

Prediction of Protein Function from Tertiary Structure of the Active Site in Heme Proteins by Convolutional Neural Network

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
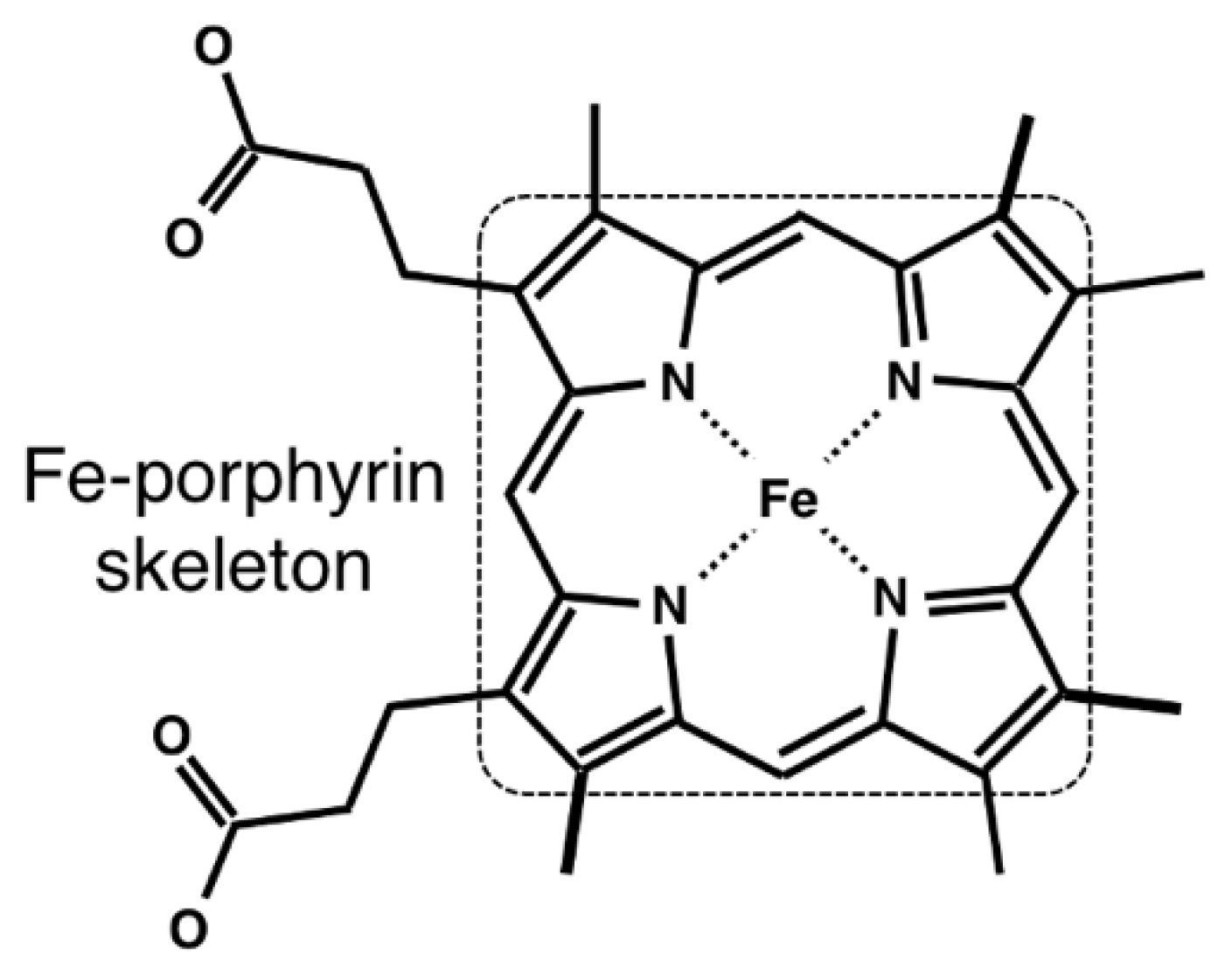
2.1. Data Collection of Heme and Its Host Proteins
2.2. Assignment of Protein Function to Each Heme Sample
- (1)
- If the protein chain(s), including the axial ligand(s), had an EC number(s), the first digit of the EC number(s) was assigned.
- (2)
- If case 1 did not apply and the protein chain(s) had GO associated with “oxygen-binding”, “oxidoreductase activity”, “electron transfer activity”, “transcription”, or “heme transport” as the molecular function or biological process, one function was assigned in order from these functions.
- (3)
- If cases 1 and 2 did not apply and the PDB entry had keywords associated with “hemophore”, “electron transfer activity”, “oxygen-binding”, “oxidoreductase activity”, “heme extraction”, “signaling protein”, “nitrophorin (NO transport)”, or “heme transport”, one function was assigned in order from these functions.
- (4)
- If cases 1–3 did not apply and the PDB entry had a description of cytochrome p460, “oxidoreductase” was assigned.
- (5)
- If cases 1–4 did not apply or there was no axial ligand, “unclassified” was assigned.
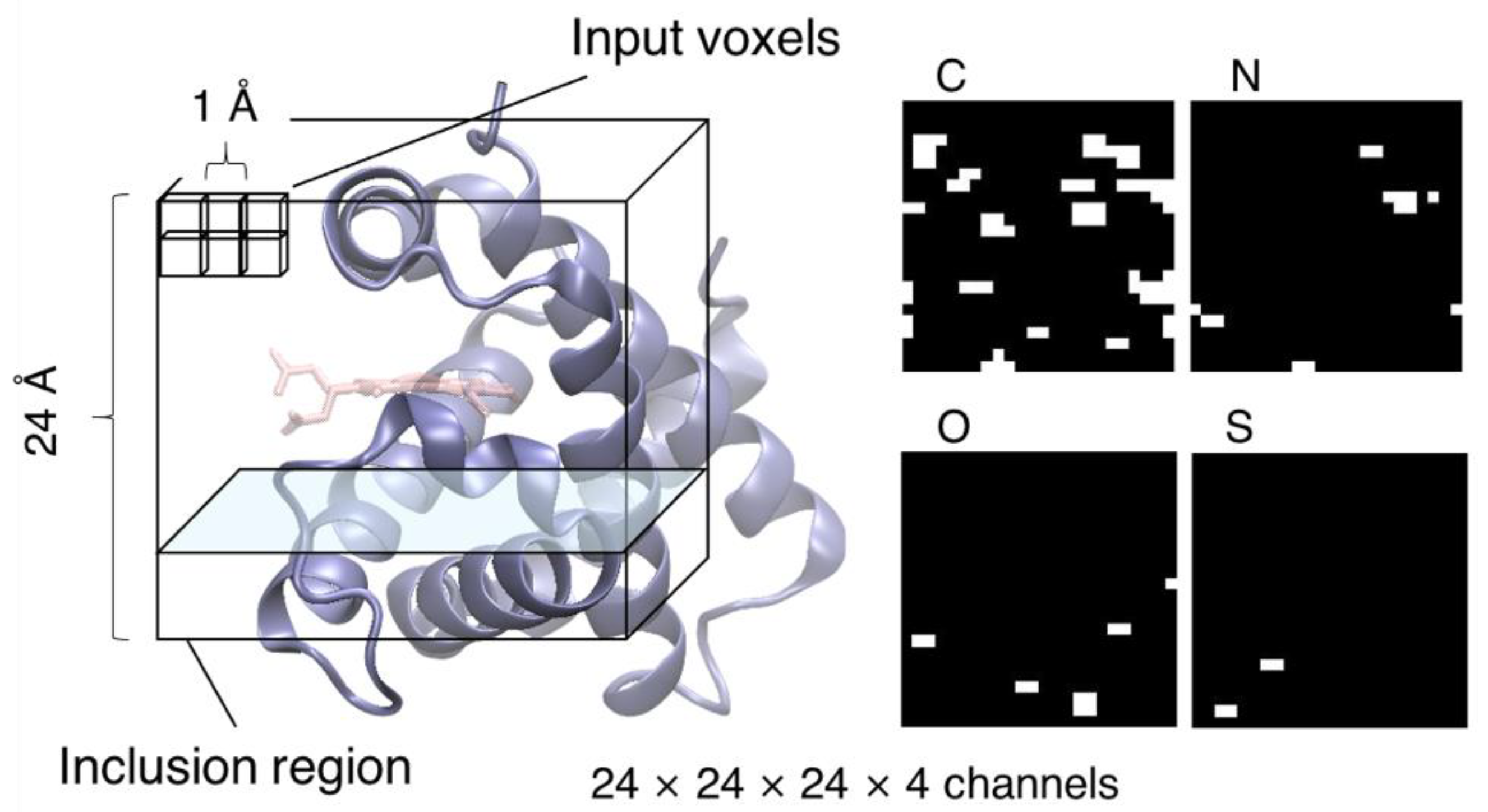
2.3. CNN Model
2.4. Analyses of Cavity of Heme-Binding Site
3. Results and Discussion
3.1. Prediction of Protein Function from the Tertiary Structure of the Heme-Binding Pocket Using a CNN Model: Two-Label Classification
3.2. Specification of Regions in Input Data Significant for Prediction
3.3. Prediction of Protein Function from the Tertiary Structure of the Heme-Binding Pocket Using a CNN Model: Three-Label Classification
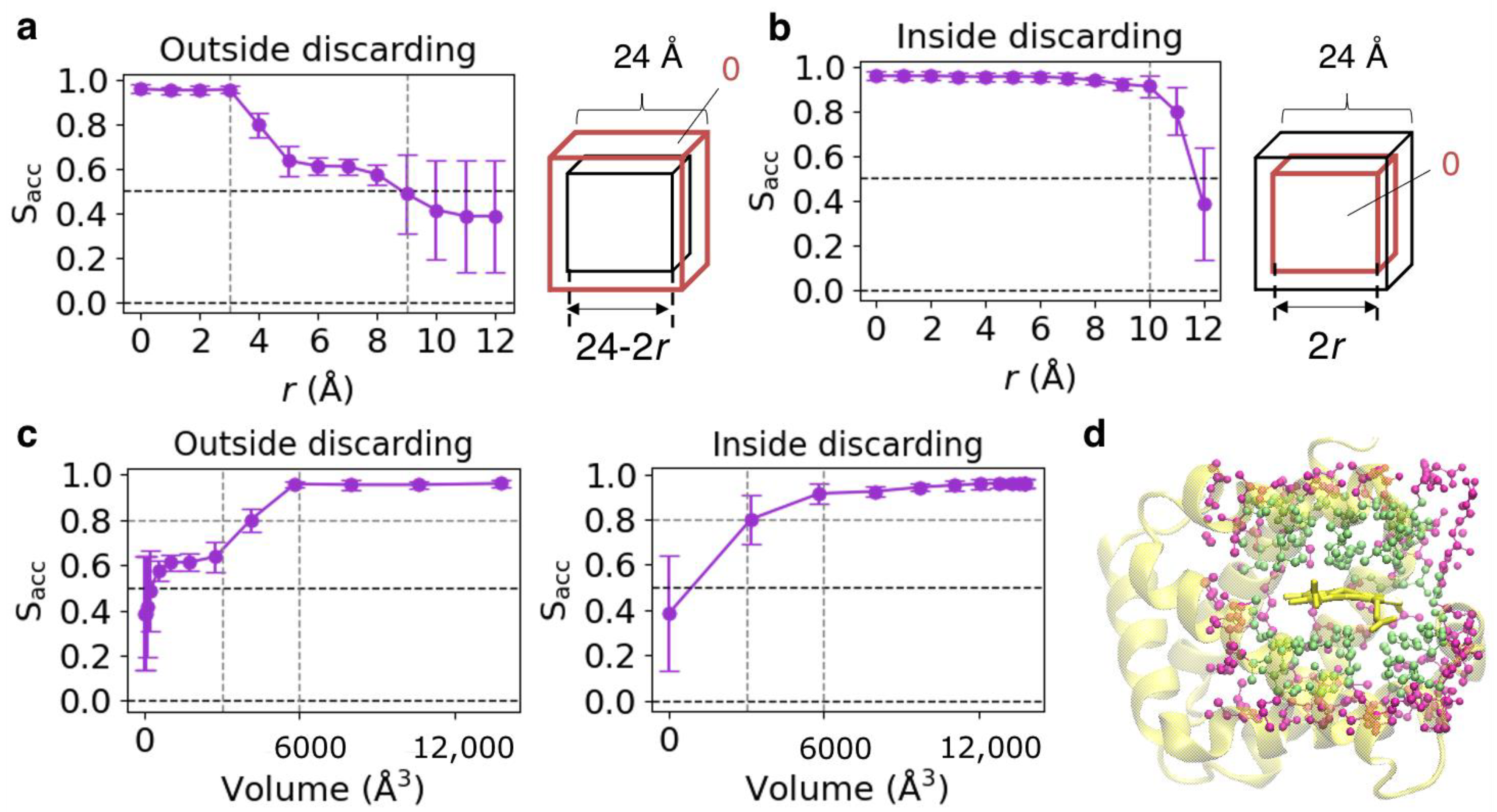
3.4. Validation of Datasets Used for CNN Model Construction
3.5. Similarity of the Structures of Heme-Binding Pockets between Proteins with the Same Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Solomon, E.I.; Szilagyi, R.K.; DeBeer George, S.; Basumallick, L. Electronic Structures of Metal Sites in Proteins and Models: Contributions to Function in Blue Copper Proteins. Chem. Rev. 2004, 104, 419–458. [Google Scholar] [CrossRef] [PubMed]
- Tosha, T.; Nomura, T.; Nishida, T.; Saeki, N.; Okubayashi, K.; Yamagiwa, R.; Sugahara, M.; Nakane, T.; Yamashita, K.; Hirata, K.; et al. Capturing an Initial Intermediate during the P450nor Enzymatic Reaction Using Time-Resolved XFEL Crystallography and Caged-Substrate. Nat. Commun. 2017, 8, 1585. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Kimura, T.; Kanematsu, Y.; Yamada, D.; Yamashita, K.; Hirata, K.; Ueno, G.; Murakami, H.; Hisano, T.; Yamagiwa, R.; et al. Short-Lived Intermediate in N2O Generation by P450 NO Reductase Captured by Time-Resolved IR Spectroscopy and XFEL Crystallography. Proc. Natl. Acad. Sci. USA 2021, 118, e2101481118. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Takano, Y. QM/MM Molecular Dynamics Simulations Revealed Catalytic Mechanism of Urease. J. Phys. Chem. B 2022, 126, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Murakawa, T.; Nakanishi, S.; Boero, M.; Shigeta, Y.; Hayashi, H.; Okajima, T. Molecular Mechanism of a Large Conformational Change of the Quinone Cofactor in the Semiquinone Intermediate of Bacterial Copper Amine Oxidase. Chem. Sci. 2022, 13, 10923–10938. [Google Scholar] [CrossRef]
- Poulos, T.L. The Janus Nature of Heme. Nat. Prod. Rep. 2007, 24, 504–510. [Google Scholar] [CrossRef]
- Louie, G.V.; Brayer, G.D. High-Resolution Refinement of Yeast Iso-1-Cytochrome c and Comparisons with Other Eukaryotic Cytochromes C. J. Mol. Biol. 1990, 214, 527–555. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef]
- Ostermeier, C. Cytochrome c Oxidase. Curr. Opin. Struct. Biol. 1996, 6, 460–466. [Google Scholar] [CrossRef]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A.C.T. Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Dickerson, R.E.; Strandberg, B.E.; Hart, R.G.; Davies, D.R.; Phillips, D.C.; Shore, V.C. Structure of Myoglobin: A Three-Dimensional Fourier Synthesis at 2 Å. Resolution. Nature 1960, 185, 422–427. [Google Scholar] [CrossRef]
- Faller, M.; Matsunaga, M.; Yin, S.; Loo, J.A.; Guo, F. Heme Is Involved in MicroRNA Processing. Nat. Struct. Mol. Biol. 2007, 14, 23–29. [Google Scholar] [CrossRef]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 Regulates Enhancer Availability of Heme Oxygenase-1 Gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Liu, H.-L.; Zhou, H.-N.; Xing, W.-M.; Zhao, J.-F.; Li, S.-X.; Huang, J.-F.; Bi, R.-C. 2.6 Å Resolution Crystal Structure of the Bacterioferritin from Azotobacter Vinelandii. FEBS Lett. 2004, 573, 93–98. [Google Scholar] [CrossRef]
- Bateman, T.J.; Shah, M.; Ho, T.P.; Shin, H.E.; Pan, C.; Harris, G.; Fegan, J.E.; Islam, E.A.; Ahn, S.K.; Hooda, Y.; et al. A Slam-Dependent Hemophore Contributes to Heme Acquisition in the Bacterial Pathogen Acinetobacter Baumannii. Nat. Commun. 2021, 12, 6270. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Rydberg, P.; Sigfridsson, E.; Ryde, U. On the Role of the Axial Ligand in Heme Proteins: A Theoretical Study. JBIC J. Biol. Inorg. Chem. 2004, 9, 203–223. [Google Scholar] [CrossRef]
- Walker, F.A. Magnetic Spectroscopic (EPR, ESEEM, Mossbauer, MCD and NMR) Studies of Low-Spin Ferriheme Centers and Their Corresponding Heme Proteins. Coord. Chem. Rev. 1999, 185–186, 471–534. [Google Scholar] [CrossRef]
- Takano, Y.; Nakamura, H. Density Functional Study of Roles of Porphyrin Ring in Electronic Structures of Heme. Int. J. Quantum Chem. 2009, 109, 3583–3591. [Google Scholar] [CrossRef]
- Kondo, H.X.; Kanematsu, Y.; Masumoto, G.; Takano, Y. PyDISH: Database and Analysis Tools for Heme Porphyrin Distortion in Heme Proteins. Database 2020, 2020, baaa066. [Google Scholar] [CrossRef]
- Bikiel, D.E.; Forti, F.; Boechi, L.; Nardini, M.; Luque, F.J.; Martí, M.A.; Estrin, D.A. Role of Heme Distortion on Oxygen Affinity in Heme Proteins: The Protoglobin Case. J. Phys. Chem. B 2010, 114, 8536–8543. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Benabbas, A.; Zeng, W.; Kleingardner, J.G.; Bren, K.L.; Champion, P.M. Investigations of Heme Distortion, Low-Frequency Vibrational Excitations, and Electron Transfer in Cytochrome C. Proc. Natl. Acad. Sci. USA 2014, 111, 6570–6575. [Google Scholar] [CrossRef]
- Imada, Y.; Nakamura, H.; Takano, Y. Density Functional Study of Porphyrin Distortion Effects on Redox Potential of Heme. J. Comput. Chem. 2018, 39, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Kondo, H.X.; Kanematsu, Y.; Imada, Y. Computational Study of Distortion Effect of Fe-Porphyrin Found as a Biological Active Site. Jpn. J. Appl. Phys. 2020, 59, 010502. [Google Scholar] [CrossRef]
- Kanematsu, Y.; Kondo, H.X.; Imada, Y.; Takano, Y. Statistical and Quantum-Chemical Analysis of the Effect of Heme Porphyrin Distortion in Heme Proteins: Differences between Oxidoreductases and Oxygen Carrier Proteins. Chem. Phys. Lett. 2018, 710, 108–112. [Google Scholar] [CrossRef]
- Kondo, H.X.; Takano, Y. Analysis of Fluctuation in the Heme-Binding Pocket and Heme Distortion in Hemoglobin and Myoglobin. Life 2022, 12, 210. [Google Scholar] [CrossRef]
- Kondo, H.X.; Fujii, M.; Tanioka, T.; Kanematsu, Y.; Yoshida, T.; Takano, Y. Global Analysis of Heme Proteins Elucidates the Correlation between Heme Distortion and the Heme-Binding Pocket. J. Chem. Inf. Model. 2022, 62, 775–784. [Google Scholar] [CrossRef]
- Kondo, H.X.; Iizuka, H.; Masumoto, G.; Kabaya, Y.; Kanematsu, Y.; Takano, Y. Elucidation of the Correlation between Heme Distortion and Tertiary Structure of the Heme-Binding Pocket Using a Convolutional Neural Network. Biomolecules 2022, 12, 1172. [Google Scholar] [CrossRef]
- Liao, F.; Yuan, H.; Du, K.-J.; You, Y.; Gao, S.-Q.; Wen, G.-B.; Lin, Y.-W.; Tan, X. Distinct Roles of a Tyrosine-Associated Hydrogen-Bond Network in Fine-Tuning the Structure and Function of Heme Proteins: Two Cases Designed for Myoglobin. Mol. Biosyst. 2016, 12, 3139–3145. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nakajima, H.; Ueno, T. Reactivities of Oxo and Peroxo Intermediates Studied by Hemoprotein Mutants. Acc. Chem. Res. 2007, 40, 554–562. [Google Scholar] [CrossRef]
- Du, J.-F.; Li, W.; Li, L.; Wen, G.-B.; Lin, Y.-W.; Tan, X. Regulating the Coordination State of a Heme Protein by a Designed Distal Hydrogen-Bonding Network. ChemistryOpen 2015, 4, 97–101. [Google Scholar] [CrossRef]
- Zhang, P.; Yuan, H.; Xu, J.; Wang, X.-J.; Gao, S.-Q.; Tan, X.; Lin, Y.-W. A Catalytic Binding Site Together with a Distal Tyr in Myoglobin Affords Catalytic Efficiencies Similar to Natural Peroxidases. ACS Catal. 2020, 10, 891–896. [Google Scholar] [CrossRef]
- Wang, C.; Lovelace, L.L.; Sun, S.; Dawson, J.H.; Lebioda, L. Structures of K42N and K42Y Sperm Whale Myoglobins Point to an Inhibitory Role of Distal Water in Peroxidase Activity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 2833–2839. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. In Advances in Neural Information Processing Systems; Pereira, F., Burges, C.J., Bottou, L., Weinberger, K.Q., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2012; Volume 25. [Google Scholar]
- Simonyan, K.; Zisserman, A. Very Deep Convolutional Networks for Large-Scale Image Recognition. arXiv 2015, arXiv:1409.1556. [Google Scholar]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.-C.; Brylinski, M. DeepDrug3D: Classification of Ligand-Binding Pockets in Proteins with a Convolutional Neural Network. PLoS Comput. Biol. 2019, 15, e1006718. [Google Scholar] [CrossRef]
- Kinjo, A.R.; Yamashita, R.; Nakamura, H. PDBj Mine: Design and Implementation of Relational Database Interface for Protein Data Bank Japan. Database 2010, 2010, baq021. [Google Scholar] [CrossRef]
- Kinjo, A.R.; Suzuki, H.; Yamashita, R.; Ikegawa, Y.; Kudou, T.; Igarashi, R.; Kengaku, Y.; Cho, H.; Standley, D.M.; Nakagawa, A.; et al. Protein Data Bank Japan (PDBj): Maintaining a Structural Data Archive and Resource Description Framework Format. Nucleic Acids Res. 2012, 40, D453–D460. [Google Scholar] [CrossRef]
- Hamelryck, T.; Manderick, B. PDB File Parser and Structure Class Implemented in Python. Bioinformatics 2003, 19, 2308–2310. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L. PISCES: A Protein Sequence Culling Server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef] [PubMed]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems 32; Wallach, H., Larochelle, H., Beygelzimer, A., Alché-Buc, F., Fox, E., Garnett, R., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2019; pp. 8024–8035. [Google Scholar]
- Wagner, J.R.; Sørensen, J.; Hensley, N.; Wong, C.; Zhu, C.; Perison, T.; Amaro, R.E. POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput. 2017, 13, 4584–4592. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Pesce, A.; Tilleman, L.; Dewilde, S.; Ascenzi, P.; Coletta, M.; Ciaccio, C.; Bruno, S.; Moens, L.; Bolognesi, M.; Nardini, M. Structural Heterogeneity and Ligand Gating in Ferric Methanosarcina Acetivorans Protoglobin Mutants. IUBMB Life 2011, 63, 287–294. [Google Scholar] [CrossRef]
- Rice, S.L.; Boucher, L.E.; Schlessman, J.L.; Preimesberger, M.R.; Bosch, J.; Lecomte, J.T.J. Structure of Chlamydomonas Reinhardtii THB1, a Group 1 Truncated Hemoglobin with a Rare Histidine–Lysine Heme Ligation. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 718–725. [Google Scholar] [CrossRef]
- Ianiri, G.; Coelho, M.A.; Ruchti, F.; Sparber, F.; McMahon, T.J.; Fu, C.; Bolejack, M.; Donovan, O.; Smutney, H.; Myler, P.; et al. HGT in the Human and Skin Commensal Malassezia: A Bacterially Derived Flavohemoglobin Is Required for NO Resistance and Host Interaction. Proc. Natl. Acad. Sci. USA 2020, 117, 15884–15894. [Google Scholar] [CrossRef]
- de Sanctis, D.; Dewilde, S.; Vonrhein, C.; Pesce, A.; Moens, L.; Ascenzi, P.; Hankeln, T.; Burmester, T.; Ponassi, M.; Nardini, M.; et al. Bishistidyl Heme Hexacoordination, a Key Structural Property in Drosophila Melanogaster Hemoglobin. J. Biol. Chem. 2005, 280, 27222–27229. [Google Scholar] [CrossRef]
- Yoon, J.; Herzik, M.A.; Winter, M.B.; Tran, R.; Olea, C.; Marletta, M.A. Structure and Properties of a Bis-Histidyl Ligated Globin from Caenorhabditis Elegans. Biochemistry 2010, 49, 5662–5670. [Google Scholar] [CrossRef]
- Walsh, I.; Fishman, D.; Garcia-Gasulla, D.; Titma, T.; Pollastri, G.; Capriotti, E.; Casadio, R.; Capella-Gutierrez, S.; Cirillo, D.; Del Conte, A.; et al. DOME: Recommendations for Supervised Machine Learning Validation in Biology. Nat. Methods 2021, 18, 1122–1127. [Google Scholar] [CrossRef]
- Rost, B. PHD: Predicting One-Dimensional Protein Structure by Profile-Based Neural Networks. Methods Enzymol. 1996, 266, 525–539. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Schwartz, D.; Chou, M.F.; Church, G.M. Predicting Protein Post-Translational Modifications Using Meta-Analysis of Proteome Scale Data Sets. Mol. Cell. Proteom. 2009, 8, 365–379. [Google Scholar] [CrossRef]
- Gligorijević, V.; Renfrew, P.D.; Kosciolek, T.; Leman, J.K.; Berenberg, D.; Vatanen, T.; Chandler, C.; Taylor, B.C.; Fisk, I.M.; Vlamakis, H.; et al. Structure-Based Protein Function Prediction Using Graph Convolutional Networks. Nat. Commun. 2021, 12, 3168. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Kondo, H.X.; Kanematsu, Y.; Takano, Y. Structure of Heme-Binding Pocket in Heme Protein Is Generally Rigid and Can Be Predicted by AlphaFold2. Chem. Lett. 2022, 51, 704–708. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Layer | Function | Filter (Kernel) | Output Dimension (Channel × Depth × Width × Height) |
|---|---|---|---|
| 1 | Conv3d | 2 × 2 × 2 with 0-padding | 64 × 21 × 21 × 21 |
| 2 | Conv3d | 2 × 2 × 2 with 0-padding | 128 × 22 × 22 × 22 |
| 3 | BatchNorm3d | - | 128 × 22 × 22 × 22 |
| 4 | Conv3d | 2 × 2 × 2 without padding | 128 × 21 × 21 × 21 |
| 5 | ReLU | - | 128 × 21 × 21 × 21 |
| 6 | BatchNorm3d | - | 128 × 21 × 21 × 21 |
| 7 | MaxPool3d | 2 × 2 × 2 stride: 2 × 2 × 2 | 128 × 10 × 10 × 10 |
| 8 | Full connection | - | 128,000 |
| 9 | Linear | - | 128 |
| 10 | ReLU | - | 128 |
| 11 | Dropout | 0.4 | 128 |
| 12 | Linear | - | 64 |
| 13 | BatchNorm1d | - | 64 |
| 14 | ReLU | - | 64 |
| 15 | Linear | - | 2 or 3 |
| 16 | Sigmoid | - | 2 or 3 |
| Predicted Value | ||||
|---|---|---|---|---|
| OB | OR | OB–OR | ||
| Observed Value | OB [190] † | 0.985 ± 0.012 (187) | 0.010 ± 0.012 (2) | 0.005 ± 0.010 (1) |
| OR [312] † | 0.010 ± 0.008 (3) | 0.990 ± 0.008 (309) | 0.000 ± 0.000 (0) | |
| OB–OR [35] † | 0.436 ± 0.248 (15) | 0.000 ± 0.000 (0) | 0.564 ± 0.248 (20) | |
| PDB ID | Protein Name | Observed Value | Predicted Value | Remark |
|---|---|---|---|---|
| 2BK9 | hemoglobin | OR | OB | misassignment |
| 3MVC | GLB-6 | OB | OR | misassignment |
| 3QZX | protoglobin | OB | OR | - |
| 4XDI | THB1 (truncated hemoglobin) | OR | OB | - |
| 6O0A | flavohemoglobin | OR | OB | - |
| 7CEZ | myoglobin (G5K/Q8K/A19K/V21K) | OB | OB–OR | detailed function unknown |
| Predicted Value | ||||||
|---|---|---|---|---|---|---|
| OB | OR | OB–OR | ET | Others † | ||
| Observed Value | OB [193] ‡ | 0.973 ± 0.016 (188) | 0.016 ± 0.013 (3) | 0.006 ± 0.012 (1) | 0.000 ± 0.000 (0) | 0.005 ± 0.010 (1) |
| OR [297] ‡ | 0.006 ± 0.007 (2) | 0.907 ± 0.054 (268) | 0.000 ± 0.000 (0) | 0.084 ± 0.049 (26) | 0.004 ± 0.007 (1) | |
| OB–OR [36] ‡ | 0.570 ± 0.296 (20) | 0.000 ± 0.000 (0) | 0.430 ± 0.296 (16) | 0.000 ± 0.000 (0) | 0.000 ± 0.000 (0) | |
| ET [371] ‡ | 0.000 ± 0.000 (0) | 0.110 ± 0.019 (40) | 0.000 ± 0.000 (0) | 0.890 ± 0.019 (331) | 0.000 ± 0.000 (0) | |
| Dataset | Class Label | Accuracy | Recall | Precision | Specificity |
|---|---|---|---|---|---|
| Dataset_25 | OB [9] † | 0.923 ± 0.069 | 0.800 ± 0.400 | 0.653 ± 0.366 | 0.925 ± 0.074 |
| OR [55] † | 0.923 ± 0.069 | 0.925 ± 0.074 | 0.983 ± 0.033 | 0.800 ± 0.400 | |
| Dataset_60 | OB [36] † | 0.934 ± 0.089 | 0.703 ± 0.381 | 0.979 ± 0.036 ‡ | 0.994 ± 0.013 |
| OR [192] † | 0.934 ± 0.089 | 0.994 ± 0.013 | 0.937 ± 0.090 | 0.703 ± 0.381 | |
| Dataset_80 | OB [59] † | 0.974 ± 0.022 | 0.932 ± 0.097 | 0.896 ± 0.106 | 0.983 ± 0.015 |
| OR [239] † | 0.974 ± 0.022 | 0.983 ± 0.015 | 0.987 ± 0.017 | 0.932 ± 0.097 | |
| Dataset_99_ without_OB-OR | OB [196] † | 0.990 ± 0.011 | 0.995 ± 0.010 | 0.981 ± 0.026 | 0.987 ± 0.020 |
| OR [308] † | 0.990 ± 0.011 | 0.987 ± 0.020 | 0.997 ± 0.007 | 0.995 ± 0.010 |
| Protein Group | Sample Number | |||
|---|---|---|---|---|
| Dataset_99 | Dataset_25 | Dataset_99 | Dataset_25 | |
| OB | 241 | 16 | 12.70 (1.78) † | 15.45 (2.23) † |
| OR | 388 | 63 | 16.88 (2.45) † | 15.86 (2.61) † |
| OB–OR | 42 | 0 | 12.44 (2.41) † | - |
| ET | 450 | 47 | 16.52 (2.88) † | 16.07 (2.05) † |
| Combined | 1121 | 126 | 16.80 (2.51) † | 15.99 (2.34) † |
| Dehaloperoxidase | 10 | 0 | 9.95 (1.21) † | - |
| DF-Myoglobin | 32 | 0 | 11.20 (2.60) † | - |
| Myoglobin (OB) | 82 | 1 | 9.99 (2.33) † | 0.00 (0.00) † |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondo, H.X.; Iizuka, H.; Masumoto, G.; Kabaya, Y.; Kanematsu, Y.; Takano, Y. Prediction of Protein Function from Tertiary Structure of the Active Site in Heme Proteins by Convolutional Neural Network. Biomolecules 2023, 13, 137. https://doi.org/10.3390/biom13010137
Kondo HX, Iizuka H, Masumoto G, Kabaya Y, Kanematsu Y, Takano Y. Prediction of Protein Function from Tertiary Structure of the Active Site in Heme Proteins by Convolutional Neural Network. Biomolecules. 2023; 13(1):137. https://doi.org/10.3390/biom13010137
Chicago/Turabian StyleKondo, Hiroko X., Hiroyuki Iizuka, Gen Masumoto, Yuichi Kabaya, Yusuke Kanematsu, and Yu Takano. 2023. "Prediction of Protein Function from Tertiary Structure of the Active Site in Heme Proteins by Convolutional Neural Network" Biomolecules 13, no. 1: 137. https://doi.org/10.3390/biom13010137
APA StyleKondo, H. X., Iizuka, H., Masumoto, G., Kabaya, Y., Kanematsu, Y., & Takano, Y. (2023). Prediction of Protein Function from Tertiary Structure of the Active Site in Heme Proteins by Convolutional Neural Network. Biomolecules, 13(1), 137. https://doi.org/10.3390/biom13010137