


cccDNA-Targeted Drug Screen Reveals a Class of Antihistamines as Suppressors of HBV Genome Levels

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid Constructs
2.3. HBV Assays
2.4. NTCP Expression
2.5. Immunofluorescence Staining
2.6. Electron Microscopy
2.7. Cell Proliferation and Cytotoxicity Assays
2.8. Quantification and Statistical Analysis
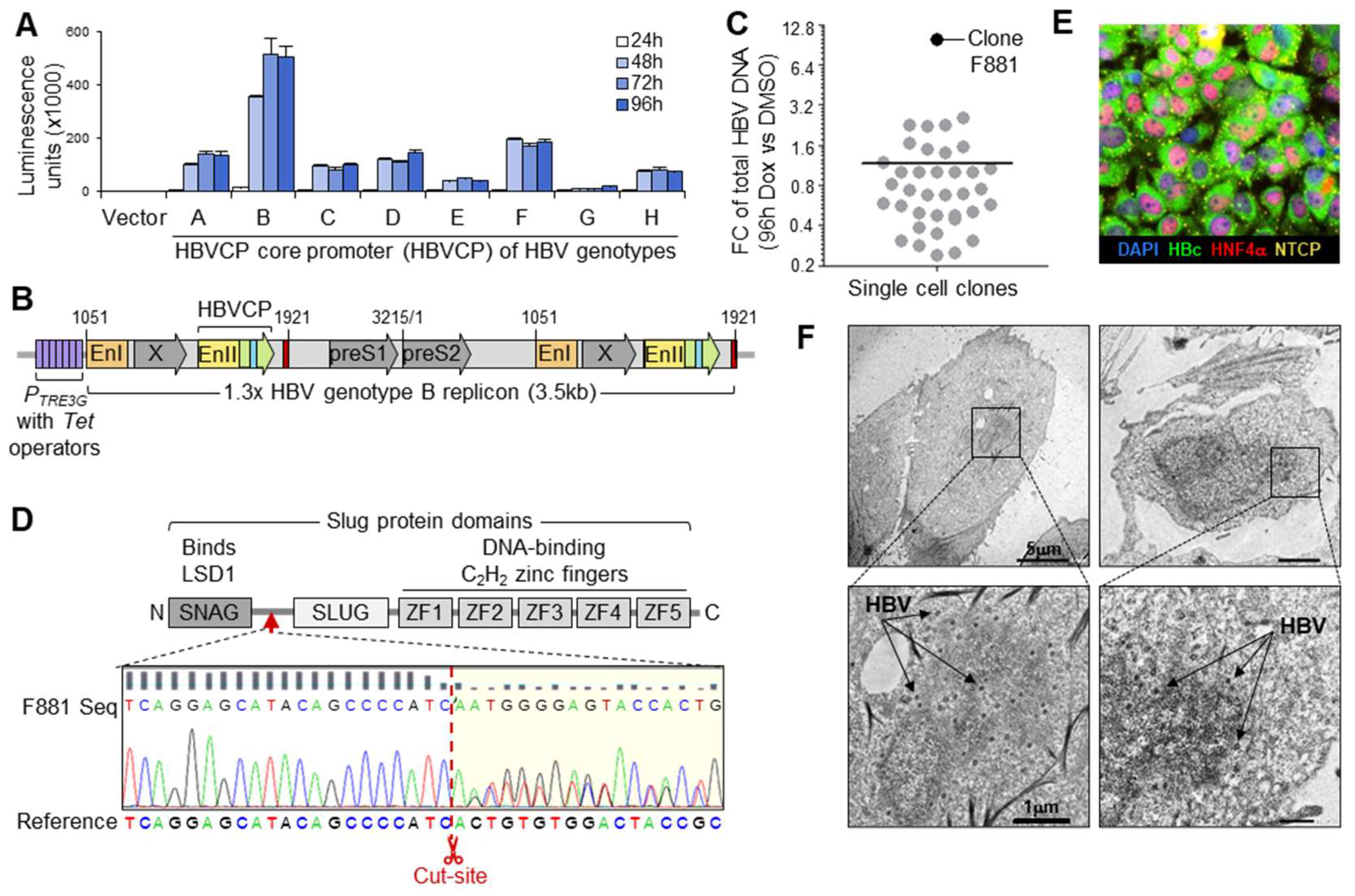
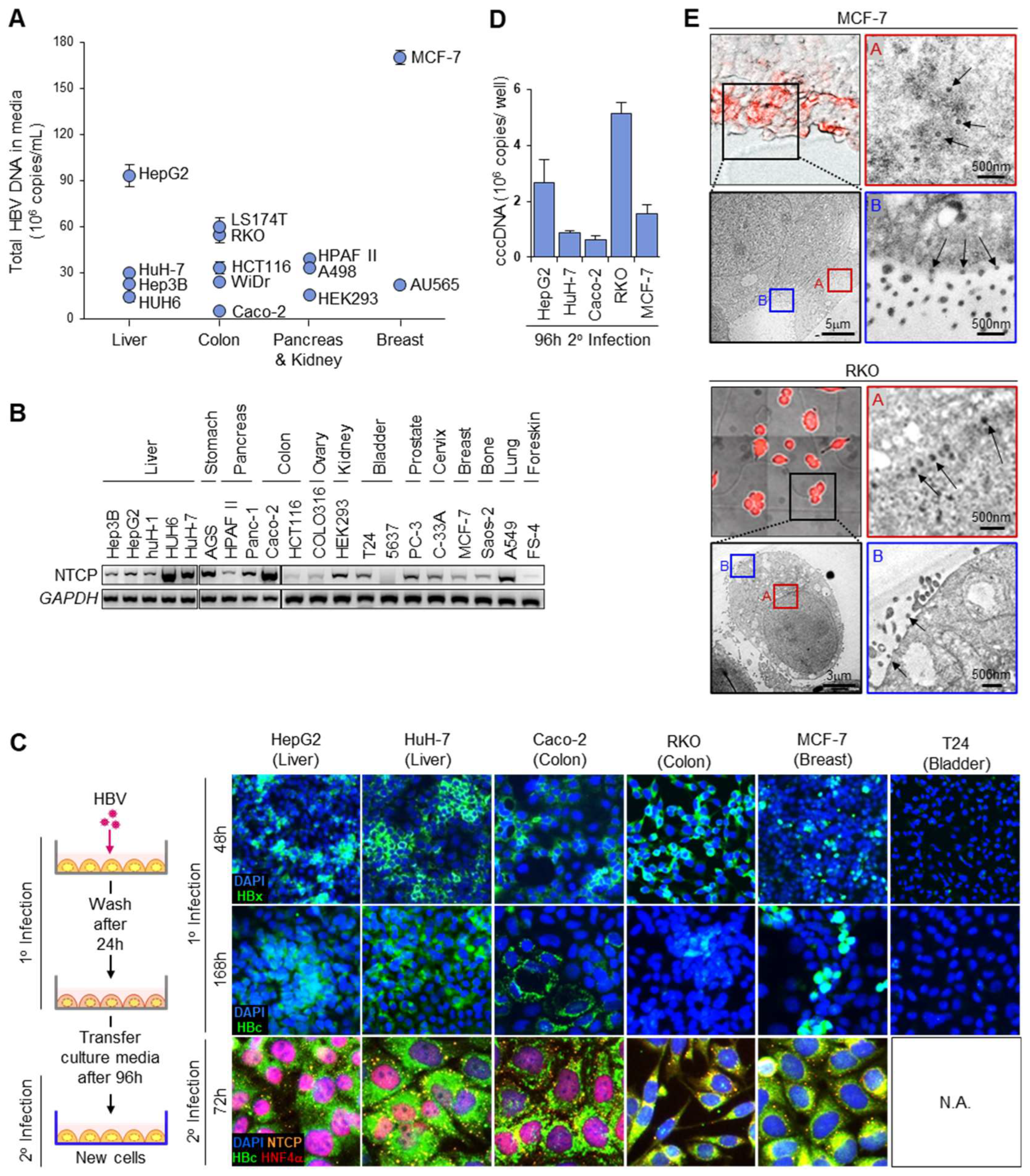
3. Results
3.1. HBV Acceptor Cell Lines
3.2. Maximizing Viral Production from HBV Generator Cell
3.3. cccDNA Accumulation through Continuous Infection
3.4. Fluorescent cccDNA Assay
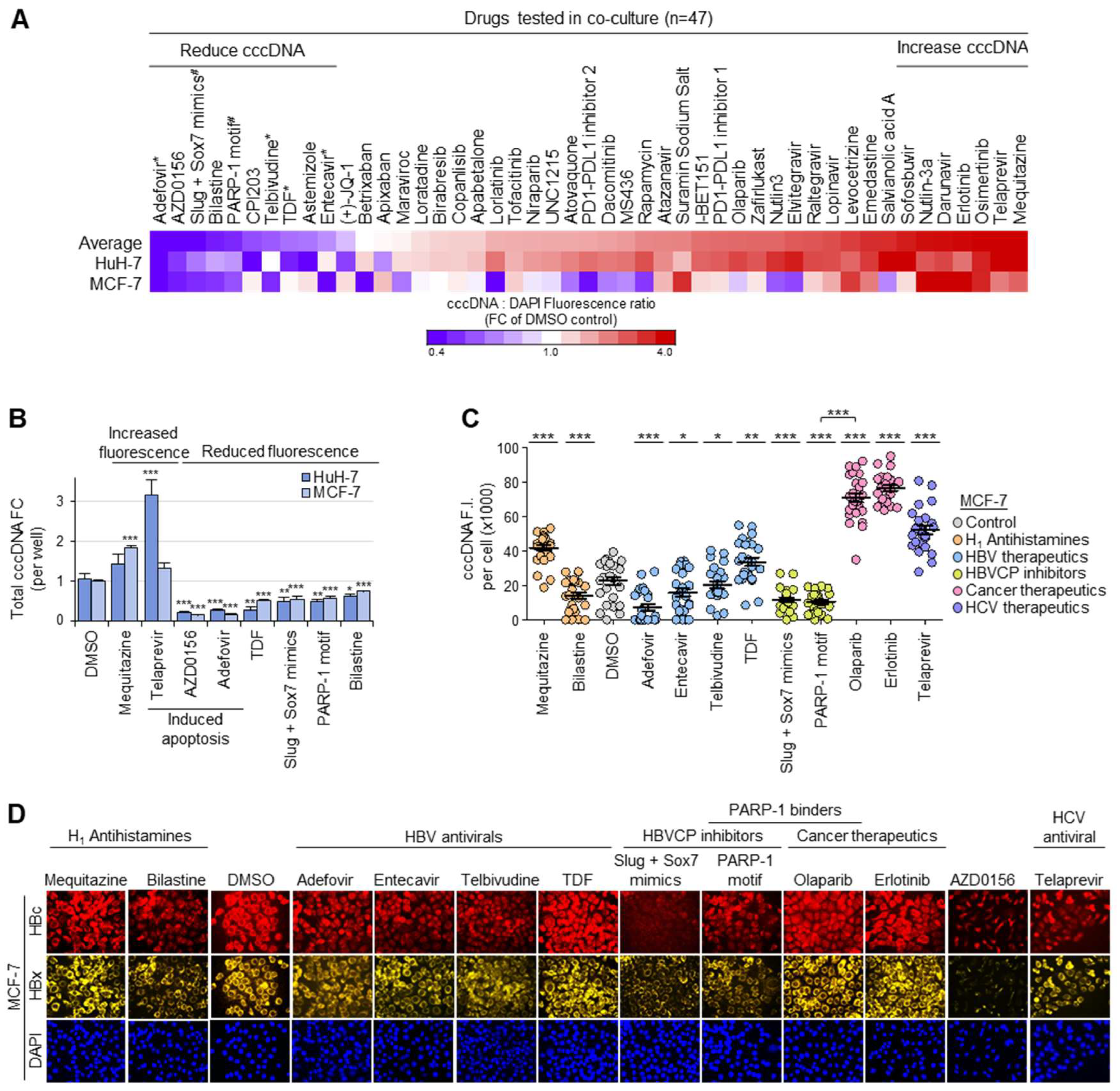
3.5. Drug Screen for Modulators of Intranuclear cccDNA Levels
3.6. Functional Validation of cccDNA-Targeted Drug Screen
3.7. Structurally Similar Antihistamines Downregulate cccDNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cccDNA | Covalently closed circular DNA |
| CLEM | Correlative light and electron microscopy |
| Dox | Doxycycline |
| HBV | Hepatitis B virus |
| HBVCP | HBV core promoter |
| HNF4α | Hepatocyte nuclear factor 4α |
| h.p.i. | Hours post-infection |
| MGE | Multiplicities of genome equivalents |
| Pol/RT | Polymerase/reverse transcriptase |
| pcRNA | pre-core RNA |
| pgRNA | pre-genomic RNA |
| rcDNA | Relaxed closed circular DNA |
| sFISH | Simplified fluorescence in situ hybridization |
| TEM | Transmission electron microscope |
References
- Yang, H.C.; Kao, J.H. Persistence of hepatitis B virus covalently closed circular DNA in hepatocytes: Molecular mechanisms and clinical significance. Emerg. Microbes Infect. 2014, 3, e64. [Google Scholar] [CrossRef]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 3, 667. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Boyd, A.; Combe, E.; Testoni, B.; Zoulim, F. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 2021, 75, 706–717. [Google Scholar] [CrossRef]
- Li, X.; Zhao, J.; Yuan, Q.; Xia, N. Detection of HBV Covalently Closed Circular DNA. Viruses 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Rumlová, M.; Ruml, T. In vitro methods for testing antiviral drugs. Biotechnol. Adv. 2018, 6, 557–576. [Google Scholar] [CrossRef]
- World Health Organization. Hepatitis B. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b/ (accessed on 28 July 2023).
- Hepatitis B Foundation. What is Hepatitis B? 2023. Available online: https://www.hepb.org/what-is-hepatitis-b/what-is-hepb/ (accessed on 5 August 2023).
- Zhang, X.; Zhou, Y.; Chen, C.; Fang, W.; Cai, X.; Zhang, X.; Zhao, M.; Zhang, B.; Jiang, W.; Lin, Z.; et al. Hepatitis B virus reactivation in cancer patients with positive Hepatitis B surface antigen undergoing PD-1 inhibition. J. Immunother. Cancer 2019, 7, 322. [Google Scholar] [CrossRef]
- Liu, S.; Lai, J.; Lyu, N.; Xie, Q.; Cao, H.; Chen, D.; He, M.; Zhang, B.; Zhao, M. Effects of Antiviral Therapy on HBV Reactivation and Survival in Hepatocellular Carcinoma Patients Undergoing Hepatic Artery Infusion Chemotherapy. Front. Oncol. 2021, 10, 582504. [Google Scholar] [CrossRef]
- Qin, B.; Zhao, K.; Wei, J.; Wang, X.; Xu, M.; Lang, J.; Sun, H.; Jin, K. Novel evidence indicates the presence and replication of hepatitis B virus in breast cancer tissue. Oncol. Rep. 2020, 43, 296–305. [Google Scholar] [CrossRef]
- Guo, P.T.; Yang, D.; Sun, Z.; Xu, H.M. Hepatitis B virus X protein plays an important role in gastric ulcers. Oncol. Rep. 2012, 28, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Wu, D.; Wang, T.; Li, T.; Xu, S.; Chen, F.; Jin, X.; Lou, G. Detection of viral antigens in renal tissue of glomerulonephritis patients without serological evidence of hepatitis B virus and hepatitis C virus infection. Int. J. Infect. Dis. 2013, 17, e535–e538. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Kim, J.W.; Shim, J.H.; Han, S.; Yu, C.S.; Choe, J.; Lee, D.; Kim, K.M.; Lim, Y.S.; Chung, Y.H.; et al. Chronic hepatitis B infection and non-hepatocellular cancers: A hospital registry-based, case-control study. PLoS ONE 2018, 13, e0193232. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Lv, J.; Guo, Y.; Bian, Z.; Si, J.; Yang, L.; Chen, Y.; Zhou, Y.; Zhang, H.; Liu, J.; et al. China Kadoorie Biobank Collaborative Group. Associations Between Hepatitis B Virus Infection and Risk of All Cancer Types. JAMA Netw. Open 2019, 2, e195718. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Song, C.I.; Jiang, L.; Dai, J.; Lin, Y.; Xu, X.; Yu, C.; Ge, Z.; Ding, Y.; Wen, Y.; et al. Hepatitis B virus infection and the risk of cancer among the Chinese population. Int. J. Cancer 2020, 147, 3075–3084. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Cai, D.; Wang, X.; Yan, R.; Mao, R.; Liu, Y.; Ji, C.; Cuconati, A.; Guo, H. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res. 2016, 132, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2020, 3, 100195. [Google Scholar] [CrossRef]
- Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Lam, T.H.; Ng, H.; Toh, J.; Wang, L.W.; Ren, E.C. Identification of Slug and SOX7 as transcriptional repressors binding to the hepatitis B virus core promoter. J. Hepatol. 2017, 68, 42–52. [Google Scholar] [CrossRef]
- Zhang, H.; Tu, T. Approaches to quantifying hepatitis B virus covalently closed circular DNA. Clin. Mol. Hepatol. 2022, 28, 135–149. [Google Scholar] [CrossRef]
- Wei, L.; Ploss, A. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat. Commun. 2021, 12, 1591. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Ni, Y.; Lempp, F.A.; Vondran, F.W.R.; Urban, S. T5 Exonuclease Hydrolysis of Hepatitis B Virus Replicative Intermediates Allows Reliable Quantification and Fast Drug Efficacy Testing of Covalently Closed Circular DNA by PCR. J. Virol. 2018, 92, e01117–e01118. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Nandi, S.; Dutta, S.; Saha, M.K. Detection of hepatitis B virus infection: A systematic review. World J. Hepatol. 2015, 7, 2482–2491. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Oswald, A.; Chakraborty, A.; Ni, Y.; Wettengel, J.M.; Urban, S.; Protzer, U. Concentration of Na+-taurocholate-cotransporting polypeptide expressed after in vitro-transcribed mRNA transfection determines susceptibility of hepatoma cells for hepatitis B virus. Sci. Rep. 2021, 11, 19799. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antiviral Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Chen, Y.; Sze, J.; He, M.L. HBV cccDNA in patients’ sera as an indicator for HBV reactivation and an early signal of liver damage. World J. Gastroenterol. 2004, 10, 82–85. [Google Scholar] [CrossRef]
- Liang, L.B.; Zhu, X.; Yan, L.B.; Du, L.Y.; Liu, C.; Liao, J.; Tang, H. Quantitative intrahepatic HBV cccDNA correlates with histological liver inflammation in chronic hepatitis B virus infection. Int. J. Infect. Dis. 2016, 52, 77–82. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef]
- Ko, H.L.; Ren, E.C. Novel poly (ADP-ribose) polymerase 1 binding motif in hepatitis B virus core promoter impairs DNA damage repair. Hepatology 2011, 54, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Ng, H.J.; Goh, E.H.; Ren, E.C. Reduced ADP-ribosylation by PARP1 natural polymorphism V762A and by PARP1 inhibitors enhance Hepatitis B virus replication. J. Viral Hepat. 2013, 20, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Corcóstegui, R.; Labeaga, L.; Innerárity, A.; Berisa, A.; Orjales, A. Preclinical pharmacology of bilastine, a new selective histamine H1 receptor antagonist: Receptor selectivity and in vitro antihistaminic activity. Drugs R D 2005, 6, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C.; Hsu, H.C.; Lin, T.M.; Chang, Y.S.; Hu, L.F.; Chen, L.F.; Lin, S.H.; Kuo, P.I.; Chen, W.S.; Lin, Y.C.; et al. H1-Antihistamines Reduce the Risk of Hepatocellular Carcinoma in Patients With Hepatitis B Virus, Hepatitis C Virus, or Dual Hepatitis B Virus-Hepatitis C Virus Infection. J. Clin. Oncol. 2022, 40, 1206–1219. [Google Scholar] [CrossRef]
- Meng, C.; Liu, T.; Liu, Y.W.; Zhang, L.Z.; Wang, Y.L. Hepatitis B Virus cccDNA in Hepatocellular Carcinoma Tissue Increases the Risk of Recurrence After Liver Transplantation. Transplant. Proc. 2019, 51, 3364–3368. [Google Scholar] [CrossRef]
- Wong, D.K.; Yuen, M.F.; Poon, R.T.; Yuen, J.C.; Fung, J.; Lai, C.L. Quantification of hepatitis B virus covalently closed circular DNA in patients with hepatocellular carcinoma. J. Hepatol. 2006, 45, 553–559. [Google Scholar] [CrossRef]
- Althaus, F.R.; Lawrence, S.D.; Sattler, G.L.; Pitot, H.C. DNA damage induced by the antihistaminic drug methapyrilene hydrochloride. Mutat. Res. 1982, 103, 213–218. [Google Scholar] [CrossRef]
- Xu, W.; Xia, S.; Pu, J.; Wang, Q.; Li, P.; Lu, L.; Jiang, S. The Antihistamine Drugs Carbinoxamine Maleate and Chlorpheniramine Maleate Exhibit Potent Antiviral Activity Against a Broad Spectrum of Influenza Viruses. Front. Microbiol. 2018, 9, 2643. [Google Scholar] [CrossRef]
- He, S.; Lin, B.; Chu, V.; Hu, Z.; Hu, X.; Xiao, J.; Wang, A.Q.; Schweitzer, C.J.; Li, Q.; Imamura, M.; et al. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Transl. Med. 2015, 7, 282ra49. [Google Scholar] [CrossRef]
- Raghavan, S.; Leo, M.D. Histamine Potentiates SARS-CoV-2 Spike Protein Entry Into Endothelial Cells. Front. Pharmacol. 2022, 13, 872736. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Jin, Y.; Son, J.K.; Lee, S.H.; Chang, H.W. Curcumin inhibits the activation of immunoglobulin e-mediated mast cells and passive systemic anaphylaxis in mice by reducing serum eicosanoid and histamine levels. Biomol. Ther. 2014, 22, 27–34. [Google Scholar] [CrossRef]
- Riza, Y.M.; Parves, M.R.; Tithi, F.A.; Alam, S. Quantum chemical calculation and binding modes of H1R: A combined study of molecular docking and DFT for suggesting therapeutically potent H1R antagonist. In Silico Pharmacol. 2019, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.Q.; Zhang, Y.H.; Ke, C.Z.; Chen, H.X.; Ren, P.; He, Y.L.; Hu, P.; Ma, D.Q.; Luo, J.; Meng, Z.J. Curcumin inhibits hepatitis B virus infection by down-regulating cccDNA-bound histone acetylation. World J. Gastroenterol. 2017, 23, 6252–6260. [Google Scholar] [CrossRef] [PubMed]
- Thongsri, P.; Pewkliang, Y.; Borwornpinyo, S.; Wongkajornsilp, A.; Hongeng, S.; Sa-Ngiamsuntorn, K. Curcumin inhibited hepatitis B viral entry through NTCP binding. Sci. Rep. 2021, 11, 19125. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG inhibitors in carcer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef]
- Wang, X.; Wang, P.; Wang, W.; Murray, J.W.; Wolkoff, A.W. The Na+-Taurocholate Cotransporting Polypeptide Traffics with the Epidermal Growth Factor Receptor. Traffic 2016, 17, 230–244. [Google Scholar] [CrossRef]
- Ibach, J.; Radon, Y.; Gelléri, M.; Sonntag, M.H.; Brunsveld, L.; Bastiaens, P.I.; Verveer, P.J. Single Particle Tracking Reveals that EGFR Signaling Activity Is Amplified in Clathrin-Coated Pits. PLoS ONE 2015, 10, e0143162. [Google Scholar] [CrossRef]
- de Wit, M.; Gao, Y.; Mercieca, D.; de Heer, I.; Valkenburg, B.; van Royen, M.E.; Aerts, J.; Smitt, P.S.; French, P. Mutation and drug-specific intracellular accumulation of EGFR predict clinical responses to tyrosine kinase inhibitors. EBioMedicine 2020, 56, 102796. [Google Scholar] [CrossRef]
- Yao, Z.H.; Liao, W.Y.; Ho, C.C.; Chen, K.Y.; Shih, J.Y.; Chen, J.S.; Lin, Z.Z.; Lin, C.C.; Yang, J.C.H.; Yu, C.J. Incidence of hepatitis B reactivation during epidermal growth factor receptor tyrosine kinase inhibitor treatment in non-small-cell lung cancer patients. Eur. J. Cancer 2019, 117, 107–115. [Google Scholar] [CrossRef]
- US Food & Drug Administration. FDA Drug Safety Communication: FDA Warns about the Risk of Hepatitis B Reactivating in Some Patients Treated with Direct-Acting Antivirals for Hepatitis C. 2016. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-about-risk-hepatitis-b-reactivating-some-patients-treated (accessed on 10 August 2023).
- Human Protein Atlas. Dataset #HPA042727. Available online: https://www.proteinatlas.org/ENSG00000100652-SLC10A1/tissue (accessed on 2 September 2023).
- Herrscher, C.; Pastor, F.; Burlaud-Gaillard, J.; Dumans, A.; Seigneuret, F.; Moreau, A.; Patient, R.; Eymieux, S.; de Rocquigny, H.; Hourioux, C.; et al. Hepatitis B virus entry into HepG2-NTCP cells requires clathrin-mediated endocytosis. Cell Microbiol. 2020, 22, e13205. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, F.; Duan, L.; Wang, B.; Ye, Y.; Li, P.; Li, D.; Yang, S.; Zhou, L.; Chen, W. E-cadherin Plays a Role in Hepatitis B Virus Entry Through Affecting Glycosylated Sodium-Taurocholate Cotransporting Polypeptide Distribution. Front. Cell Infect. Microbiol. 2020, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zong, L.; Sureau, C.; Barker, L.; Wands, J.R.; Tong, S. Unusual Features of Sodium Taurocholate Cotransporting Polypeptide as a Hepatitis B Virus Receptor. J. Virol. 2016, 90, 8302–8313. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, E.C.; Zhuo, N.Z.; Goh, Z.Y.; Bonne, I.; Malleret, B.; Ko, H.L. cccDNA-Targeted Drug Screen Reveals a Class of Antihistamines as Suppressors of HBV Genome Levels. Biomolecules 2023, 13, 1438. https://doi.org/10.3390/biom13101438
Ren EC, Zhuo NZ, Goh ZY, Bonne I, Malleret B, Ko HL. cccDNA-Targeted Drug Screen Reveals a Class of Antihistamines as Suppressors of HBV Genome Levels. Biomolecules. 2023; 13(10):1438. https://doi.org/10.3390/biom13101438
Chicago/Turabian StyleRen, Ee Chee, Nicole Ziyi Zhuo, Zhi Yi Goh, Isabelle Bonne, Benoît Malleret, and Hui Ling Ko. 2023. "cccDNA-Targeted Drug Screen Reveals a Class of Antihistamines as Suppressors of HBV Genome Levels" Biomolecules 13, no. 10: 1438. https://doi.org/10.3390/biom13101438
APA StyleRen, E. C., Zhuo, N. Z., Goh, Z. Y., Bonne, I., Malleret, B., & Ko, H. L. (2023). cccDNA-Targeted Drug Screen Reveals a Class of Antihistamines as Suppressors of HBV Genome Levels. Biomolecules, 13(10), 1438. https://doi.org/10.3390/biom13101438