
The Important Role of Zinc in Neurological Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Homeostasis of Zinc in Physiological Conditions
3. Distribution of Zinc in the Brain
3.1. Role of Zinc in Neurogenesis
3.2. Role of Zinc in Promoting Redox Homeostasis
3.3. Role of Zinc on Immunity in the CNS
4. The Role of Zinc in Stroke
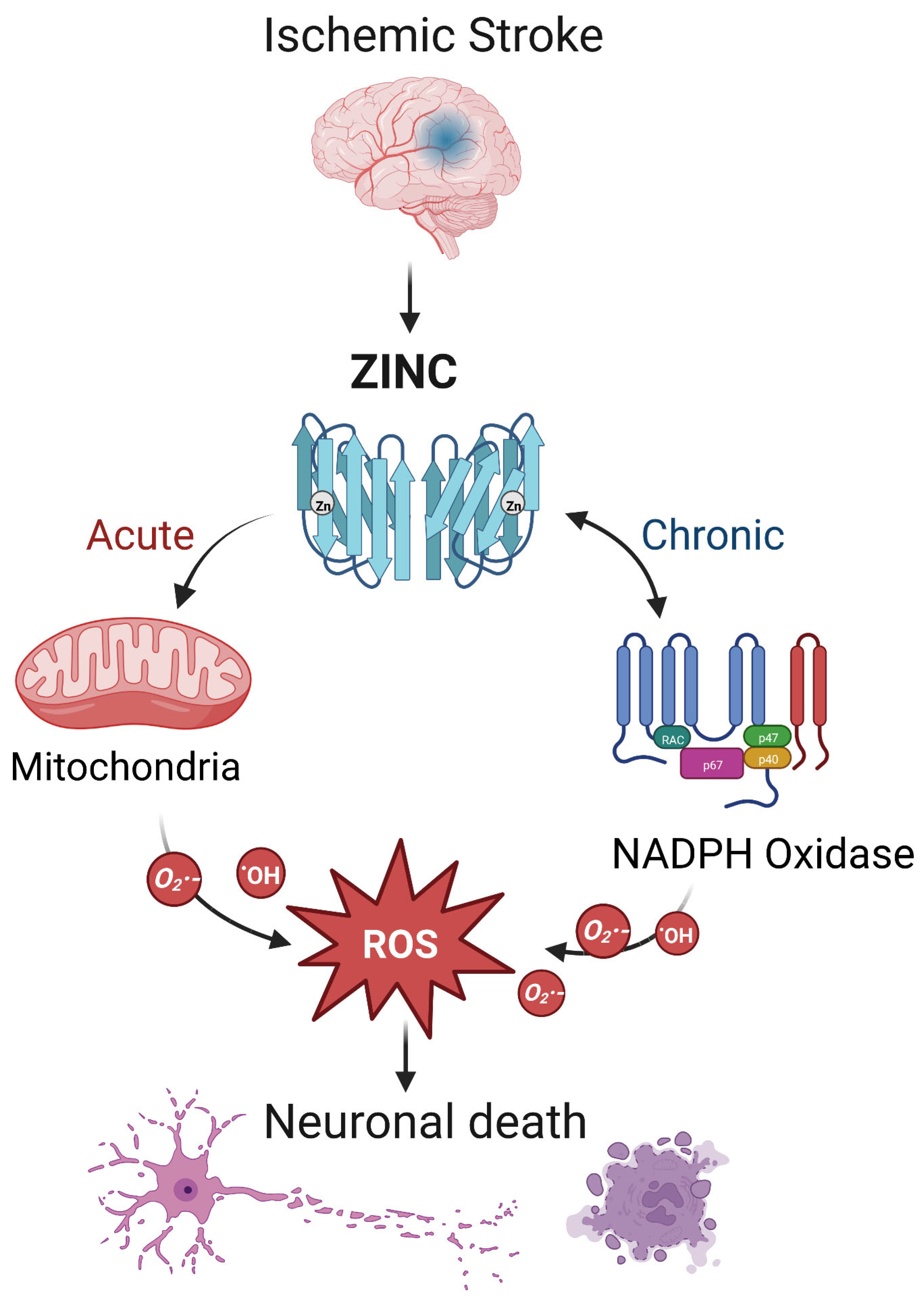
4.1. Ischemic Stroke
4.2. Intracerebral Hemorrhage (Hemorrhagic Stroke)
5. Zinc and Other Disorders of the CNS
5.1. Alzheimer’s Disease
5.2. Traumatic Brain Injury
5.3. Epilepsy
5.4. Depression
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prasad, A.S. Discovery of human zinc deficiency: Its impact on human health and disease. Adv. Nutr. 2013, 4, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Sanna, A.; Firinu, D.; Zavattari, P.; Valera, P. Zinc Status and Autoimmunity: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 68. [Google Scholar] [CrossRef]
- McAllister, B.B.; Dyck, R.H. A new role for zinc in the brain. eLife 2017, 6, e31816. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, S.; Mori, K.; Shoji, T.; Emoto, M. Association of Zinc Deficiency with Development of CVD Events in Patients with CKD. Nutrients 2021, 13, 1680. [Google Scholar] [CrossRef] [PubMed]
- Begum, F.; Me, H.M.; Christov, M. The Role of Zinc in Cardiovascular Disease. Cardiol. Rev. 2022, 30, 100–108. [Google Scholar] [CrossRef]
- Vujasinovic, M.; Hedstrom, A.; Maisonneuve, P.; Valente, R.; von Horn, H.; Lohr, J.M.; Haas, S.L. Zinc deficiency in patients with chronic pancreatitis. World J. Gastroenterol. 2019, 25, 600–607. [Google Scholar] [CrossRef]
- Swardfager, W.; Herrmann, N.; McIntyre, R.S.; Mazereeuw, G.; Goldberger, K.; Cha, D.S.; Schwartz, Y.; Lanctot, K.L. Potential roles of zinc in the pathophysiology and treatment of major depressive disorder. Neurosci. Biobehav. Rev. 2013, 37, 911–929. [Google Scholar] [CrossRef]
- Wessells, K.R.; Brown, K.H. Estimating the global prevalence of zinc deficiency: Results based on zinc availability in national food supplies and the prevalence of stunting. PLoS ONE 2012, 7, e50568. [Google Scholar] [CrossRef]
- Adamo, A.M.; Zago, M.P.; Mackenzie, G.G.; Aimo, L.; Keen, C.L.; Keenan, A.; Oteiza, P.I. The role of zinc in the modulation of neuronal proliferation and apoptosis. Neurotox. Res. 2010, 17, 1. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef] [PubMed]
- Bertoni-Freddari, C.; Fattoretti, P.; Casoli, T.; Di Stefano, G.; Giorgetti, B.; Balietti, M. Brain aging: The zinc connection. Exp. Gerontol. 2008, 43, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A. Zinc homeostasis and functions of zinc in the brain. Biometals 2001, 14, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Sekler, I.; Silverman, W.F. Zinc homeostasis and signaling in glia. Glia 2012, 60, 843–850. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Khmeleva, S.A.; Mezentsev, Y.V.; Kozin, S.A.; Tsvetkov, P.O.; Ivanov, A.S.; Bodoev, N.V.; Makarov, A.A.; Radko, S.P. Zinc-induced interaction of the metal-binding domain of amyloid-beta peptide with DNA. J. Alzheimers Dis. 2013, 36, 633–636. [Google Scholar] [CrossRef]
- Zhou, X.; Cooper, K.L.; Sun, X.; Liu, K.J.; Hudson, L.G. Selective Sensitization of Zinc Finger Protein Oxidation by Reactive Oxygen Species through Arsenic Binding. J. Biol. Chem. 2015, 290, 18361–18369. [Google Scholar] [CrossRef]
- Stefanidou, M.; Maravelias, C.; Dona, A.; Spiliopoulou, C. Zinc: A multipurpose trace element. Arch. Toxicol. 2006, 80, 1–9. [Google Scholar] [CrossRef]
- Micheletti, A.; Rossi, R.; Rufini, S. Zinc status in athletes: Relation to diet and exercise. Sports Med. 2001, 31, 577–582. [Google Scholar] [CrossRef]
- Marger, L.; Schubert, C.R.; Bertrand, D. Zinc: An underappreciated modulatory factor of brain function. Biochem. Pharmacol. 2014, 91, 426–435. [Google Scholar] [CrossRef]
- Oteiza, P.I.; Mackenzie, G.G. Zinc, oxidant-triggered cell signaling, and human health. Mol. Asp. Med. 2005, 26, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Barceloux, D.G. Zinc. J. Toxicol. Clin. Toxicol. 1999, 37, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.S.; Dyck, R.H. Zinc and cortical plasticity. Brain Res. Rev. 2009, 59, 347–373. [Google Scholar] [CrossRef]
- Plum, L.M.; Rink, L.; Haase, H. The essential toxin: Impact of zinc on human health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; Obeid, S.; Ahlenstiel, C.; Ahlenstiel, G. The Role of Zinc in Antiviral Immunity. Adv. Nutr. 2019, 10, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef]
- Solomons, N.W. Update on zinc biology. Ann Nutr Metab 2013, 62 (Suppl. 1), 8–17. [Google Scholar] [CrossRef]
- King, J.C.; Shames, D.M.; Woodhouse, L.R. Zinc homeostasis in humans. J. Nutr. 2000, 130, 1360S–1366S. [Google Scholar] [CrossRef]
- Bitanihirwe, B.K.; Cunningham, M.G. Zinc: The brain’s dark horse. Synapse 2009, 63, 1029–1049. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Wei, R.; Khan, S.; Zhang, R.; Zhang, Y.; Yong, V.W.; Xue, M. Iron Neurotoxicity and Protection by Deferoxamine in Intracerebral Hemorrhage. Front. Mol. Neurosci. 2022, 15, 927334. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989, 31, 145–238. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Won, S.J.; Hamby, A.M.; Yoo, B.H.; Fan, Y.; Sheline, C.T.; Tamano, H.; Takeda, A.; Liu, J. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J. Cereb. Blood Flow Metab. 2009, 29, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y. Zinc and disease of the brain. Mol. Neurobiol. 2001, 24, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Parisi, A.F.; Vallee, B.L. Zinc metalloenzymes: Characteristics and significance in biology and medicine. Am. J. Clin. Nutr. 1969, 22, 1222–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, Y.; Li, Z.; Yong, V.W.; Xue, M. Extracellular matrix metalloproteinase inducer in brain ischemia and intracerebral hemorrhage. Front. Immunol. 2022, 13, 986469. [Google Scholar] [CrossRef]
- Toth, K. Zinc in neurotransmission. Annu. Rev. Nutr. 2011, 31, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.S.; Ferguson, E.L. Assessment of dietary zinc in a population. Am. J. Clin. Nutr. 1998, 68, 430S–434S. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Canzoniero, L.M.; Yu, S.P.; Ling, C.; Choi, D.W. Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J. Physiol. 2000, 528 Pt 1, 39–52. [Google Scholar] [CrossRef]
- Koh, J.Y.; Choi, D.W. Zinc toxicity on cultured cortical neurons: Involvement of N-methyl-D-aspartate receptors. Neuroscience 1994, 60, 1049–1057. [Google Scholar] [CrossRef]
- Marin, P.; Israel, M.; Glowinski, J.; Premont, J. Routes of zinc entry in mouse cortical neurons: Role in zinc-induced neurotoxicity. Eur. J. Neurosci. 2000, 12, 8–18. [Google Scholar] [CrossRef]
- Karakas, E.; Simorowski, N.; Furukawa, H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009, 28, 3910–3920. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Sensi, S.L. Ca2+-Zn2+ permeable AMPA or kainate receptors: Possible key factors in selective neurodegeneration. Trends Neurosci. 2000, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, A.; Singh, K.; Avasthi, K.; Kim, J.J. Neurobiology of zinc and its role in neurogenesis. Eur. J. Nutr. 2021, 60, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Levenson, C.W.; Morris, D. Zinc and neurogenesis: Making new neurons from development to adulthood. Adv. Nutr. 2011, 2, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, Y.; Wright, K.S.; Murray, E.A. Hippocampal lesions in rhesus monkeys disrupt emotional responses but not reinforcer devaluation effects. Biol. Psychiatr. 2008, 63, 1084–1091. [Google Scholar] [CrossRef]
- Goeldner, C.; Reiss, D.; Wichmann, J.; Meziane, H.; Kieffer, B.L.; Ouagazzal, A.M. Nociceptin receptor impairs recognition memory via interaction with NMDA receptor-dependent mitogen-activated protein kinase/extracellular signal-regulated kinase signaling in the hippocampus. J. Neurosci. 2008, 28, 2190–2198. [Google Scholar] [CrossRef]
- Piechal, A.; Blecharz-Klin, K.; Pyrzanowska, J.; Widy-Tyszkiewicz, E. Maternal zinc supplementation improves spatial memory in rat pups. Biol. Trace Elem. Res. 2012, 147, 299–308. [Google Scholar] [CrossRef]
- National Research Council. Recommended Dietary Allowances, 10th ed.; Reports funded by National Institutes of Health; The National Academies Collection: Washington, DC, USA, 1989. [Google Scholar] [PubMed]
- Yu, X.; Chen, W.; Wei, Z.; Ren, T.; Yang, X.; Yu, X. Effects of maternal mild zinc deficiency and different ways of zinc supplementation for offspring on learning and memory. Food Nutr. Res. 2016, 60, 29467. [Google Scholar] [CrossRef]
- Wang, H.; Hu, Y.F.; Hao, J.H.; Chen, Y.H.; Su, P.Y.; Wang, Y.; Yu, Z.; Fu, L.; Xu, Y.Y.; Zhang, C.; et al. Maternal zinc deficiency during pregnancy elevates the risks of fetal growth restriction: A population-based birth cohort study. Sci. Rep. 2015, 5, 11262. [Google Scholar] [CrossRef]
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Drlica, K. Reactive oxygen species and the bacterial response to lethal stress. Curr. Opin. Microbiol. 2014, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. The redox biology of redox-inert zinc ions. Free Radic. Biol. Med. 2019, 134, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Camacho, J.D.; Vicente-Garcia, C.; Parsons, D.S.; Navas-Enamorado, I. Zinc at the crossroads of exercise and proteostasis. Redox Biol. 2020, 35, 101529. [Google Scholar] [CrossRef] [PubMed]
- Hempe, J.M.; Cousins, R.J. Cysteine-rich intestinal protein binds zinc during transmucosal zinc transport. Proc. Natl. Acad. Sci. USA 1991, 88, 9671–9674. [Google Scholar] [CrossRef]
- Baltaci, A.K.; Yuce, K.; Mogulkoc, R. Zinc Metabolism and Metallothioneins. Biol. Trace Elem. Res. 2018, 183, 22–31. [Google Scholar] [CrossRef]
- Maret, W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox Sign. 2006, 8, 1419–1441. [Google Scholar] [CrossRef]
- Mehta, A.J.; Yeligar, S.M.; Elon, L.; Brown, L.A.; Guidot, D.M. Alcoholism causes alveolar macrophage zinc deficiency and immune dysfunction. Am. J. Respir. Crit. Care Med. 2013, 188, 716–723. [Google Scholar] [CrossRef]
- Joshi, P.C.; Mehta, A.; Jabber, W.S.; Fan, X.; Guidot, D.M. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am. J. Respir. Cell Mol. Biol. 2009, 41, 207–216. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Appukuttan, A.T.; Renoir, T.; Foliaki, S.; Chen, F.; Adlard, P.A.; Hannan, A.J.; Bush, A.I. Brain Zinc Deficiency Exacerbates Cognitive Decline in the R6/1 Model of Huntington’s Disease. Neurotherapeutics 2020, 17, 243–251. [Google Scholar] [CrossRef]
- Ibs, K.H.; Rink, L. Zinc-altered immune function. J. Nutr. 2003, 133, 1452S–1456S. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Maywald, M.; Rink, L. Zinc as a Gatekeeper of Immune Function. Nutrients 2017, 9, 1286. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.Z.; Rink, L. Zinc in Infection and Inflammation. Nutrients 2017, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Haase, H.; Engelhardt, G.; Rink, L.; Uciechowski, P. Zinc deficiency induces production of the proinflammatory cytokines IL-1beta and TNFalpha in promyeloid cells via epigenetic and redox-dependent mechanisms. J. Nutr. Biochem. 2013, 24, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Kahmann, L.; Uciechowski, P.; Warmuth, S.; Plumakers, B.; Gressner, A.M.; Malavolta, M.; Mocchegiani, E.; Rink, L. Zinc supplementation in the elderly reduces spontaneous inflammatory cytokine release and restores T cell functions. Rejuvenation Res. 2008, 11, 227–237. [Google Scholar] [CrossRef]
- Xue, M.; Yong, V.W. Neuroinflammation in intracerebral haemorrhage: Immunotherapies with potential for translation. Lancet Neurol. 2020, 19, 1023–1032. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Khan, S.; Zhang, R.; Wei, R.; Zhang, Y.; Xue, M.; Yong, V.W. Neuroprotection of minocycline by inhibition of extracellular matrix metalloproteinase inducer expression following intracerebral hemorrhage in mice. Neurosci. Lett. 2021, 764, 136297. [Google Scholar] [CrossRef]
- Haase, H.; Rink, L. Functional significance of zinc-related signaling pathways in immune cells. Annu. Rev. Nutr. 2009, 29, 133–152. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Xu, S.; Zhang, M.; Wu, Z.; Zhang, X.; Xu, Y.; Chen, Y. Delayed PARP-1 Inhibition Alleviates Post-stroke Inflammation in Male Versus Female Mice: Differences and Similarities. Front. Cell. Neurosci 2020, 14, 77. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.H.; Gan, L.; Swanson, R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J. Neuroinflamm. 2011, 8, 152. [Google Scholar] [CrossRef]
- Maida, C.D.; Norrito, R.L.; Daidone, M.; Tuttolomondo, A.; Pinto, A. Neuroinflammatory Mechanisms in Ischemic Stroke: Focus on Cardioembolic Stroke, Background, and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 6454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Wang, F.; Liu, Y.; Yong, V.W.; Xue, M. Necrosulfonamide Alleviates Acute Brain Injury of Intracerebral Hemorrhage via Inhibiting Inflammation and Necroptosis. Front. Mol. Neurosci. 2022, 15, 916249. [Google Scholar] [CrossRef] [PubMed]
- Hennig, B.; Toborek, M.; McClain, C.J. Antiatherogenic properties of zinc: Implications in endothelial cell metabolism. Nutrition 1996, 12, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Grungreiff, K.; Gottstein, T.; Reinhold, D. Zinc Deficiency-An Independent Risk Factor in the Pathogenesis of Haemorrhagic Stroke? Nutrients 2020, 12, 3548. [Google Scholar] [CrossRef]
- Hennig, B.; Wang, Y.; Ramasamy, S.; McClain, C.J. Zinc deficiency alters barrier function of cultured porcine endothelial cells. J. Nutr. 1992, 122, 1242–1247. [Google Scholar] [CrossRef]
- Shuttleworth, C.W.; Weiss, J.H. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol. Sci. 2011, 32, 480–486. [Google Scholar] [CrossRef]
- Bu, S.; Lv, Y.; Liu, Y.; Qiao, S.; Wang, H. Zinc Finger Proteins in Neuro-Related Diseases Progression. Front. Neurosci. 2021, 15, 760567. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Braun, A. Zinc Homeostasis in Platelet-Related Diseases. Int. J. Mol. Sci. 2019, 20, 5258. [Google Scholar] [CrossRef]
- Zhang, Y.; Khan, S.; Liu, Y.; Siddique, R.; Zhang, R.; Yong, V.W.; Xue, M. Gap Junctions and Hemichannels Composed of Connexins and Pannexins Mediate the Secondary Brain Injury Following Intracerebral Hemorrhage. Biology 2021, 11, 27. [Google Scholar] [CrossRef]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn(2+): A novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 2000, 21, 395–401. [Google Scholar] [CrossRef]
- Lee, J.M.; Zipfel, G.J.; Park, K.H.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience 2002, 115, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Liuyang, Z.Y.; Lei, P.; Yan, X.; Shentu, Y.P.; Liang, J.W.; Zhou, H.; Pei, L.; Xiong, Y.; Hou, T.Y.; et al. Zinc induces CDK5 activation and neuronal death through CDK5-Tyr15 phosphorylation in ischemic stroke. Cell Death Dis. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Qi, Z.; Liang, J.; Shi, W.; Zhao, Y.; Luo, Y.; Ji, X.; Liu, K.J. Reduction of zinc accumulation in mitochondria contributes to decreased cerebral ischemic injury by normobaric hyperoxia treatment in an experimental stroke model. Exp. Neurol. 2015, 272, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Babu, S.; Kaul, S.; Shafi, G.; Rajeshwar, K.; Alladi, S.; Jyothy, A. Depletion of serum zinc in ischemic stroke patients. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Mattern, L.; Chen, C.; McClure, L.A.; Brockman, J.; Cushman, M.; Judd, S.; Kahe, K. Serum Zinc Levels and Incidence of Ischemic Stroke: The Reasons for Geographic and Racial Differences in Stroke Study. Stroke 2021, 52, 3953–3960. [Google Scholar] [CrossRef]
- Howard, V.J.; Madsen, T.E.; Kleindorfer, D.O.; Judd, S.E.; Rhodes, J.D.; Soliman, E.Z.; Kissela, B.M.; Safford, M.M.; Moy, C.S.; McClure, L.A.; et al. Sex and Race Differences in the Association of Incident Ischemic Stroke With Risk Factors. JAMA Neurol. 2019, 76, 179–186. [Google Scholar] [CrossRef]
- Shao, Z.; Tu, S.; Shao, A. Pathophysiological Mechanisms and Potential Therapeutic Targets in Intracerebral Hemorrhage. Front. Pharmacol. 2019, 10, 1079. [Google Scholar] [CrossRef]
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11, 720–731. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Wei, R.; Khan, S.; Xue, M.; Yong, V.W. The combination of deferoxamine and minocycline strengthens neuroprotective effect on acute intracerebral hemorrhage in rats. Neurol. Res. 2021, 43, 854–864. [Google Scholar] [CrossRef]
- Hanley, D.F.; Thompson, R.E.; Rosenblum, M.; Yenokyan, G.; Lane, K.; McBee, N.; Mayo, S.W.; Bistran-Hall, A.J.; Gandhi, D.; Mould, W.A.; et al. Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): A randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet 2019, 393, 1021–1032. [Google Scholar] [CrossRef]
- Bai, Q.; Xue, M.; Yong, V.W. Microglia and macrophage phenotypes in intracerebral haemorrhage injury: Therapeutic opportunities. Brain 2020, 143, 1297–1314. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bai, Q.; Yong, V.W.; Xue, M. EMMPRIN Promotes the Expression of MMP-9 and Exacerbates Neurological Dysfunction in a Mouse Model of Intracerebral Hemorrhage. Neurochem. Res. 2022, 47, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Levenson, C.W. Trace metal regulation of neuronal apoptosis: From genes to behavior. Physiol. Behav. 2005, 86, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Khan, S.; Liu, Y.; Wu, G.; Yong, V.W.; Xue, M. Oxidative Stress Following Intracerebral Hemorrhage: From Molecular Mechanisms to Therapeutic Targets. Front. Immunol. 2022, 13, 847246. [Google Scholar] [CrossRef]
- Bai, Q.; Sheng, Z.; Liu, Y.; Zhang, R.; Yong, V.W.; Xue, M. Intracerebral haemorrhage: From clinical settings to animal models. Stroke Vasc. Neurol. 2020, 5, 388–395. [Google Scholar] [CrossRef]
- Bernardo, M.M.; Day, D.E.; Halvorson, H.R.; Olson, S.T.; Shore, J.D. Surface-independent acceleration of factor XII activation by zinc ions. II. Direct binding and fluorescence studies. J. Biol. Chem. 1993, 268, 12477–12483. [Google Scholar] [CrossRef]
- Taylor, K.A.; Pugh, N. The contribution of zinc to platelet behaviour during haemostasis and thrombosis. Metallomics 2016, 8, 144–155. [Google Scholar] [CrossRef]
- Vu, T.T.; Fredenburgh, J.C.; Weitz, J.I. Zinc: An important cofactor in haemostasis and thrombosis. Thromb. Haemost. 2013, 109, 421–430. [Google Scholar] [CrossRef]
- Karadas, S.; Sayin, R.; Aslan, M.; Gonullu, H.; Kati, C.; Dursun, R.; Duran, L.; Gonullu, E.; Demir, H. Serum levels of trace elements and heavy metals in patients with acute hemorrhagic stroke. J. Membr. Biol. 2014, 247, 175–180. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, J.; Zhang, Y.; Li, H.; Zhang, H.; Huo, Y.; Li, J.; Liu, X.; Wang, X.; Qin, X.; et al. Baseline Plasma Zinc and Risk of First Stroke in Hypertensive Patients: A Nested Case-Control Study. Stroke 2019, 50, 3255–3258. [Google Scholar] [CrossRef]
- Arleth, T.; Olsen, M.H.; Orre, M.; Rasmussen, R.; Bache, S.; Eskesen, V.; Frikke-Schmidt, R.; Moller, K. Hypozincaemia is associated with severity of aneurysmal subarachnoid haemorrhage: A retrospective cohort study. Acta Neurochir. (Wien) 2020, 162, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wu, H.; Zhao, J. Multifunctional roles of zinc in Alzheimer’s disease. Neurotoxicology 2020, 80, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Hodson, R. Alzheimer’s disease. Nature 2018, 559, S1. [Google Scholar] [CrossRef]
- Xu, Y.; Xiao, G.; Liu, L.; Lang, M. Zinc transporters in Alzheimer’s disease. Mol. Brain 2019, 12, 106. [Google Scholar] [CrossRef]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- Religa, D.; Strozyk, D.; Cherny, R.A.; Volitakis, I.; Haroutunian, V.; Winblad, B.; Naslund, J.; Bush, A.I. Elevated cortical zinc in Alzheimer disease. Neurology 2006, 67, 69–75. [Google Scholar] [CrossRef]
- Lyubartseva, G.; Smith, J.L.; Markesbery, W.R.; Lovell, M.A. Alterations of zinc transporter proteins ZnT-1, ZnT-4 and ZnT-6 in preclinical Alzheimer’s disease brain. Brain Pathol. 2010, 20, 343–350. [Google Scholar] [CrossRef]
- Zhang, L.H.; Wang, X.; Zheng, Z.H.; Ren, H.; Stoltenberg, M.; Danscher, G.; Huang, L.; Rong, M.; Wang, Z.Y. Altered expression and distribution of zinc transporters in APP/PS1 transgenic mouse brain. Neurobiol. Aging 2010, 31, 74–87. [Google Scholar] [CrossRef]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef]
- Lovell, M.A. A potential role for alterations of zinc and zinc transport proteins in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Wang, X.; Stoltenberg, M.; Danscher, G.; Huang, L.; Wang, Z.Y. Abundant expression of zinc transporters in the amyloid plaques of Alzheimer’s disease brain. Brain Res. Bull. 2008, 77, 55–60. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s disease facts and figures. Alzheimers Deme. 2020, 16, 391–460. [CrossRef] [PubMed]
- Craven, K.M.; Kochen, W.R.; Hernandez, C.M.; Flinn, J.M. Zinc Exacerbates Tau Pathology in a Tau Mouse Model. J. Alzheimers Dis. 2018, 64, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Park, E.J.; Seo, J.; Ko, S.J.; Lee, J.; Kim, C.H. Zinc stimulates tau S214 phosphorylation by the activation of Raf/mitogen-activated protein kinase-kinase/extracellular signal-regulated kinase pathway. Neuroreport 2011, 22, 839–844. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef]
- Roozenbeek, B.; Maas, A.I.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef]
- Selassie, A.W.; Zaloshnja, E.; Langlois, J.A.; Miller, T.; Jones, P.; Steiner, C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. J. Head Trauma Rehabil. 2008, 23, 123–131. [Google Scholar] [CrossRef]
- Salmond, C.H.; Sahakian, B.J. Cognitive outcome in traumatic brain injury survivors. Curr. Opin. Crit. Care 2005, 11, 111–116. [Google Scholar] [CrossRef]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Sandsmark, D.K.; Diaz-Arrastia, R. Advances in traumatic brain injury research in 2020. Lancet Neurol. 2021, 20, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Listiack, K.; Bell, B.; Chen, J.; Motamedi, M.; Silva, D.; Danscher, G.; Whetsell, W.; Thompson, R.; Frederickson, C. Detection of pathological zinc accumulation in neurons: Methods for autopsy, biopsy, and cultured tissue. J. Histochem. Cytochem. 1999, 47, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014, 28, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, Q.; Ma, S.; Zhang, Y.; Liang, P. TPEN Attenuates Neural Autophagy Induced by Synaptically-released Zinc Translocation and Improves Histological Outcomes after Traumatic Brain Injury in Rats. Ann. Clin. Lab. Sci. 2018, 48, 446–452. [Google Scholar]
- Dominguez, M.I.; Blasco-Ibanez, J.M.; Crespo, C.; Marques-Mari, A.I.; Martinez-Guijarro, F.J. Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience 2003, 116, 791–806. [Google Scholar] [CrossRef]
- Corniola, R.S.; Tassabehji, N.M.; Hare, J.; Sharma, G.; Levenson, C.W. Zinc deficiency impairs neuronal precursor cell proliferation and induces apoptosis via p53-mediated mechanisms. Brain Res. 2008, 1237, 52–61. [Google Scholar] [CrossRef]
- Seth, R.; Corniola, R.S.; Gower-Winter, S.D.; Morgan, T.J., Jr.; Bishop, B.; Levenson, C.W. Zinc deficiency induces apoptosis via mitochondrial p53- and caspase-dependent pathways in human neuronal precursor cells. J. Trace Elem. Med. Biol. 2015, 30, 59–65. [Google Scholar] [CrossRef]
- Gao, H.L.; Zheng, W.; Xin, N.; Chi, Z.H.; Wang, Z.Y.; Chen, J.; Wang, Z.Y. Zinc deficiency reduces neurogenesis accompanied by neuronal apoptosis through caspase-dependent and -independent signaling pathways. Neurotox. Res. 2009, 16, 416–425. [Google Scholar] [CrossRef]
- Young, B.; Ott, L.; Kasarskis, E.; Rapp, R.; Moles, K.; Dempsey, R.J.; Tibbs, P.A.; Kryscio, R.; McClain, C. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J. Neurotrauma 1996, 13, 25–34. [Google Scholar] [CrossRef]
- Cope, E.C.; Morris, D.R.; Scrimgeour, A.G.; Levenson, C.W. Use of zinc as a treatment for traumatic brain injury in the rat: Effects on cognitive and behavioral outcomes. Neurorehabil. Neural Repair 2012, 26, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Manford, M. Recent advances in epilepsy. J. Neurol. 2017, 264, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshe, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Pathak, H.R.; Weissinger, F.; Terunuma, M.; Carlson, G.C.; Hsu, F.C.; Moss, S.J.; Coulter, D.A. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J. Neurosci. 2007, 27, 14012–14022. [Google Scholar] [CrossRef]
- Gower-Winter, S.D.; Levenson, C.W. Zinc in the central nervous system: From molecules to behavior. Biofactors 2012, 38, 186–193. [Google Scholar] [CrossRef]
- Harrison, B.K.; Asplund, C. Sudden unexplained death in epilepsy during physical activity. Curr. Sports Med. Rep. 2007, 6, 13–15. [Google Scholar] [CrossRef]
- Baraka, A.M.; Hassab El Nabi, W.; El Ghotni, S. Investigating the role of zinc in a rat model of epilepsy. CNS Neurosci. Ther. 2012, 18, 327–333. [Google Scholar] [CrossRef]
- Chen, N.N.; Zhao, D.J.; Sun, Y.X.; Wang, D.D.; Ni, H. Long-Term Effects of Zinc Deficiency and Zinc Supplementation on Developmental Seizure-Induced Brain Damage and the Underlying GPR39/ZnT-3 and MBP Expression in the Hippocampus. Front. Neurosci. 2019, 13, 920. [Google Scholar] [CrossRef]
- Foresti, M.L.; Arisi, G.M.; Fernandes, A.; Tilelli, C.Q.; Garcia-Cairasco, N. Chelatable zinc modulates excitability and seizure duration in the amygdala rapid kindling model. Epilepsy Res. 2008, 79, 166–172. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Lee, B.E.; Lee, S.H.; Kho, A.R.; Sohn, M.; Suh, S.W. Zinc plus cyclo-(His-Pro) promotes hippocampal neurogenesis in rats. Neuroscience 2016, 339, 634–643. [Google Scholar] [CrossRef]
- Seven, M.; Basaran, S.Y.; Cengiz, M.; Unal, S.; Yuksel, A. Deficiency of selenium and zinc as a causative factor for idiopathic intractable epilepsy. Epilepsy Res. 2013, 104, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, S.L.; Ghosh, B.M.; Serotte, J.C.; Cranwell, V. Psychotic Depression: Diagnosis, Differential Diagnosis, and Treatment. Psychother. Psychosom. 2021, 90, 160–177. [Google Scholar] [CrossRef] [PubMed]
- McCarron, R.M.; Shapiro, B.; Rawles, J.; Luo, J. Depression. Ann. Intern. Med. 2021, 174, ITC65–ITC80. [Google Scholar] [CrossRef] [PubMed]
- Hammen, C. Risk Factors for Depression: An Autobiographical Review. Annu. Rev. Clin. Psychol. 2018, 14, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.E.; Lobo, M.K. The molecular and cellular mechanisms of depression: A focus on reward circuitry. Mol. Psychiatr. 2019, 24, 1798–1815. [Google Scholar] [CrossRef]
- Szewczyk, B.; Kubera, M.; Nowak, G. The role of zinc in neurodegenerative inflammatory pathways in depression. Prog. Neuropsychopharmacol. Biol. Psychiatr. 2011, 35, 693–701. [Google Scholar] [CrossRef]
- Takeda, A. Zinc signaling in the hippocampus and its relation to pathogenesis of depression. Mol. Neurobiol. 2011, 44, 166–174. [Google Scholar] [CrossRef]
- Little, K.Y.; Castellanos, X.; Humphries, L.L.; Austin, J. Altered zinc metabolism in mood disorder patients. Biol. Psychiatr. 1989, 26, 646–648. [Google Scholar] [CrossRef]
- Tamano, H.; Kan, F.; Kawamura, M.; Oku, N.; Takeda, A. Behavior in the forced swim test and neurochemical changes in the hippocampus in young rats after 2-week zinc deprivation. Neurochem. Int. 2009, 55, 536–541. [Google Scholar] [CrossRef]
- Tassabehji, N.M.; Corniola, R.S.; Alshingiti, A.; Levenson, C.W. Zinc deficiency induces depression-like symptoms in adult rats. Physiol. Behav. 2008, 95, 365–369. [Google Scholar] [CrossRef]
- Whittle, N.; Lubec, G.; Singewald, N. Zinc deficiency induces enhanced depression-like behaviour and altered limbic activation reversed by antidepressant treatment in mice. Amino Acids 2009, 36, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Rafalo-Ulinska, A.; Pochwat, B.; Misztak, P.; Bugno, R.; Kryczyk-Poprawa, A.; Opoka, W.; Muszynska, B.; Poleszak, E.; Nowak, G.; Szewczyk, B. Zinc Deficiency Blunts the Effectiveness of Antidepressants in the Olfactory Bulbectomy Model of Depression in Rats. Nutrients 2022, 14, 2746. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Herrmann, N.; Mazereeuw, G.; Goldberger, K.; Harimoto, T.; Lanctot, K.L. Zinc in depression: A meta-analysis. Biol. Psychiatr. 2013, 74, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Stanislawska, M.; Szkup-Jablonska, M.; Jurczak, A.; Wieder-Huszla, S.; Samochowiec, A.; Jasiewicz, A.; Nocen, I.; Augustyniuk, K.; Brodowska, A.; Karakiewicz, B.; et al. The severity of depressive symptoms vs. serum Mg and Zn levels in postmenopausal women. Biol. Trace Elem. Res. 2014, 157, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Amani, R.; Saeidi, S.; Nazari, Z.; Nematpour, S. Correlation between dietary zinc intakes and its serum levels with depression scales in young female students. Biol. Trace Elem. Res. 2010, 137, 150–158. [Google Scholar] [CrossRef]
- Roozbeh, J.; Sharifian, M.; Ghanizadeh, A.; Sahraian, A.; Sagheb, M.M.; Shabani, S.; Hamidian Jahromi, A.; Kashfi, M.; Afshariani, R. Association of zinc deficiency and depression in the patients with end-stage renal disease on hemodialysis. J. Ren. Nutr. 2011, 21, 184–187. [Google Scholar] [CrossRef]
- Maserejian, N.N.; Hall, S.A.; McKinlay, J.B. Low dietary or supplemental zinc is associated with depression symptoms among women, but not men, in a population-based epidemiological survey. J. Affect. Disord. 2012, 136, 781–788. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Liu, Y.; Wei, R.; Yong, V.W.; Xue, M. The Important Role of Zinc in Neurological Diseases. Biomolecules 2023, 13, 28. https://doi.org/10.3390/biom13010028
Li Z, Liu Y, Wei R, Yong VW, Xue M. The Important Role of Zinc in Neurological Diseases. Biomolecules. 2023; 13(1):28. https://doi.org/10.3390/biom13010028
Chicago/Turabian StyleLi, Zhe, Yang Liu, Ruixue Wei, V. Wee Yong, and Mengzhou Xue. 2023. "The Important Role of Zinc in Neurological Diseases" Biomolecules 13, no. 1: 28. https://doi.org/10.3390/biom13010028
APA StyleLi, Z., Liu, Y., Wei, R., Yong, V. W., & Xue, M. (2023). The Important Role of Zinc in Neurological Diseases. Biomolecules, 13(1), 28. https://doi.org/10.3390/biom13010028