley, M.C. Cell Lines in Inner Ear Research. J. Neurobiol. 2002, 53, 306–318. [Google Scholar] [CrossRef]
- Lim, D.J.; Moon, S.K. Establishment of Cell Lines from the Human Middle and Inner Ear Epithelial Cells. Adv. Exp. Med. Biol. 2011, 720, 15–25. [Google Scholar] [CrossRef]
- Haque, K.D.; Pandey, A.K.; Kelley, M.W.; Puligilla, C. Culture of Embryonic Mouse Cochlear Explants and Gene Transfer by Electroporation. JoVE 2015, 95, e52260. [Google Scholar] [CrossRef]
- Mazzarda, F.; D’Elia, A.; Massari, R.; De Ninno, A.; Bertani, F.R.; Businaro, L.; Ziraldo, G.; Zorzi, V.; Nardin, C.; Peres, C.; et al. Organ-on-Chip Model Shows That ATP Release through Connexin Hemichannels Drives Spontaneous Ca2+ Signaling in Non-Sensory Cells of the Greater Epithelial Ridge in the Developing Cochlea. Lab Chip 2020, 20, 3011–3023. [Google Scholar] [CrossRef]
- Roccio, M.; Edge, A.S.B. Inner Ear Organoids: New Tools to Understand Neurosensory Cell Development, Degeneration and Regeneration. Development 2019, 146, dev177188. [Google Scholar] [CrossRef]
- Lin, X.; Luo, J.; Tan, J.; Yang, L.; Wang, M.; Li, P. Experimental Animal Models of Drug-Induced Sensorineural Hearing Loss: A Narrative Review. Ann. Transl. Med. 2021, 9, 1393. [Google Scholar] [CrossRef]
- Legan, P.K.; Rau, A.; Keen, J.N.; Richardson, G.P. The Mouse Tectorins. Modular Matrix Proteins of the Inner Ear Homologous to Components of the Sperm-Egg Adhesion System. J. Biol. Chem. 1997, 272, 8791–8801. [Google Scholar] [CrossRef]
- Verhoeven, K.; Van Laer, L.; Kirschhofer, K.; Legan, P.K.; Hughes, D.C.; Schatteman, I.; Verstreken, M.; Van Hauwe, P.; Coucke, P.; Chen, A.; et al. Mutations in the Human Alpha-Tectorin Gene Cause Autosomal Dominant Non-Syndromic Hearing Impairment. Nat. Genet. 1998, 19, 60–62. [Google Scholar] [CrossRef]
- Vreugde, S.; Erven, A.; Kros, C.J.; Marcotti, W.; Fuchs, H.; Kurima, K.; Wilcox, E.R.; Friedman, T.B.; Griffith, A.J.; Bailing, R.; et al. Beethoven, a Mouse Model for Dominant, Progressive Hearing Loss DFNA36. Nat. Genet. 2002, 30, 257–258. [Google Scholar] [CrossRef]
- Friedman, L.M.; Dror, A.A.; Avraham, K.B. Mouse Models to Study Inner Ear Development and Hereditary Hearing Loss. Int. J. Dev. Biol. 2007, 51, 609–631. [Google Scholar] [CrossRef]
- Lanvers-Kaminsky, C.; Zehnhoff-Dinnesen, A.A.; Parfitt, R.; Ciarimboli, G. Drug-Induced Ototoxicity: Mechanisms, Pharmacogenetics, and Protective Strategies. Clin. Pharmacol. Ther. 2017, 101, 491–500. [Google Scholar] [CrossRef]
- Le Prell, C.G.; Hammill, T.L.; Murphy, W.J. Noise-Induced Hearing Loss: Translating Risk from Animal Models to Real-World Environments. J. Acoust. Soc. Am. 2019, 146, 3646–3651. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 Therapeutics: Progress and Prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef]
- Akram, F.; Sahreen, S.; Aamir, F.; Haq, I.U.; Malik, K.; Imtiaz, M.; Naseem, W.; Nasir, N.; Waheed, H.M. An Insight into Modern Targeted Genome-Editing Technologies with a Special Focus on CRISPR/Cas9 and Its Applications. Mol. Biotechnol. 2023, 65, 227–242. [Google Scholar] [CrossRef]
- Zou, B.; Mittal, R.; Grati, M.; Lu, Z.; Shu, Y.; Tao, Y.; Feng, Y.; Xie, D.; Kong, W.; Yang, S.; et al. The Application of Genome Editing in Studying Hearing Loss. Hear. Res. 2015, 327, 102–108. [Google Scholar] [CrossRef]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.H.; Pan, B.; Hu, Y.J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of Autosomal Dominant Hearing Loss by in Vivo Delivery of Genome Editing Agents. Nature 2017, 553, 217–221. [Google Scholar] [CrossRef]
- Farooq, R.; Hussain, K.; Tariq, M.; Farooq, A.; Mustafa, M. CRISPR/Cas9: Targeted Genome Editing for the Treatment of Hereditary Hearing Loss. J. Appl. Genet. 2020, 61, 51–65. [Google Scholar] [CrossRef]
- Wu, J.; Tao, Y.; Deng, D.; Meng, Z.; Zhao, Y. The Applications of CRISPR/Cas-Mediated Genome Editing in Genetic Hearing Loss. Cell Biosci. 2023, 13, 93. [Google Scholar] [CrossRef]
- Rathinam, R.; Rosati, R.; Jamesdaniel, S. CRISPR/Cas9-Mediated Knockout of Lim-Domain Only Four Retards Organ of Corti Cell Growth. J. Cell. Biochem. 2018, 119, 3545–3553. [Google Scholar] [CrossRef]
- Gu, X.; Wang, D.; Xu, Z.; Wang, J.; Guo, L.; Chai, R.; Li, G.; Shu, Y.; Li, H. Prevention of Acquired Sensorineural Hearing Loss in Mice by in Vivo Htra2 Gene Editing. Genome Biol. 2021, 22, 86. [Google Scholar] [CrossRef]
- Liu, J.; Duan, X.; Li, M.; Liu, D.; Bai, X. CRISPR/Cas9-Mediated Pou4f3 Knockout Induces Defects in the Development of the Zebrafish Inner Ear. J. BioX Res. 2021, 4, 163–170. [Google Scholar] [CrossRef]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 911861. [Google Scholar] [CrossRef]
- Kong, S.; Li, R.; Tian, Y.; Zhang, Y.; Lu, Y.; Ou, Q.; Gao, P.; Li, K.; Zhang, Y. Single-Cell Omics: A New Direction for Functional Genetic Research in Human Diseases and Animal Models. Front. Genet. 2023, 13, 1100016. [Google Scholar] [CrossRef]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-Omics: Novel Approaches to Probe Cell Heterogeneity and Extracellular Matrix Biology. Matrix Biol. 2020, 91–92, 152–166. [Google Scholar] [CrossRef]
- Zheng, Q.Y.; Rozanas, C.R.; Thalmann, I.; Chance, M.R.; Alagramam, K.N. Inner Ear Proteomics of Mouse Models for Deafness, a Discovery Strategy. Brain Res. 2006, 1091, 113–121. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature 2011, 472, 90–95. [Google Scholar] [CrossRef]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-Coverage Single-Cell MRNA Sequencing Reveals Cellular Heterogeneity and Activated Signaling Pathways in Developing Cerebral Cortex. Nat. Biotechnol. 2014, 32, 1053–1058. [Google Scholar] [CrossRef]
- Ealy, M.; Ellwanger, D.C.; Kosaric, N.; Stapper, A.P.; Heller, S. Single-Cell Analysis Delineates a Trajectory toward the Human Early Otic Lineage. Proc. Natl. Acad. Sci. USA 2016, 113, 8508–8513. [Google Scholar] [CrossRef]
- Tasic, B. Single Cell Transcriptomics in Neuroscience: Cell Classification and Beyond. Curr. Opin. Neurobiol. 2018, 50, 242–249. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Wu, L.; Guo, J.; Song, Y.; Tian, T.; Wang, W.; Zhu, Z.; Yang, C. Microfluidic Single-Cell Omics Analysis. Small 2020, 16, 1903905. [Google Scholar] [CrossRef]
- Menze, L.; Duarte, P.A.; Haddon, L.; Chu, M.; Chen, J. Selective Single-Cell Sorting Using a Multisectorial Electroactive Nanowell Platform. ACS Nano 2022, 16, 211–220. [Google Scholar] [CrossRef]
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef]
- Mincarelli, L.; Lister, A.; Lipscombe, J.; Macaulay, I.C. Defining Cell Identity with Single-Cell Omics. Proteomics 2018, 18, 1700312. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-Cell Triple Omics Sequencing Reveals Genetic, Epigenetic, and Transcriptomic Heterogeneity in Hepatocellular Carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Raphael, Y.; Altschuler, R.A. Structure and Innervation of the Cochlea. Brain Res. Bull. 2003, 60, 397–422. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, A.; Chana, I. Computational Techniques and Tools for Omics Data Analysis: State-of-the-Art, Challenges, and Future Directions. Arch. Comput. Methods Eng. 2021, 28, 4595–4631. [Google Scholar] [CrossRef]
- Hie, B.; Peters, J.; Nyquist, S.K.; Shalek, A.K.; Berger, B.; Bryson, B.D. Computational Methods for Single-Cell RNA Sequencing. Annu. Rev. Biomed. Data Sci. 2020, 3, 339–364. [Google Scholar] [CrossRef]
- Stanojevic, S.; Li, Y.; Ristivojevic, A.; Garmire, L.X. Computational Methods for Single-Cell Multi-Omics Integration and Alignment. Genom. Proteom. Bioinform. 2022, 20, 836–849. [Google Scholar] [CrossRef]
- Li, Y.; Stanojevic, S.; Garmire, L.X. Emerging Artificial Intelligence Applications in Spatial Transcriptomics Analysis. Comput. Struct. Biotechnol. J. 2022, 20, 2895–2908. [Google Scholar] [CrossRef]
- Efremova, M.; Teichmann, S.A. Computational Methods for Single-Cell Omics across Modalities. Nat. Methods 2020, 17, 14–17. [Google Scholar] [CrossRef]
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaría, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of Deep Learning: Concepts, CNN Architectures, Challenges, Applications, Future Directions. J. Big Data 2021, 8, 53. [Google Scholar] [CrossRef]
- Wang, H.; Guo, F.; Du, M.; Wang, G.; Cao, C. A Novel Method for Drug-Target Interaction Prediction Based on Graph Transformers Model. BMC Bioinform. 2022, 23, 459. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, T.; Yang, B.; Su, J.; Song, Q. SpaCI: Deciphering Spatial Cellular Communications through Adaptive Graph Model. Brief. Bioinform. 2023, 24, bbac563. [Google Scholar] [CrossRef]
- Tang, Z.; Li, Z.; Hou, T.; Zhang, T.; Yang, B.; Su, J.; Song, Q. SiGra: Single-Cell Spatial Elucidation through an Image-Augmented Graph Transformer. Nat. Commun. 2023, 14, 5618. [Google Scholar] [CrossRef]
- Rehman, A.U.; Morell, R.J.; Belyantseva, I.A.; Khan, S.Y.; Boger, E.T.; Shahzad, M.; Ahmed, Z.M.; Riazuddin, S.; Khan, S.N.; Riazuddin, S.; et al. Targeted Capture and Next-Generation Sequencing Identifies C9orf75, Encoding Taperin, as the Mutated Gene in Nonsyndromic Deafness DFNB79. Am. J. Hum. Genet. 2010, 86, 378–388. [Google Scholar] [CrossRef]
- Yan, D.; Tekin, M.; Blanton, S.H.; Liu, X.Z. Next-Generation Sequencing in Genetic Hearing Loss. Genet. Test. Mol. Biomark. 2013, 17, 581–587. [Google Scholar] [CrossRef]
- Shearer, A.E.; Smith, R.J.H. Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss. Otolaryngol.–Head Neck Surg. 2015, 153, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 2018, 9, 314696. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Santani, A.; Mao, R.; Ferreira-Gonzalez, A.; Weck, K.E.; Voelkerding, K.V. Development and Validation of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline Variants in Inherited Disease. Arch. Pathol. Lab. Med. 2017, 141, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Ascari, G.; Rendtorff, N.D.; De Bruyne, M.; De Zaeytijd, J.; Van Lint, M.; Bauwens, M.; Van Heetvelde, M.; Arno, G.; Jacob, J.; Creytens, D.; et al. Long-Read Sequencing to Unravel Complex Structural Variants of CEP78 Leading to Cone-Rod Dystrophy and Hearing Loss. Front. Cell Dev. Biol. 2021, 9, 664317. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef]
- Drmanac, R.; Drmanac, S.; Chui, G.; Diaz, R.; Hou, A.; Jin, H.; Jin, P.; Kwon, S.; Lacy, S.; Moeur, B.; et al. Sequencing by Hybridization (SBH): Advantages, Achievements, and Opportunities. Adv. Biochem. Eng. Biotechnol. 2002, 77, 75–101. [Google Scholar] [CrossRef]
- Blazej, R.G.; Kumaresan, P.; Mathies, R.A. Microfabricated Bioprocessor for Integrated Nanoliter-Scale Sanger DNA Sequencing. Proc. Natl. Acad. Sci. USA 2006, 103, 7240–7245. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A Tale of Three next Generation Sequencing Platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq Sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved Data Analysis for the MinION Nanopore Sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef]
- Evrony, G.D.; Hinch, A.G.; Luo, C. Applications of Single-Cell DNA Sequencing. Annu. Rev. Genom. Hum. Genet. 2021, 22, 171. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-Cell Genome Sequencing: Current State of the Science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Yang, C.; Li, W.; Bai, X.; Zhou, X.; Xie, H.; Wen, L.; Tang, F. SMOOTH-Seq: Single-Cell Genome Sequencing of Human Cells on a Third-Generation Sequencing Platform. Genome Biol. 2021, 22, 195. [Google Scholar] [CrossRef]
- Tang, L. Spatially Resolved DNA Sequencing. Nat. Methods 2022, 19, 139. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.X.; Kun, S.; Jing, Q.; Jing, C.; Denise, Y. Non-Syndromic Hearing Loss and High-Throughput Strategies to Decipher Its Genetic Heterogeneity. J. Otol. 2013, 8, 6–24. [Google Scholar] [CrossRef]
- Aldè, M.; Cantarella, G.; Zanetti, D.; Pignataro, L.; Mantia, I.L.; Maiolino, L.; Ferlito, S.; Mauro, P. Di; Cocuzza, S.; Lechien, J.R.; et al. Autosomal Dominant Non-Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review. Biomedicines 2023, 11, 1616. [Google Scholar] [CrossRef]
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss—Syndromic. Otolaryngol. Clin. N. Am. 2015, 48, 1041. [Google Scholar] [CrossRef]
- Bai, X.; Nian, S.; Feng, L.; Ruan, Q.; Luo, X.; Wu, M.; Yan, Z. Identification of Novel Variants in MYO15A, OTOF, and RDX with Hearing Loss by next-Generation Sequencing. Mol. Genet. Genom. Med. 2019, 7, e808. [Google Scholar] [CrossRef]
- Cheng, H.; Capponi, S.; Wakeling, E.; Marchi, E.; Li, Q.; Zhao, M.; Weng, C.; Stefan, P.G.; Ahlfors, H.; Kleyner, R.; et al. Missense Variants in TAF1 and Developmental Phenotypes: Challenges of Determining Pathogenicity. Hum. Mutat. 2020, 41, 449–464. [Google Scholar] [CrossRef]
- Brownstein, Z.; Gulsuner, S.; Walsh, T.; Martins, F.T.A.; Taiber, S.; Isakov, O.; Lee, M.K.; Bordeynik-Cohen, M.; Birkan, M.; Chang, W.; et al. Spectrum of Genes for Inherited Hearing Loss in the Israeli Jewish Population, Including the Novel Human Deafness Gene ATOH1. Clin. Genet. 2020, 98, 353–364. [Google Scholar] [CrossRef]
- Costales, M.; Diñeiro, M.; Cifuentes, G.A.; Capín, R.; Otero, A.; Viejo-Díaz, M.; Plasencia, A.; Núñez, F.; Gómez, J.R.; Llorente, J.L.; et al. Utilidad Clínica de La Secuenciación de Nueva Generación En El Diagnóstico Etiológico de La Hipoacusia Neurosensorial En Una Unidad de Hipoacusia Infantil. Acta Otorrinolaringol. Esp. 2020, 71, 166–174. [Google Scholar] [CrossRef]
- Van Heurck, R.; Carminho-rodrigues, M.T.; Ranza, E.; Stafuzza, C.; Quteineh, L.; Gehrig, C.; Hammar, E.; Guipponi, M.; Abramowicz, M.; Senn, P.; et al. Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes 2021, 12, 1277. [Google Scholar] [CrossRef] [PubMed]
- Tropitzsch, A.; Schade-Mann, T.; Gamerdinger, P.; Dofek, S.; Schulte, B.; Schulze, M.; Battke, F.; Fehr, S.; Biskup, S.; Heyd, A.; et al. Diagnostic Yield of Targeted Hearing Loss Gene Panel Sequencing in a Large German Cohort with a Balanced Age Distribution from a Single Diagnostic Center: An Eight-Year Study. Ear Hear. 2022, 43, 1049–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hu, L.; Yang, L.; Wang, L.; Lu, Y.; Dong, X.; Xiao, T.; Xu, Z.; Wu, B.; Zhou, W. Association Between Expanded Genomic Sequencing Combined with Hearing Screening and Detection of Hearing Loss Among Newborns in a Neonatal Intensive Care Unit. JAMA Netw. Open 2022, 5, e2220986. [Google Scholar] [CrossRef]
- Dong, Z.C.; Chen, Y. Transcriptomics: Advances and Approaches. Sci. China Life Sci. 2013, 56, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, T.; Jenjaroenpun, P.; Wanchai, V.; Nookaew, I. Native RNA or CDNA Sequencing for Transcriptomic Analysis: A Case Study on Saccharomyces Cerevisiae. Front. Bioeng. Biotechnol. 2022, 10, 842299. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y.; Correspondence, Y.; Luo, D.; Jovic, L. Single-Cell RNA Sequencing Technologies and Applications: A Brief Overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, A.M.; Mazutis, L. Single-Cell Barcoding and Sequencing Using Droplet Microfluidics. Nat. Protoc. 2016, 12, 44–73. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Christopher Love, J.; Shalek, A.K. Seq-Well: Portable, Low-Cost RNA Sequencing of Single Cells at High Throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-Sequencing from Single Nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [PubMed]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A Single-Cell and Single-Nucleus RNA-Seq Toolbox for Fresh and Frozen Human Tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef]
- Chen, W.; Guillaume-Gentil, O.; Rainer, P.Y.; Gäbelein, C.G.; Saelens, W.; Gardeux, V.; Klaeger, A.; Dainese, R.; Zachara, M.; Zambelli, T.; et al. Live-Seq Enables Temporal Transcriptomic Recording of Single Cells. Nature 2022, 608, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Fangma, Y.; Liu, M.; Liao, J.; Chen, Z.; Zheng, Y. Dissecting the Brain with Spatially Resolved Multi-Omics. J. Pharm. Anal. 2023, 13, 694–710. [Google Scholar] [CrossRef]
- Payne, A.C.; Chiang, Z.D.; Reginato, P.L.; Mangiameli, S.M.; Murray, E.M.; Yao, C.C.; Markoulaki, S.; Earl, A.S.; Labade, A.S.; Jaenisch, R.; et al. In Situ Genome Sequencing Resolves DNA Sequence and Structure in Intact Biological Samples. Science 2021, 371, eaay3446. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An Introduction to Spatial Transcriptomics for Biomedical Research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-Cell in Situ RNA Profiling by Sequential Hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Borm, L.E.; Mossi Albiach, A.; Mannens, C.C.A.; Janusauskas, J.; Özgün, C.; Fernández-García, D.; Hodge, R.; Castillo, F.; Hedin, C.R.H.; Villablanca, E.J.; et al. Scalable in Situ Single-Cell Profiling by Electrophoretic Capture of MRNA Using EEL FISH. Nat. Biotechnol. 2022, 41, 222–231. [Google Scholar] [CrossRef]
- Wang, Y.; Eddison, M.; Fleishman, G.; Weigert, M.; Xu, S.; Wang, T.; Rokicki, K.; Goina, C.; Henry, F.E.; Lemire, A.L.; et al. EASI-FISH for Thick Tissue Defines Lateral Hypothalamus Spatio-Molecular Organization. Cell 2021, 184, 6361–6377.e24. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In Situ Sequencing for RNA Analysis in Preserved Tissue and Cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.y.; Takumi, Y.; Usami, S. ichi Laser-Capture Micro Dissection Combined with next-Generation Sequencing Analysis of Cell Type-Specific Deafness Gene Expression in the Mouse Cochlea. Hear. Res. 2017, 348, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kruse, F.; Junker, J.P.; van Oudenaarden, A.; Bakkers, J. Tomo-Seq: A Method to Obtain Genome-Wide Expression Data with Spatial Resolution. Methods Cell Biol. 2016, 135, 299–307. [Google Scholar] [CrossRef]
- Chen, J.; Suo, S.; Tam, P.P.; Han, J.D.J.; Peng, G.; Jing, N. Spatial Transcriptomic Analysis of Cryosectioned Tissue Samples with Geo-Seq. Nat. Protoc. 2017, 12, 566–580. [Google Scholar] [CrossRef]
- Hernandez, S.; Lazcano, R.; Serrano, A.; Powell, S.; Kostousov, L.; Mehta, J.; Khan, K.; Lu, W.; Solis, L.M. Challenges and Opportunities for Immunoprofiling Using a Spatial High-Plex Technology: The NanoString GeoMx® Digital Spatial Profiler. Front. Oncol. 2022, 12, 890410. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex Digital Spatial Profiling of Proteins and RNA in Fixed Tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- Schede, H.H.; Schneider, C.G.; Stergiadou, J.; Borm, L.E.; Ranjak, A.; Yamawaki, T.M.; David, F.P.A.; Lönnerberg, P.; Tosches, M.A.; Codeluppi, S.; et al. Spatial Tissue Profiling by Imaging-Free Molecular Tomography. Nat. Biotechnol. 2021, 39, 968–977. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L.; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-Scale Spatial Gene Expression in the Human Dorsolateral Prefrontal Cortex. Nat. Neurosci. 2021, 24, 425–436. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal Transcriptomic Atlas of Mouse Organogenesis Using DNA Nanoball-Patterned Arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly Sensitive Spatial Transcriptomics at Near-Cellular Resolution with Slide-SeqV2. Nat. Biotechnol. 2020, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665. [Google Scholar] [CrossRef] [PubMed]
- Schimmang, T.; Maconochie, M. Gene Expression Profiling of the Inner Ear. J. Anat. 2016, 228, 255–269. [Google Scholar] [CrossRef]
- Ramírez-Gordillo, D.; Powers, T.R.; Van Velkinburgh, J.C.; Trujillo-Provencio, C.; Schilkey, F.; Serrano, E.E. RNA-Seq and Microarray Analysis of the Xenopus Inner Ear Transcriptome Discloses Orthologous OMIM® Genes for Hereditary Disorders of Hearing and Balance. BMC Res. Notes 2015, 8, 691. [Google Scholar] [CrossRef][Green Version]
- Schrauwen, I.; Hasin-Brumshtein, Y.; Corneveaux, J.J.; Ohmen, J.; White, C.; Allen, A.N.; Lusis, A.J.; Van Camp, G.; Huentelman, M.J.; Friedman, R.A. A Comprehensive Catalogue of the Coding and Non-Coding Transcripts of the Human Inner Ear. Hear. Res. 2016, 333, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Takechi, M.; Wang, X.; Furutera, T.; Nojiri, T.; Koyabu, D.; Li, J. Temporal and Regulatory Dynamics of the Inner Ear Transcriptome during Development in Mice. Sci. Rep. 2022, 12, 21196. [Google Scholar] [CrossRef]
- Liu, H.; Giffen, K.P.; Chen, L.; Henderson, H.J.; Cao, T.A.; Kozeny, G.A.; Beisel, K.W.; Li, Y.; He, D.Z. Molecular and Cytological Profiling of Biological Aging of Mouse Cochlear Inner and Outer Hair Cells. Cell Rep. 2022, 39, 110665. [Google Scholar] [CrossRef]
- Cederroth, C.R.; Park, J.s.; Basinou, V.; Weger, B.D.; Tserga, E.; Sarlus, H.; Magnusson, A.K.; Kadri, N.; Gachon, F.; Canlon, B. Circadian Regulation of Cochlear Sensitivity to Noise by Circulating Glucocorticoids. Curr. Biol. 2019, 29, 2477–2487.e6. [Google Scholar] [CrossRef]
- Lush, M.E.; Diaz, D.C.; Koenecke, N.; Baek, S.; Boldt, H.; St Peter, M.K.; Gaitan-Escudero, T.; Romero-Carvajal, A.; Busch-Nentwich, E.M.; Perera, A.G.; et al. Scrna-Seq Reveals Distinct Stem Cell Populations That Drive Hair Cell Regeneration after Loss of Fgf and Notch Signaling. eLife 2019, 8, e44431. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Scheibinger, M.; Benkafadar, N.; Kirti, S.; Ellwanger, D.C.; Heller, S. Cell-Type Identity of the Avian Cochlea. Cell Rep. 2021, 34, 108900. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Noda, T.; Mulvaney, J.F.; Lin, V.Y.W.; Edge, A.S.B.; Dabdoub, A. Comprehensive Expression of Wnt Signaling Pathway Genes during Development and Maturation of the Mouse Cochlea. PLoS ONE 2016, 11, e0148339. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, M.M.M.; Wan, G.; Zhang, L.L.; Gigliello, A.R.; McInnis, J.J.; Zhang, Y.; Bergles, D.; Zuo, J.; Corfas, G. Spontaneous Regeneration of Cochlear Supporting Cells after Neonatal Ablation Ensures Hearing in the Adult Mouse. Proc. Natl. Acad. Sci. USA 2014, 111, 16919–16924. [Google Scholar] [CrossRef]
- Ranum, P.T.; Goodwin, A.T.; Yoshimura, H.; Kolbe, D.L.; Walls, W.D.; Koh, J.Y.; He, D.Z.Z.; Smith, R.J.H. Insights into the Biology of Hearing and Deafness Revealed by Single-Cell RNA Sequencing. Cell Rep. 2019, 26, 3160–3171.e3. [Google Scholar] [CrossRef]
- Tang, P.C.; Alex, A.L.; Nie, J.; Lee, J.; Roth, A.A.; Booth, K.T.; Koehler, K.R.; Hashino, E.; Nelson, R.F. Defective Tmprss3-Associated Hair Cell Degeneration in Inner Ear Organoids. Stem Cell Rep. 2019, 13, 147–162. [Google Scholar] [CrossRef]
- Milon, B.; Shulman, E.D.; So, K.S.; Cederroth, C.R.; Lipford, E.L.; Sperber, M.; Sellon, J.B.; Sarlus, H.; Pregernig, G.; Shuster, B.; et al. A Cell-Type-Specific Atlas of the Inner Ear Transcriptional Response to Acoustic Trauma. Cell Rep. 2021, 36, 109758. [Google Scholar] [CrossRef]
- Yang, L.M.; Cheah, K.S.E.; Huh, S.H.; Ornitz, D.M. Sox2 and FGF20 Interact to Regulate Organ of Corti Hair Cell and Supporting Cell Development in a Spatially-Graded Manner. PLoS Genet. 2019, 15, e1008254. [Google Scholar] [CrossRef]
- Huang, G.; Eckrich, S. Quantitative Fluorescent in Situ Hybridization Reveals Differential Transcription Profile Sharpening of Endocytic Proteins in Cochlear Hair Cells Upon Maturation. Front. Cell Neurosci. 2021, 15, 643517. [Google Scholar] [CrossRef]
- Hoa, M.; Olszewski, R.; Li, X.; Taukulis, I.; Gu, S.; DeTorres, A.; Lopez, I.A.; Linthicum, F.H.; Ishiyama, A.; Martin, D.; et al. Characterizing Adult Cochlear Supporting Cell Transcriptional Diversity Using Single-Cell RNA-Seq: Validation in the Adult Mouse and Translational Implications for the Adult Human Cochlea. Front. Mol. Neurosci. 2020, 13, 491389. [Google Scholar] [CrossRef]
- Munnamalai, V.; Fekete, D.M. Notch-Wnt-Bmp Crosstalk Regulates Radial Patterning in the Mouse Cochlea in a Spatiotemporal Manner. Development 2016, 143, 4003–4015. [Google Scholar] [CrossRef] [PubMed]
- Mehrmohamadi, M.; Sepehri, M.H.; Nazer, N.; Norouzi, M.R. A Comparative Overview of Epigenomic Profiling Methods. Front. Cell Dev. Biol. 2021, 9, 714687. [Google Scholar] [CrossRef] [PubMed]
- Oakes, C.C.; La Salle, S.; Robaire, B.; Trasler, J.M. Evaluation of a Quantitative DNA Methylation Analysis Technique Using Methylation-Sensitive/Dependent Restriction Enzymes and Real-Time PCR. Epigenetics 2006, 1, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-Wide and Promoter-Specific Analyses Identify Sites of Differential DNA Methylation in Normal and Transformed Human Cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-Seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef]
- Pajoro, A.; Muiño, J.M.; Angenent, G.C.; Kaufmann, K. Profiling Nucleosome Occupancy by MNase-Seq: Experimental Protocol and Computational Analysis. Methods Mol. Biol. 2018, 1675, 167–181. [Google Scholar] [CrossRef]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-Seq and FAIRE-Seq Open Chromatin Profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef]
- Li, Y. Modern Epigenetics Methods in Biological Research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, O.; Tanay, A. Single-Cell Epigenomics: Techniques and Emerging Applications. Nat. Rev. Genet. 2015, 16, 716–726. [Google Scholar] [CrossRef]
- Han, L.; Wu, H.J.; Zhu, H.; Kim, K.Y.; Marjani, S.L.; Riester, M.; Euskirchen, G.; Zi, X.; Yang, J.; Han, J.; et al. Bisulfite-Independent Analysis of CpG Island Methylation Enables Genome-Scale Stratification of Single Cells. Nucleic Acids Res. 2017, 45, e77. [Google Scholar] [CrossRef] [PubMed]
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly Scalable Generation of DNA Methylation Profiles in Single Cells. Nat. Biotechnol. 2018, 36, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-Cell Epigenomics: Powerful New Methods for Understanding Gene Regulation and Cell Identity. Genome Biol. 2016, 17, 72. [Google Scholar] [CrossRef]
- Su, J.H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020, 182, 1641–1659.e26. [Google Scholar] [CrossRef]
- Takei, Y.; Yun, J.; Zheng, S.; Ollikainen, N.; Pierson, N.; White, J.; Shah, S.; Thomassie, J.; Suo, S.; Eng, C.H.L.; et al. Integrated Spatial Genomics Reveals Global Architecture of Single Nuclei. Nature 2021, 590, 344–350. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially Resolved Chromatin Modification Profiling at the Cellular Level. Science 2022, 375, 681–686. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial Profiling of Chromatin Accessibility in Mouse and Human Tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef]
- Lu, T.; Ang, C.E.; Zhuang, X. Spatially Resolved Epigenomic Profiling of Single Cells in Complex Tissues. Cell 2022, 185, 4448–4464.e17. [Google Scholar] [CrossRef]
- Bhamidipati, T.; Sinha, M.; Sen, C.K.; Singh, K. Laser Capture Microdissection in the Spatial Analysis of Epigenetic Modifications in Skin: A Comprehensive Review. Oxid. Med. Cell Longev. 2022, 2022, 4127238. [Google Scholar] [CrossRef] [PubMed]
- Yeming, X.; Fengying, R.; Yaning, L.; Meng, L.; Chen, Z.; Zhichao, C.; Zhe, X.; Zhe, W.; Weitian, C.; Wenfang, C.; et al. Spatial Chromatin Accessibility Sequencing Resolves High-Order Spatial Interactions of Epigenomic Markers. eLife 2023, 12, RP87868. [Google Scholar] [CrossRef]
- Klein, C.J.; Bird, T.; Ertekin-Taner, N.; Lincoln, S.; Hjorth, R.; Wu, Y.; Kwok, J.; Mer, G.; Dyck, P.J.; Nicholson, G.A. DNMT1 Mutation Hot Spot Causes Varied Phenotypes of HSAN1 with Dementia and Hearing Loss. Neurology 2013, 80, 824–828. [Google Scholar] [CrossRef]
- Balendran, V.; Ritter, K.E.; Martin, D.M. Epigenetic Mechanisms of Inner Ear Development. Hear. Res. 2022, 426, 108440. [Google Scholar] [CrossRef] [PubMed]
- Seyama, R.; Tsuchida, N.; Okada, Y.; Sakata, S.; Hamada, K.; Azuma, Y.; Hamanaka, K.; Fujita, A.; Koshimizu, E.; Miyatake, S.; et al. Two Families with TET3-Related Disorder Showing Neurodevelopmental Delay with Craniofacial Dysmorphisms. J. Hum. Genet. 2022, 67, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Streit, A. Lsd1 Interacts with CMyb to Demethylate Repressive Histone Marks and Maintain Inner Ear Progenitor Identity. Development 2018, 145, dev160325. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.O.; Lee, J.J.; Kim, M.; Chung, Y.W.; Min, H.; Kim, J.Y.; Kim, H.P.; Bok, J. CTCF Regulates Otic Neurogenesis via Histone Modification in the Neurog1 Locus. Moleucles Cells 2018, 41, 695–702. [Google Scholar] [CrossRef]
- Tao, L.; Yu, H.V.; Llamas, J.; Trecek, T.; Wang, X.; Stojanova, Z.; Groves, A.K.; Segil, N. Enhancer Decommissioning Imposes an Epigenetic Barrier to Sensory Hair Cell Regeneration. Dev. Cell 2021, 56, 2471–2485.e5. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; Van Ravenswaaij, C.M.A.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.A.; Janssen, I.M.; Van Der Vliet, W.A.; Huys, E.H.L.P.G.; De Jong, P.J.; Hamel, B.C.J.; et al. Mutations in a New Member of the Chromodomain Gene Family Cause CHARGE Syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef]
- Dawe, C.E.; Kooistra, M.K.; Fairbridge, N.A.; Pisio, A.C.; McDermid, H.E. Role of Chromatin Remodeling Gene Cecr2 in Neurulation and Inner Ear Development. Dev. Dyn. 2011, 240, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.A.; Hosamani, I.; Nguyen, J.D.; Cai, T.; Singh, S.; McGovern, M.M.; Beyer, L.; Zhang, H.; Jen, H.I.; Yousaf, R.; et al. Cellular Reprogramming with ATOH1, GFI1, and POU4F3 Implicate Epigenetic Changes and Cell-Cell Signaling as Obstacles to Hair Cell Regeneration in Mature Mammals. eLife 2022, 11, e79712. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.; Slevin, C.C.; Song, W.; Chen, Z.; Frederickson, S.C.; Gildea, D.; Wu, W.; Elkahloun, A.G.; Ovcharenko, I.; Burgess, S.M. A Regulatory Network of Sox and Six Transcription Factors Initiate a Cell Fate Transformation during Hearing Regeneration in Adult Zebrafish. Cell Genom. 2022, 2, 100170. [Google Scholar] [CrossRef]
- Song, Z.; Jadali, A.; Fritzsch, B.; Kwan, K.Y. NEUROG1 Regulates CDK2 to Promote Proliferation in Otic Progenitors. Stem Cell Rep. 2017, 9, 1516–1529. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yuan, S.S.; Zhang, L.J.; Ji, Z.L.; Quan, X.J. Atonal BHLH Transcription Factor 1 Is an Important Factor for Maintaining the Balance of Cell Proliferation and Differentiation in Tumorigenesis (Review). Oncol. Lett. 2020, 20, 2595–2605. [Google Scholar] [CrossRef]
- Kalra, G.; Lenz, D.; Abdul-Aziz, D.; Hanna, C.; Herb, B.R.; Colantuoni, C.; Milon, B.; Saxena, M.; Shetty, A.C.; Hertzano, R.P.; et al. Cochlear Organoids Reveal Epigenetic and Transcriptional Programs of Postnatal Hair Cell Differentiation from Supporting Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Webster, M.; Webster, D.B. Spiral Ganglion Neuron Loss Following Organ of Corti Loss: A Quantitative Study. Brain Res. 1981, 212, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.K.; Hardie, N.A. Deafness-Induced Changes in the Auditory Pathway: Implications for Cochlear Implants. Audiol. Neurotol. 2002, 6, 305–318. [Google Scholar] [CrossRef]
- Versnel, H.; Agterberg, M.J.H.; de Groot, J.C.M.J.; Smoorenburg, G.F.; Klis, S.F.L. Time Course of Cochlear Electrophysiology and Morphology after Combined Administration of Kanamycin and Furosemide. Hear. Res. 2007, 231, 1–12. [Google Scholar] [CrossRef]
- FX-322 in Adults with Acquired Sensorineural Hearing Loss. Available online: https://clinicaltrials.gov/study/NCT05086276 (accessed on 30 September 2023).
- Mclean, W.J.; Hinton, A.S.; Herby, J.T.J.; Salt, A.N.; Hartsock, J.J.; Wilson, S.; Lucchino, D.L.; Lenarz, T.; Warnecke, A.; Prenzler, N.; et al. Improved Speech Intelligibility in Subjects with Stable Sensorineural Hearing Loss following Intratympanic Dosing of FX-322 in a Phase 1b Study. Otol. Neurotol. 2021, 42, e849. [Google Scholar] [CrossRef]
- Gupta, S.; Maheshwari, S.; Kirtane, M.; Shrivastav, N. Pictorial Review of MRI/CT Scan in Congenital Temporal Bone Anomalies, in Patients for Cochlear Implant. Indian J. Radiol. Imaging 2009, 19, 99. [Google Scholar] [CrossRef]
- Burwood, G.W.S.; Fridberger, A.; Wang, R.K.; Nuttall, A.L. Revealing the Morphology and Function of the Cochlea and Middle Ear with Optical Coherence Tomography. Quant. Imaging Med. Surg. 2019, 9, 85881. [Google Scholar] [CrossRef]
- Rask-Andersen, H.; Liu, W.; Erixon, E.; Kinnefors, A.; Pfaller, K.; Schrott-Fischer, A.; Glueckert, R. Human Cochlea: Anatomical Characteristics and Their Relevance for Cochlear Implantation. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2012, 295, 1791–1811. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Ramekers, D.; Flati, V.; Versnel, H.; Maccarone, R. MTOR Signaling in BDNF-Treated Guinea Pigs after Ototoxic Deafening. Biomedicines 2022, 10, 2935. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Rovers, J.; Vink, H.A.; Ramekers, D.; Maccarone, R.; Versnel, H. No Protective Effects of Hair Cells or Supporting Cells in Ototoxically Deafened Guinea Pigs upon Administration of BDNF. Brain Sci. 2021, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Pujol, R.; Lavigne-rebillard, M.; Uziel, A. Development of the Human Cochlea. Acta Oto-Laryngol. 2009, 111, 7–13. [Google Scholar] [CrossRef]
- Jean, P.; Tai, F.W.J.; Singh-Estivalet, A.; Lelli, A.; Scandola, C.; Megharba, S.; Schmutz, S.; Roux, S.; Mechaussier, S.; Sudres, M.; et al. Single-Cell Transcriptomic Profiling of the Mouse Cochlea: An Atlas for Targeted Therapies. Proc. Natl. Acad. Sci. USA 2023, 120, e2221744120. [Google Scholar] [CrossRef]
- Waldhaus, J.; Durruthy-Durruthy, R.; Heller, S. Quantitative High-Resolution Cellular Map of the Organ of Corti. Cell Rep. 2015, 11, 1385–1399. [Google Scholar] [CrossRef]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and Applications for Single-Cell and Spatial Multi-Omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Lee, S.G.; Huang, M.; Obholzer, N.D.; Sun, S.; Li, W.; Petrillo, M.; Dai, P.; Zhou, Y.; Cotanche, D.A.; Megason, S.G.; et al. Myc and Fgf Are Required for Zebrafish Neuromast Hair Cell Regeneration. PLoS ONE 2016, 11, e0157768. [Google Scholar] [CrossRef]
- Jiang, L.; Jin, R.; Xu, J.; Ji, Y.; Zhang, M.; Zhang, X.; Zhang, X.; Han, Z.; Zeng, S. Hair Cell Regeneration or the Expression of Related Factors That Regulate the Fate Specification of Supporting Cells in the Cochlear Ducts of Embryonic and Posthatch Chickens. Hear. Res. 2016, 332, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Romero-Carvajal, A.; Haug, J.S.; Seidel, C.W.; Piotrowski, T. Gene-Expression Analysis of Hair Cell Regeneration in the Zebrafish Lateral Line. Proc. Natl. Acad. Sci. USA 2014, 111, E1383–E1392. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Bi, Z.; Sugino, K.; Wang, G.; Zhu, T.; Liu, Z. Comprehensive Transcriptome Analysis of Cochlear Spiral Ganglion Neurons at Multiple Ages. eLife 2020, 9, e50491. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, D.; Chen, J.; Hou, S.; He, B.; Li, Y.; Li, S.; Zhang, F.; Sun, X.; Jin, Y.; et al. Pseudo-Temporal Analysis of Single-Cell RNA Sequencing Reveals Trans-Differentiation Potential of Greater Epithelial Ridge Cells Into Hair Cells During Postnatal Development of Cochlea in Rats. Front. Mol. Neurosci. 2022, 15, 832813. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Randle, M.R.; Cuppini, C.L.; Graves, K.A.; Cox, B.C. Multiple Supporting Cell Subtypes Are Capable of Spontaneous Hair Cell Regeneration in the Neonatal Mouse Cochlea. Development 2019, 146, dev171009. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Wang, D.; Wang, Y.; Zhou, Z.; Ma, X.; Liu, X.; Dong, Y. Cisplatin-Induced Ototoxicity: From Signaling Network to Therapeutic Targets. Biomed. Pharmacother. 2023, 157, 114045. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, J.; Wang, H.; He, H.; Li, S.; Zhang, Y.; Wang, Y.; Xu, X.; Wang, S. Gene Therapy: An Emerging Therapy for Hair Cells Regeneration in the Cochlea. Front. Neurosci. 2023, 17, 1177791. [Google Scholar] [CrossRef]
- Waissbluth, S.; Maass, J.C.; Sanchez, H.A.; Martínez, A.D. Supporting Cells and Their Potential Roles in Cisplatin-Induced Ototoxicity. Front. Neurosci. 2022, 16, 867034. [Google Scholar] [CrossRef]
- Xu, J.; Ueno, H.; Xu, C.Y.; Chen, B.; Weissman, I.L.; Xu, P.X. Identification of Mouse Cochlear Progenitors That Develop Hair and Supporting Cells in the Organ of Corti. Nat. Commun. 2017, 8, 15046. [Google Scholar] [CrossRef]




| Sequencing Technology | Category | Principle | Read Length | Reference |
|---|---|---|---|---|
| NGS | Cyclic-array sequencing (Illumina and Ion Torrent) | Repeated cycles of enzymatic catalytic reactions. | Short | [67] |
| NGS | Hybridization-based sequencing | Multiple oligonucleotides are hybridized with complementary sequences of the target genome/transcriptome. | Short | [68] |
| NGS | Microelectrophoretic-based | Lab-on-a-chip level which combines all the Sanger sequencing steps together for a more efficient sequencing. | Short | [69] |
| TGS | Pacific Biosciences (PacBio) | Laser-induced fluorescence signals that are activated during the incorporation of dNTPs into DNA, alongside recording the color and duration of the signals in real time. | Long | [70] |
| TGS | Oxford nanopore technology (ONT) | Nanopore-based technology in which sequencing is allowed by determination of current change induced by nucleotides passing through the nanopore. | Long | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisi, A.; Palaniappan, S.; Maccarrone, M. Advanced Omics Techniques for Understanding Cochlear Genome, Epigenome, and Transcriptome in Health and Disease. Biomolecules 2023, 13, 1534. https://doi.org/10.3390/biom13101534
Tisi A, Palaniappan S, Maccarrone M. Advanced Omics Techniques for Understanding Cochlear Genome, Epigenome, and Transcriptome in Health and Disease. Biomolecules. 2023; 13(10):1534. https://doi.org/10.3390/biom13101534
Chicago/Turabian StyleTisi, Annamaria, Sakthimala Palaniappan, and Mauro Maccarrone. 2023. "Advanced Omics Techniques for Understanding Cochlear Genome, Epigenome, and Transcriptome in Health and Disease" Biomolecules 13, no. 10: 1534. https://doi.org/10.3390/biom13101534
APA StyleTisi, A., Palaniappan, S., & Maccarrone, M. (2023). Advanced Omics Techniques for Understanding Cochlear Genome, Epigenome, and Transcriptome in Health and Disease. Biomolecules, 13(10), 1534. https://doi.org/10.3390/biom13101534