


FK506-Binding Protein 2 Participates in Proinsulin Folding

 , , , , , , , , , , , , and
, , , , , , , , , , , , and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of FKBP2 CRISPR/Cas-9 Mediated Knockout INS-1E Cell Lines
2.3. FKBP2 Expression in Pancreatic Endocrine Cells
2.4. Immunoprecipitations and Mass Spectrometry Analysis
2.5. Size Exclusion Chromatography
2.6. Immunoblotting
2.7. Single-Cell RNA Sequencing
2.8. In Silico Modeling of Protein Interactions and Protein Sequences Analysis
2.9. FKBP2 Protein Activity Assay
2.10. qRT-PCR
2.11. Apoptosis Assay
2.12. Glucose-Stimulated Insulin-Secretion (GSIS)
2.13. Statistics
3. Results
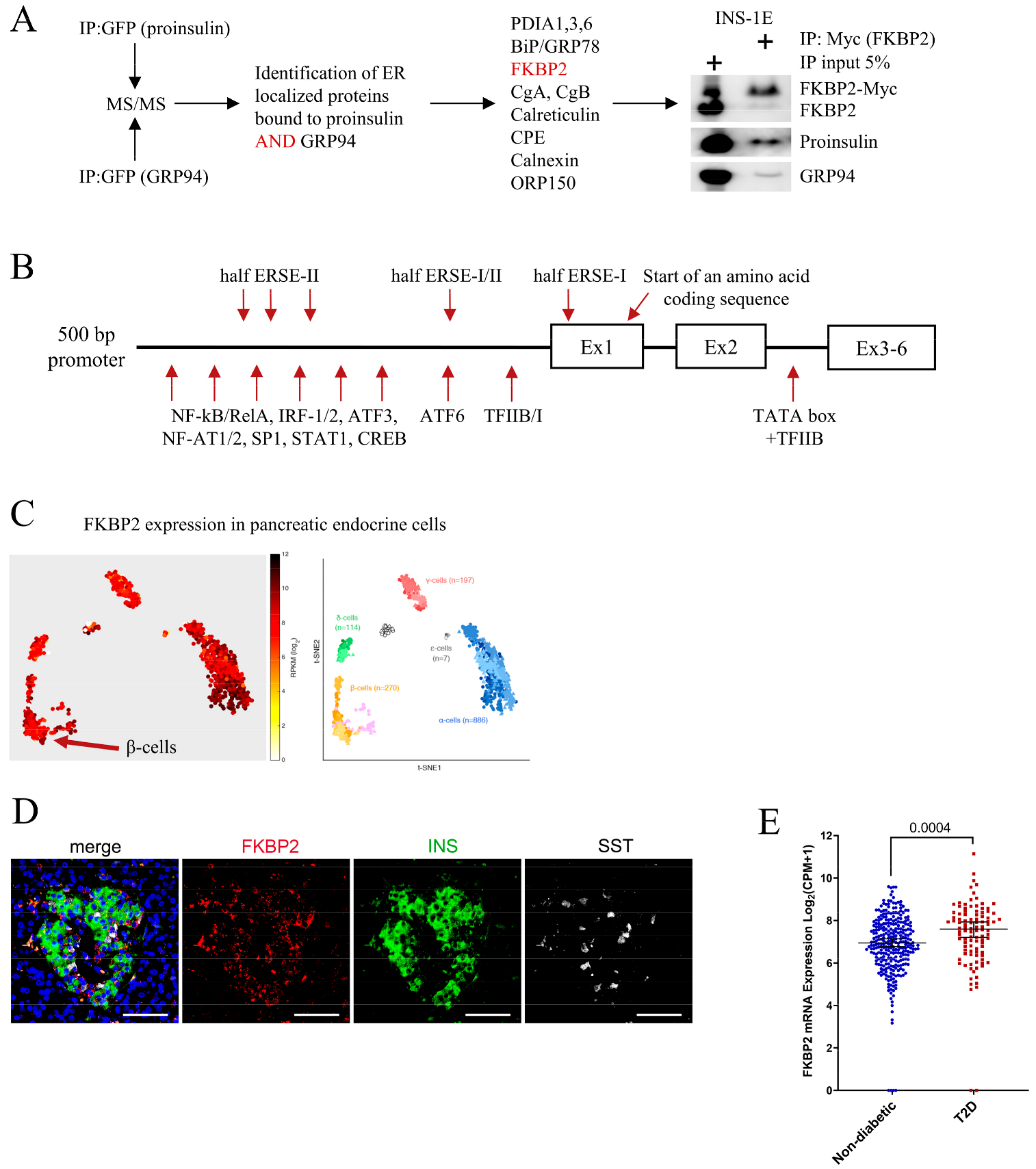
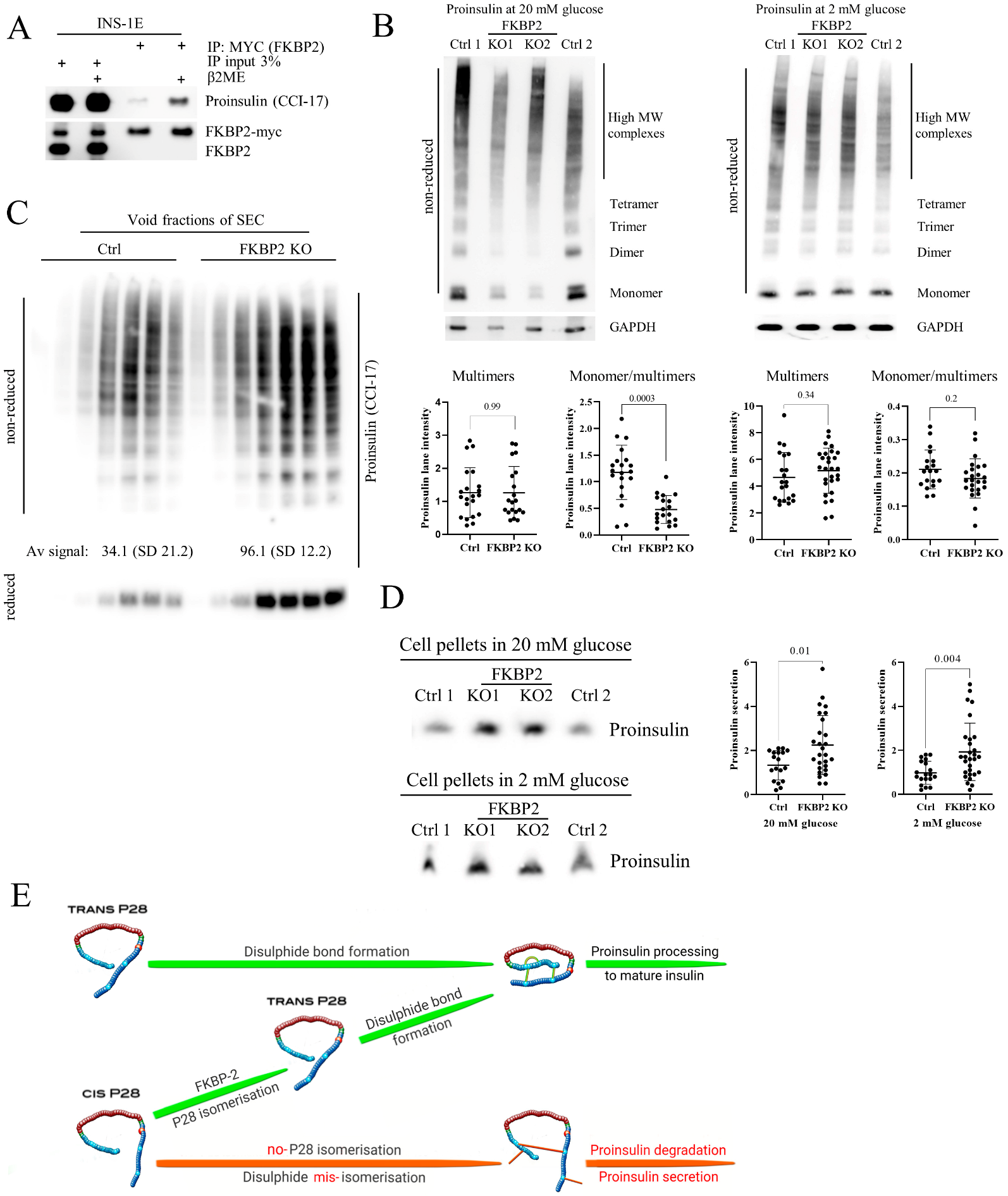
3.1. FKBP2 Directly Interacts with Proinsulin, and FKBP2 mRNA Is Overexpressed in Islets from Type 2 Diabetes Patients
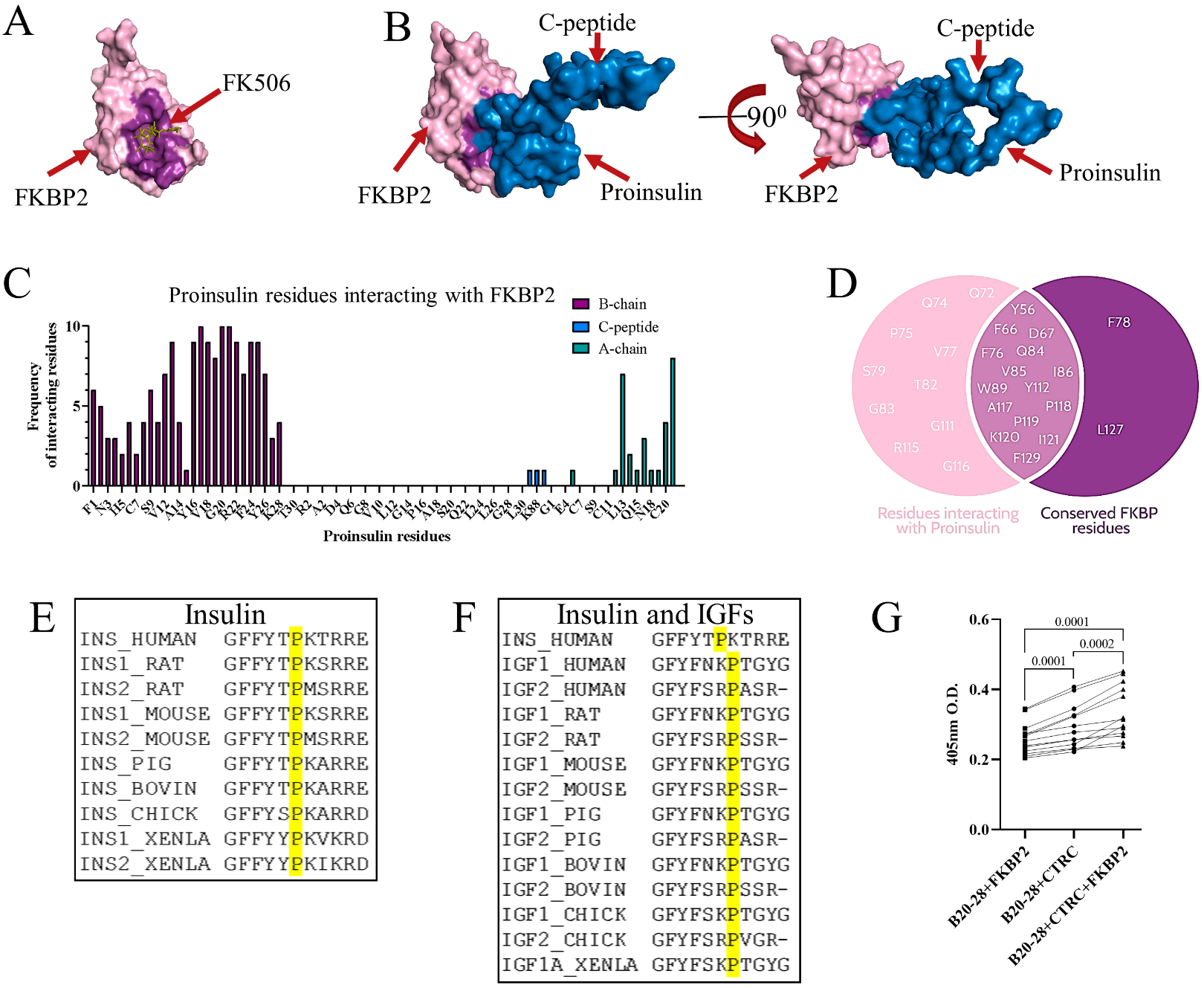
3.2. Proinsulin P28 Is a Potential Target of FKBP2-Dependent Isomerization
3.3. Diminished Intracellular Proinsulin and Insulin Contents after FKBP2 KO
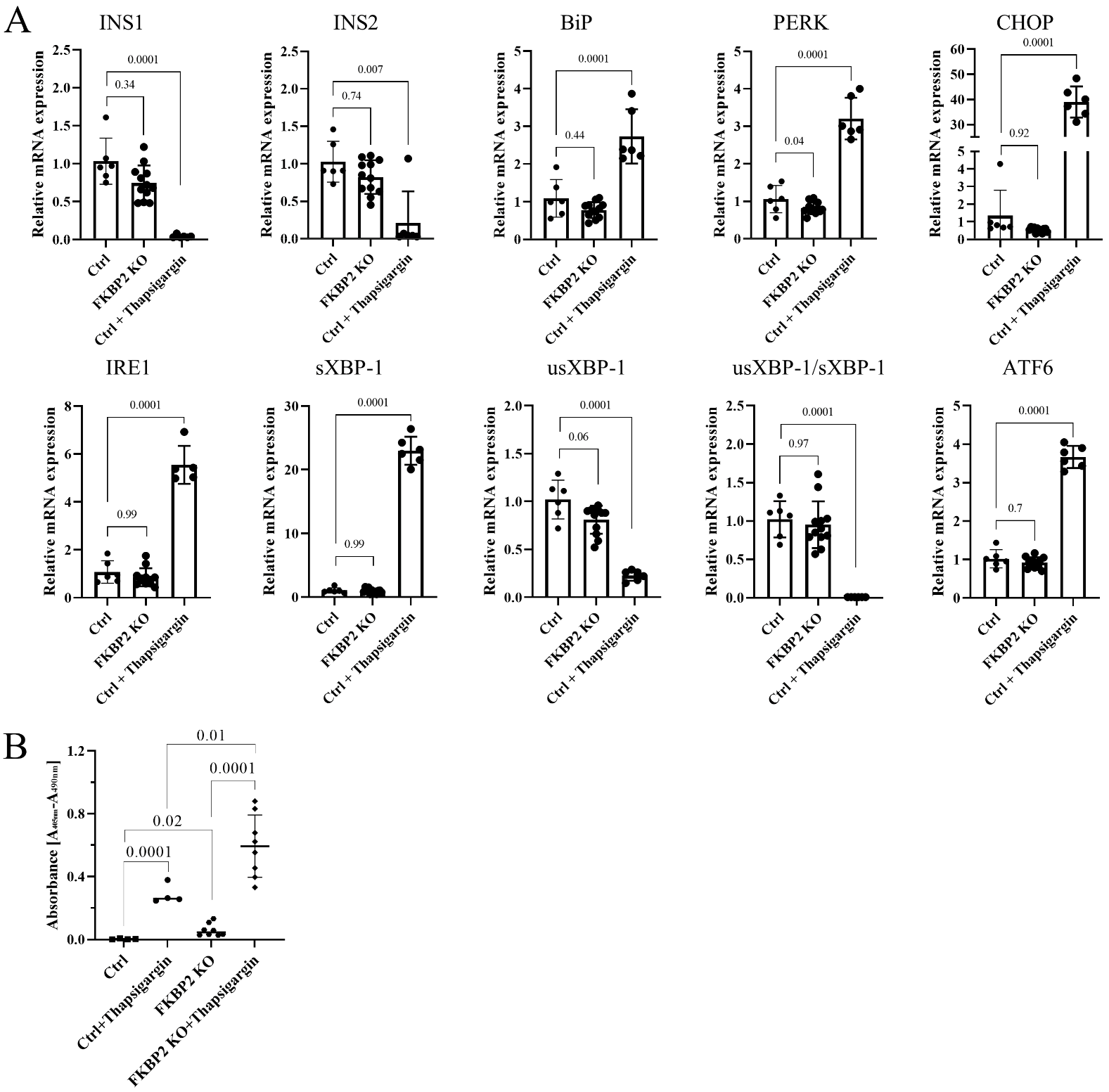
3.4. FKBP2 KO Does Not Lead to ER Stress but Induces β-Cell Apoptosis
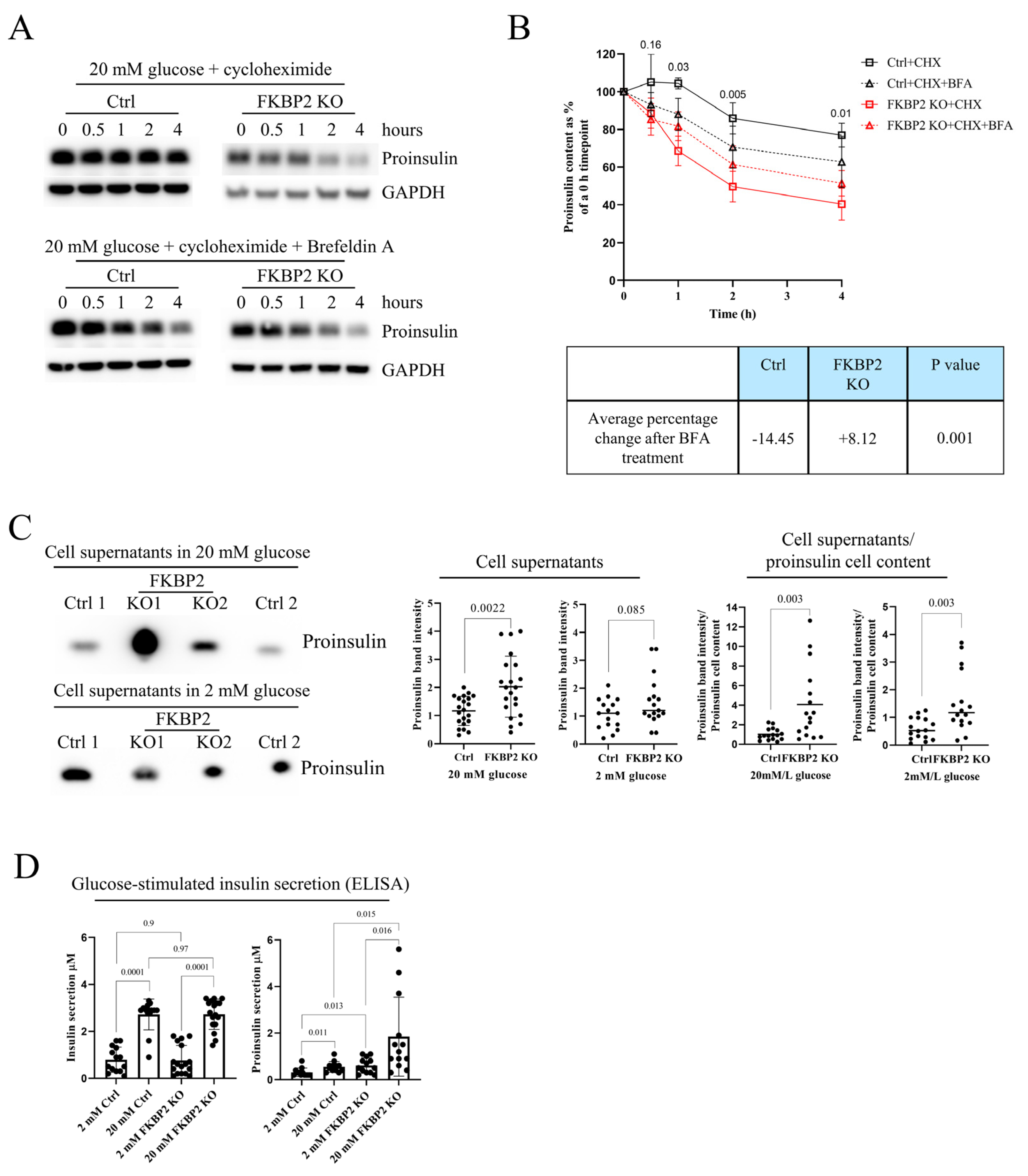
3.5. Proinsulin Turnover Is Increased in FKBP2 KO Cells Accompanied by Increased Proinsulin Secretion
3.6. FKBP2 KO Induces Proinsulin Misfolding and Decreases Its Solubility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. Elife 2019, 8, e44532. [Google Scholar] [CrossRef]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27, S60–S68. [Google Scholar] [CrossRef]
- Alam, M.; Arunagiri, A.; Haataja, L.; Torres, M.; Larkin, D.; Kappler, J.; Jin, N.; Arvan, P. Predisposition to Proinsulin Misfolding as a Genetic Risk to Diet-Induced Diabetes. Diabetes 2021, 70, 2580–2594. [Google Scholar] [CrossRef]
- Shrestha, N.; De Franco, E.; Arvan, P.; Cnop, M. Pathological beta-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front. Endocrinol. 2021, 12, 650158. [Google Scholar] [CrossRef]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic Reticulum Chaperone Glucose-Regulated Protein 94 Is Essential for Proinsulin Handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef]
- Liu, M.; Li, Y.; Cavener, D.; Arvan, P. Proinsulin Disulfide Maturation and Misfolding in the Endoplasmic Reticulum*. J. Biol. Chem. 2005, 280, 13209–13212. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, C.L.; Parsley, N.C.; Asimgil, H.; Lee, H.W.; Ahlbach, C.; Michael, A.K.; Xu, H.; Williams, O.L.; Davis, T.L.; Liu, A.C.; et al. A Slow Conformational Switch in the BMAL1 Transactivation Domain Modulates Circadian Rhythms. Mol. Cell 2017, 66, 447–457.e7. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.K.; Young, P.E.; Sullivan, C.E.; Torchia, D.A. Detection of cis and trans X-Pro peptide bonds in proteins by 13C NMR: Application to collagen. Proc. Natl. Acad. Sci. USA 1984, 81, 4800–4803. [Google Scholar] [CrossRef]
- Schmid, F.X. Prolyl isomerase: Enzymatic catalysis of slow protein-folding reactions. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Roderer, D.J.A.; Schärer, M.A.; Rubini, M.; Glockshuber, R. Acceleration of protein folding by four orders of magnitude through a single amino acid substitution. Sci. Rep. 2015, 5, 11840. [Google Scholar] [CrossRef]
- Yang, Y.; Hua, Q.X.; Liu, J.; Shimizu, E.H.; Choquette, M.H.; Mackin, R.B.; Weiss, M.A. Solution structure of proinsulin: Connecting domain flexibility and prohormone processing. J. Biol. Chem. 2010, 285, 7847–7851. [Google Scholar] [CrossRef]
- Schmid, F.X. Proline isomerization during refolding of ribonuclease A is accelerated by the presence of folding intermediates. FEBS Lett. 1986, 198, 217–220. [Google Scholar] [CrossRef]
- Jakob, R.P.; Schmid, F.X. Energetic coupling between native-state prolyl isomerization and conformational protein folding. J. Mol. Biol. 2008, 377, 1560–1575. [Google Scholar] [CrossRef]
- Fischer, G.; Aumuller, T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 2003, 148, 105–150. [Google Scholar] [CrossRef]
- Johnson, J.D.; Ao, Z.; Ao, P.; Li, H.; Dai, L.J.; He, Z.; Tee, M.; Potter, K.J.; Klimek, A.M.; Meloche, R.M.; et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transpl. 2009, 18, 833–845. [Google Scholar] [CrossRef]
- D’Amico, E.; Hui, H.; Khoury, N.; Di Mario, U.; Perfetti, R. Pancreatic beta-cells expressing GLP-1 are resistant to the toxic effects of immunosuppressive drugs. J. Mol. Endocrinol. 2005, 34, 377–390. [Google Scholar] [CrossRef]
- Zhang, N.; Su, D.; Qu, S.; Tse, T.; Bottino, R.; Balamurugan, A.N.; Xu, J.; Bromberg, J.S.; Dong, H.H. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes 2006, 55, 2429–2436. [Google Scholar] [CrossRef]
- Wei, X.; Zhu, D.; Feng, C.; Chen, G.; Mao, X.; Wang, Q.; Wang, J.; Liu, C. Inhibition of peptidyl-prolyl cis-trans isomerase B mediates cyclosporin A-induced apoptosis of islet beta cells. Exp. Ther. Med. 2018, 16, 3959–3964. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Walker, J.T.; Shostak, A.; Padgett, A.; Spears, E.; Wisniewski, S.; Poffenberger, G.; Aramandla, R.; Dean, E.D.; Prasad, N.; et al. Tacrolimus- and sirolimus-induced human β cell dysfunction is reversible and preventable. JCI Insight 2020, 5, e130770. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Saint-Martin, C.; Xu, J.; Ding, L.; Wang, R.; Feng, W.; Liu, M.; Shu, H.; Fan, Z.; Haataja, L.; et al. Biological behaviors of mutant proinsulin contribute to the phenotypic spectrum of diabetes associated with insulin gene mutations. Mol. Cell. Endocrinol. 2020, 518, 111025. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.; Jin, Y.; Jin, M.; Bush, K.; Bierer, B.; Burakoff, S. Localization of the FK506-binding protein, FKBP 13, to the lumen of the endoplasmic reticulum. Biochem. J. 1993, 294, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Hendrickson, B.; Nigam, S. Induction of the FK506-binding protein, FKBP13, under conditions which misfold proteins in the endoplasmic reticulum. Biochem. J. 1994, 303, 705–708. [Google Scholar] [CrossRef]
- Ostrovsky, O.; Makarewich, C.A.; Snapp, E.L.; Argon, Y. An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc. Natl. Acad. Sci. USA 2009, 106, 11600–11605. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Hawkes, C.P.; Eletto, D.; Boyle, S.; Rosenfeld, R.; Hwa, V.; Wit, J.M.; van Duyvenvoorde, H.A.; Oostdijk, W.; Losekoot, M.; et al. A Human Variant of Glucose-Regulated Protein 94 That Inefficiently Supports IGF Production. Endocrinology 2016, 157, 1914–1928. [Google Scholar] [CrossRef]
- Camunas-Soler, J.; Dai, X.Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; MacDonald, P.E. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020, 31, 1017–1031.e1014. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Q.; Camunas-Soler, J.; Briant, L.J.B.; Dos Santos, T.; Spigelman, A.F.; Walker, E.M.; Arrojo, E.D.R.; Bautista, A.; Jones, R.C.; Avrahami, D.; et al. Heterogenous impairment of alpha cell function in type 2 diabetes is linked to cell maturation state. Cell Metab. 2022, 34, 256–268.e5. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Walker, J.R.; Neculai, D.; Davis, T.; Butler-Cole, C.; Sicheri, F.; Weigelt, J.; Sundstrom, M.; Arrowsmith, C.H.; Edwards, A.M.; Bochkarev, A.; et al. 2PBC: FK506-binding protein 2. Struct. Genom. Consort. 2007. [Google Scholar] [CrossRef]
- Available online: https://pymolwiki.org/index.php/InterfaceResidues (accessed on 1 April 2020).
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands—Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andréasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef]
- Lin, L.N.; Brandts, J.F. Isomer-specific proteolysis of model substrates: Influence that the location of the proline residue exerts on cis/trans specificity. Biochemistry 1985, 24, 6533–6538. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://pdb101.rcsb.org/global-health/diabetes-mellitus/drugs/insulin/insulin-lispro (accessed on 1 February 2020).
- Vajo, Z.; Duckworth, W.C. Genetically engineered insulin analogs: Diabetes in the new millenium. Pharmacol. Rev. 2000, 52, 1–9. [Google Scholar] [PubMed]
- Jin, Y.; Kotler, J.L.M.; Wang, S.; Huang, B.; Halpin, J.C.; Street, T.O. The ER Chaperones BiP and Grp94 Regulate the Formation of Insulin-Like Growth Factor 2 (IGF2) Oligomers. J. Mol. Biol. 2021, 433, 166963. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-Y.; Kim, Y.D.; Lee, K.-M.; Min, A.-K.; Kim, M.-K.; Kim, H.-S.; Won, K.-C.; Park, J.-Y.; Lee, K.-U.; Choi, H.-S.; et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology 2008, 149, 3832–3841. [Google Scholar] [CrossRef]
- Pirot, P.; Naamane, N.; Libert, F.; Magnusson, N.E.; Ørntoft, T.F.; Cardozo, A.K.; Eizirik, D.L. Global profiling of genes modified by endoplasmic reticulum stress in pancreatic beta cells reveals the early degradation of insulin mRNAs. Diabetologia 2007, 50, 1006–1014. [Google Scholar] [CrossRef]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef]
- Sanger, F. Oxidation of Insulin by Performic Acid. Nature 1947, 160, 295–296. [Google Scholar] [CrossRef]
- Tran, D.T.; Pottekat, A.; Mir, S.A.; Loguercio, S.; Jang, I.; Campos, A.R.; Scully, K.M.; Lahmy, R.; Liu, M.; Arvan, P.; et al. Unbiased Profiling of the Human Proinsulin Biosynthetic Interaction Network Reveals a Role for Peroxiredoxin 4 in Proinsulin Folding. Diabetes 2020, 69, 1723–1734. [Google Scholar] [CrossRef]
- Schultz, L.W.; Martin, P.K.; Liang, J.; Schreiber, S.L.; Clardy, J. Atomic Structure of the Immunophilin FKBP13-FK506 Complex: Insights into the Composite Binding Surface for Calcineurin. J. Am. Chem. Soc. 1994, 116, 3129–3130. [Google Scholar] [CrossRef]
- Prakash, A.; Rajan, S.; Yoon, H.S. Crystal structure of the FK506 binding domain of human FKBP25 in complex with FK506. Protein Sci. A Publ. Protein Soc. 2016, 25, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Lieblich, S.A.; Fang, K.Y.; Cahn, J.K.B.; Rawson, J.; LeBon, J.; Ku, H.T.; Tirrell, D.A. 4S-Hydroxylation of Insulin at ProB28 Accelerates Hexamer Dissociation and Delays Fibrillation. J. Am. Chem. Soc. 2017, 139, 8384–8387. [Google Scholar] [CrossRef]
- Brange, J.; Owens, D.R.; Kang, S.; Volund, A. Monomeric insulins and their experimental and clinical implications. Diabetes Care 1990, 13, 923–954. [Google Scholar] [CrossRef] [PubMed]
- Rege, N.K.; Liu, M.; Yang, Y.; Dhayalan, B.; Wickramasinghe, N.P.; Chen, Y.S.; Rahimi, L.; Guo, H.; Haataja, L.; Sun, J.; et al. Evolution of insulin at the edge of foldability and its medical implications. Proc. Natl. Acad. Sci. USA 2020, 117, 29618–29628. [Google Scholar] [CrossRef]
- Markussen, J.A.N. Comparative reduction/oxidation studies with single chain des-(B30) insulin and porcine proinsulin. Int. J. Pept. Protein Res. 1985, 25, 431–434. [Google Scholar] [CrossRef]
- Baldwin, R.L. The search for folding intermediates and the mechanism of protein folding. Annu. Rev. Biophys. 2008, 37, 1–21. [Google Scholar] [CrossRef]
- Morgan, A.A.; Rubenstein, E. Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef]
- Kuliawat, R.; Prabakaran, D.; Arvan, P. Proinsulin endoproteolysis confers enhanced targeting of processed insulin to the regulated secretory pathway. Mol. Biol. Cell 2000, 11, 1959–1972. [Google Scholar] [CrossRef]
- Ward, W.K.; LaCava, E.C.; Paquette, T.L.; Beard, J.C.; Wallum, B.J.; Porte, D., Jr. Disproportionate elevation of immunoreactive proinsulin in type 2 (non-insulin-dependent) diabetes mellitus and in experimental insulin resistance. Diabetologia 1987, 30, 698–702. [Google Scholar] [CrossRef]
- Truyen, I.; De Pauw, P.; Jørgensen, P.N.; Van Schravendijk, C.; Ubani, O.; Decochez, K.; Vandemeulebroucke, E.; Weets, I.; Mao, R.; Pipeleers, D.G.; et al. Proinsulin levels and the proinsulin:c-peptide ratio complement autoantibody measurement for predicting type 1 diabetes. Diabetologia 2005, 48, 2322–2329. [Google Scholar] [CrossRef] [PubMed]
- Wasserfall, C.; Nick, H.S.; Campbell-Thompson, M.; Beachy, D.; Haataja, L.; Kusmartseva, I.; Posgai, A.; Beery, M.; Rhodes, C.; Bonifacio, E.; et al. Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metab. 2017, 26, 568–575.e563. [Google Scholar] [CrossRef]
- Lathe, G.H.; Ruthven, C.R. The separation of substances and estimation of their relative molecular sizes by the use of colums of starch in water. Biochem. J. 1956, 62, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M. Solubility of proteins. Admet Dmpk 2020, 8, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Maiti, N.C.; Phillips, N.B.; Carey, P.R.; Weiss, M.A. Structure-specific effects of protein topology on cross-beta assembly: Studies of insulin fibrillation. Biochemistry 2006, 45, 10278–10293. [Google Scholar] [CrossRef]
- Menting, J.G.; Yang, Y.; Chan, S.J.; Phillips, N.B.; Smith, B.J.; Whittaker, J.; Wickramasinghe, N.P.; Whittaker, L.J.; Pandyarajan, V.; Wan, Z.L.; et al. Protective hinge in insulin opens to enable its receptor engagement. Proc. Natl. Acad. Sci. USA 2014, 111, E3395–E3404. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Sawaya, M.R.; Wall, J.S.; Eisenberg, D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA 2009, 106, 18990–18995. [Google Scholar] [CrossRef]
- Gorden, P.; Skarulis, M.C.; Roach, P.; Comi, R.J.; Fraker, D.L.; Norton, J.A.; Alexander, H.R.; Doppman, J.L. Plasma proinsulin-like component in insulinoma: A 25-year experience. J. Clin. Endocrinol. Metab. 1995, 80, 2884–2887. [Google Scholar] [CrossRef]
- Labriola, L.; Peters, M.G.; Krogh, K.; Stigliano, I.; Terra, L.F.; Buchanan, C.; Machado, M.C.; Bal de Kier Joffé, E.; Puricelli, L.; Sogayar, M.C. Generation and characterization of human insulin-releasing cell lines. BMC Cell Biol. 2009, 10, 49. [Google Scholar] [CrossRef]
- Liu, M.; Wright, J.; Guo, H.; Xiong, Y.; Arvan, P. Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. Vitam. Horm. 2014, 95, 35–62. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Encina, G.; Soto, C.; Hetz, C. Abnormal calcium homeostasis and protein folding stress at the ER: A common factor in familial and infectious prion disorders. Commun. Integr. Biol. 2011, 4, 258–261. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Lubben, T.H.; Gatenby, A.A.; Donaldson, G.K.; Lorimer, G.H.; Viitanen, P.V. Identification of a groES-like chaperonin in mitochondria that facilitates protein folding. Proc. Natl. Acad. Sci. USA 1990, 87, 7683–7687. [Google Scholar] [CrossRef]
- Kennedy, D.; Jäger, R.; Mosser, D.D.; Samali, A. Regulation of apoptosis by heat shock proteins. IUBMB Life 2014, 66, 327–338. [Google Scholar] [CrossRef]
- Haataja, L.; Snapp, E.; Wright, J.; Liu, M.; Hardy, A.B.; Wheeler, M.B.; Markwardt, M.L.; Rizzo, M.A.; Arvan, P. Proinsulin intermolecular interactions during secretory trafficking in pancreatic beta cells. J. Biol. Chem. 2013, 288, 1896–1906. [Google Scholar] [CrossRef]
- Rykær, M.; Svensson, B.; Davies, M.J.; Hagglund, P. Unrestricted Mass Spectrometric Data Analysis for Identification, Localization, and Quantification of Oxidative Protein Modifications. J. Proteome Res. 2017, 16, 3978–3988. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoefner, C.; Bryde, T.H.; Pihl, C.; Tiedemann, S.N.; Bresson, S.E.; Hotiana, H.A.; Khilji, M.S.; Santos, T.D.; Puglia, M.; Pisano, P.; et al. FK506-Binding Protein 2 Participates in Proinsulin Folding. Biomolecules 2023, 13, 152. https://doi.org/10.3390/biom13010152
Hoefner C, Bryde TH, Pihl C, Tiedemann SN, Bresson SE, Hotiana HA, Khilji MS, Santos TD, Puglia M, Pisano P, et al. FK506-Binding Protein 2 Participates in Proinsulin Folding. Biomolecules. 2023; 13(1):152. https://doi.org/10.3390/biom13010152
Chicago/Turabian StyleHoefner, Carolin, Tenna Holgersen Bryde, Celina Pihl, Sylvia Naiga Tiedemann, Sophie Emilie Bresson, Hajira Ahmed Hotiana, Muhammad Saad Khilji, Theodore Dos Santos, Michele Puglia, Paola Pisano, and et al. 2023. "FK506-Binding Protein 2 Participates in Proinsulin Folding" Biomolecules 13, no. 1: 152. https://doi.org/10.3390/biom13010152
APA StyleHoefner, C., Bryde, T. H., Pihl, C., Tiedemann, S. N., Bresson, S. E., Hotiana, H. A., Khilji, M. S., Santos, T. D., Puglia, M., Pisano, P., Majewska, M., Durzynska, J., Klindt, K., Klusek, J., Perone, M. J., Bucki, R., Hägglund, P. M., Gourdon, P. E., Gotfryd, K., ... Marzec, M. T. (2023). FK506-Binding Protein 2 Participates in Proinsulin Folding. Biomolecules, 13(1), 152. https://doi.org/10.3390/biom13010152