
Effects of Chronic Caffeine Consumption on Synaptic Function, Metabolism and Adenosine Modulation in Different Brain Areas
 , , , , ,
, , , , ,  ,
,  , and
, and 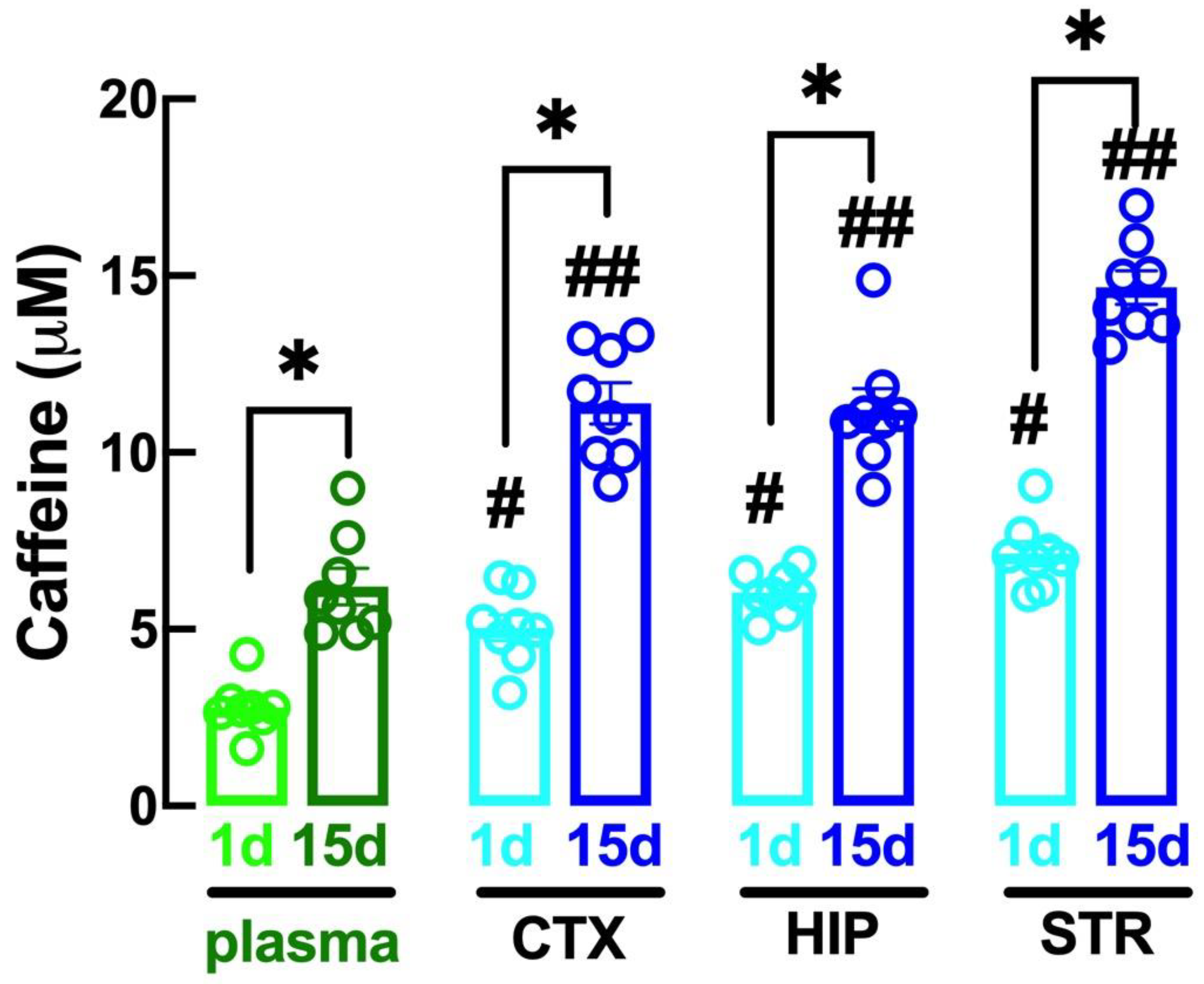{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Altered Caffeine Levels upon Regular Intake of Caffeine over Time
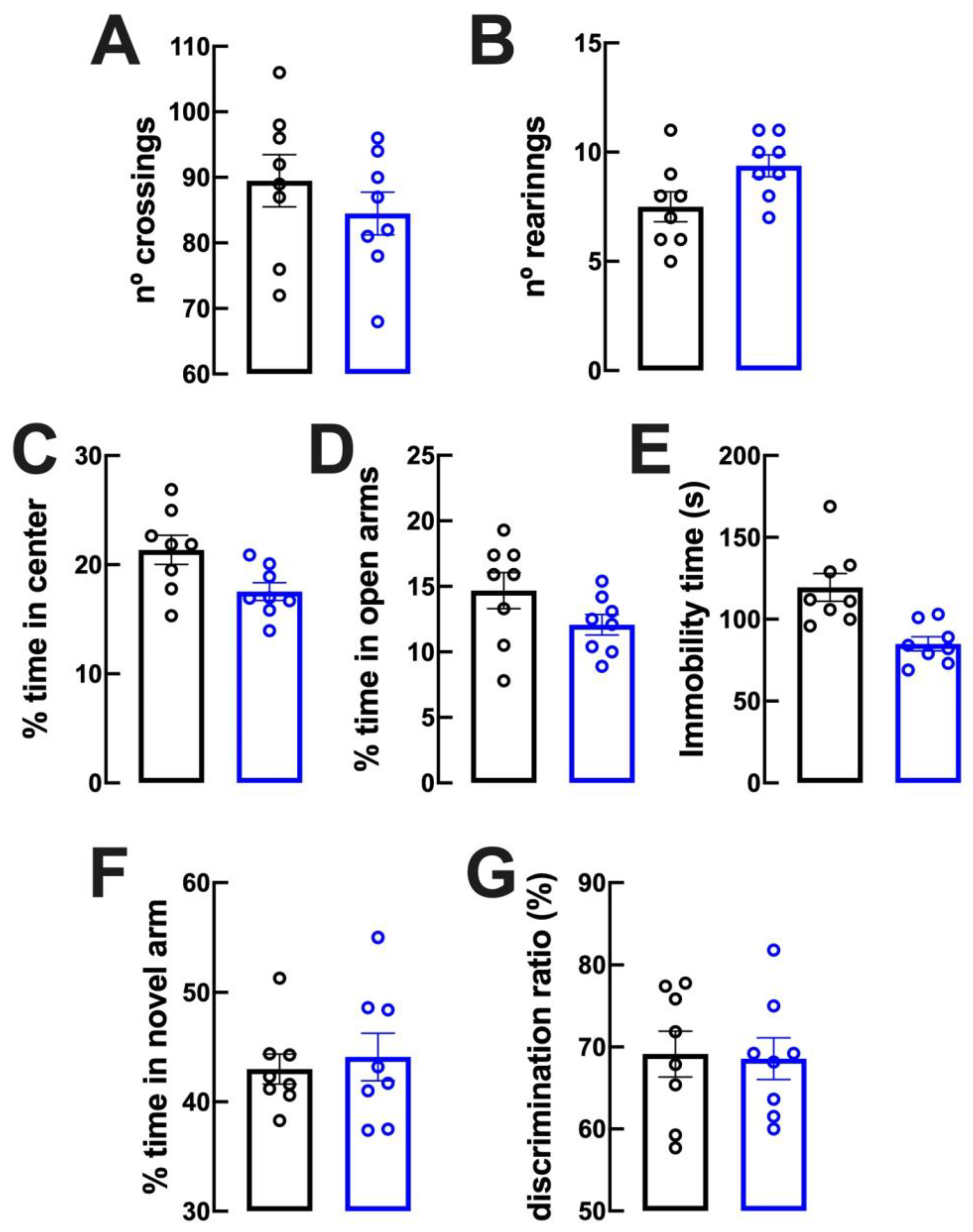
2.2. Lack of Evident Behavioral Modifications upon Caffeine Intake
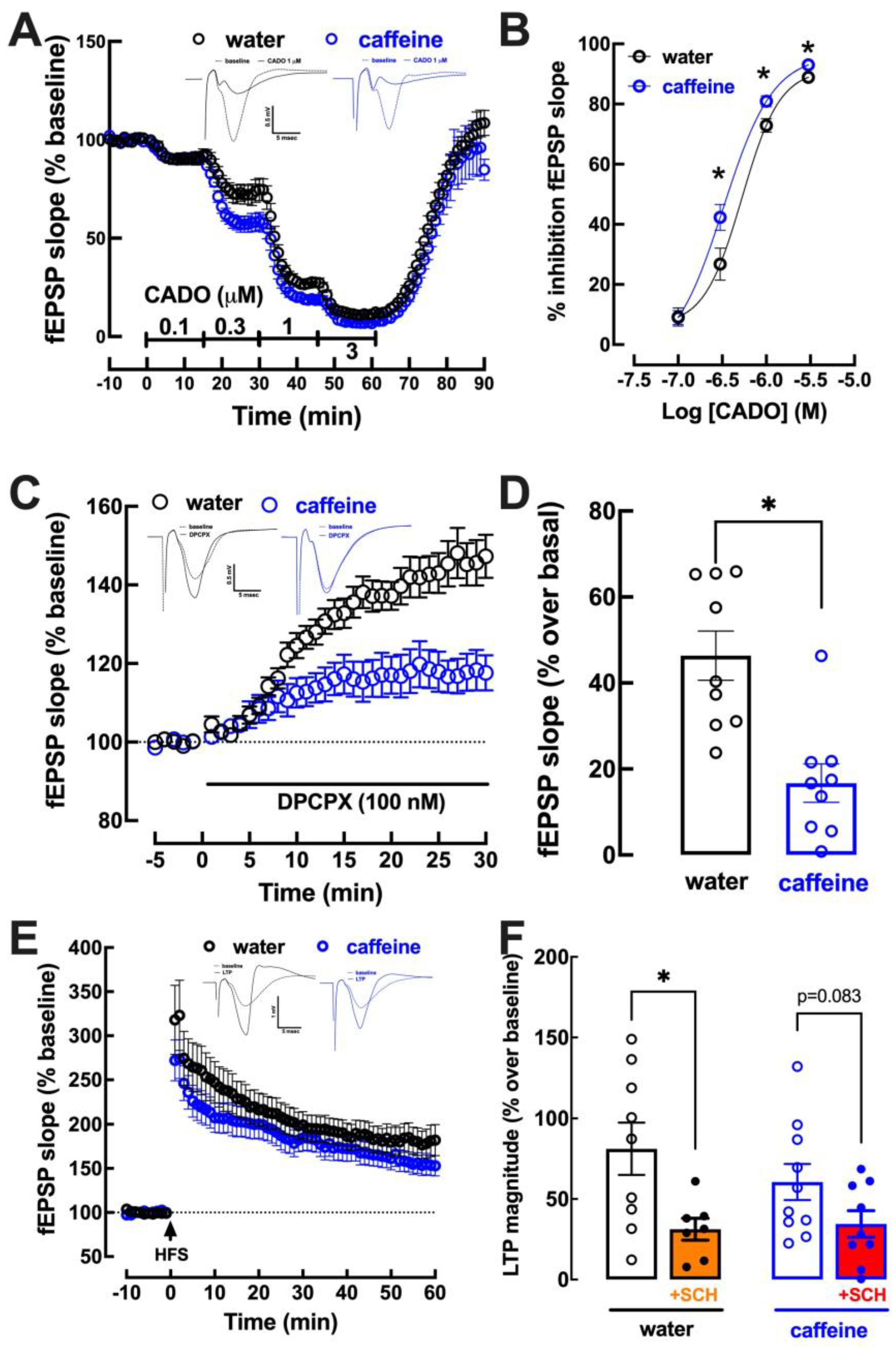
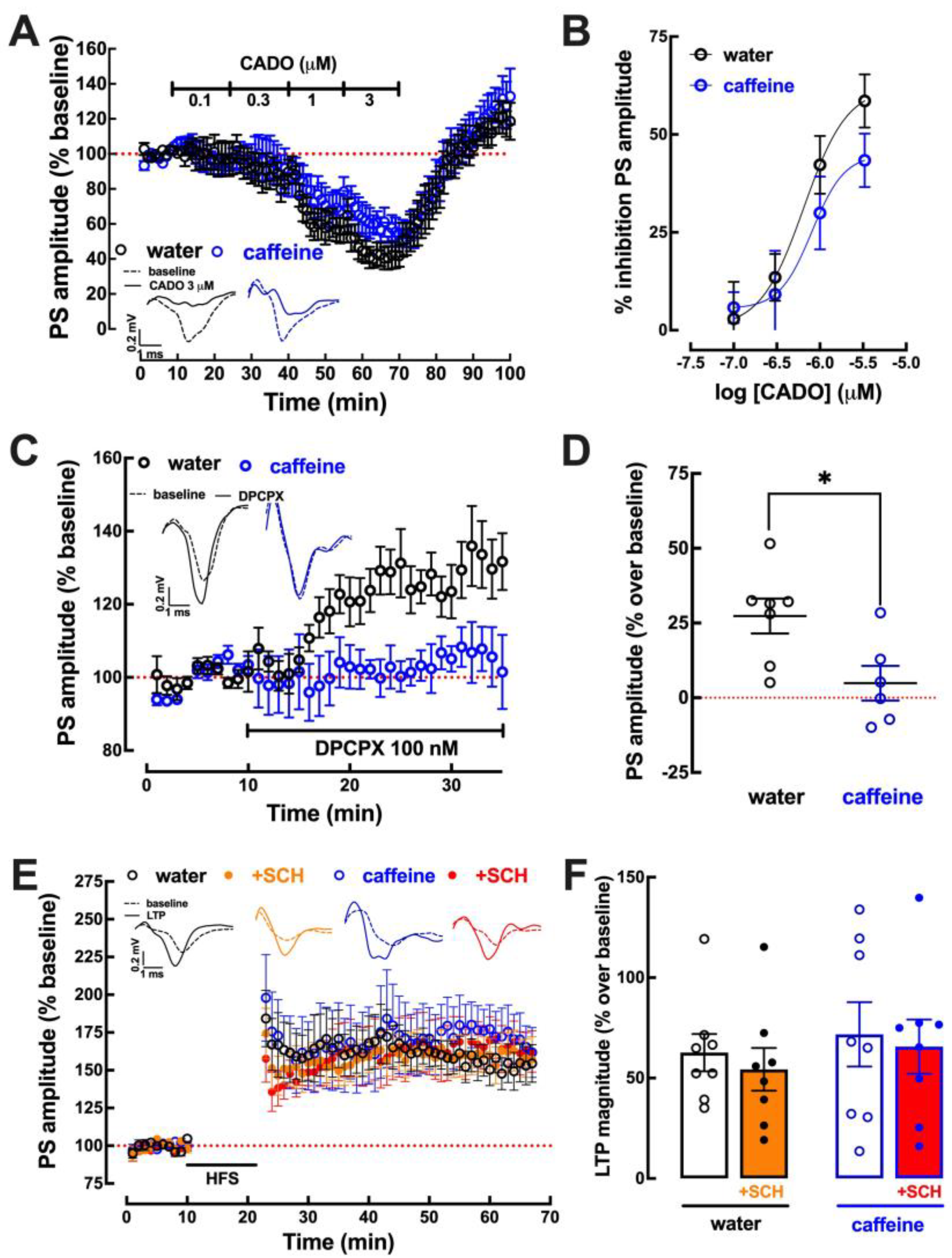
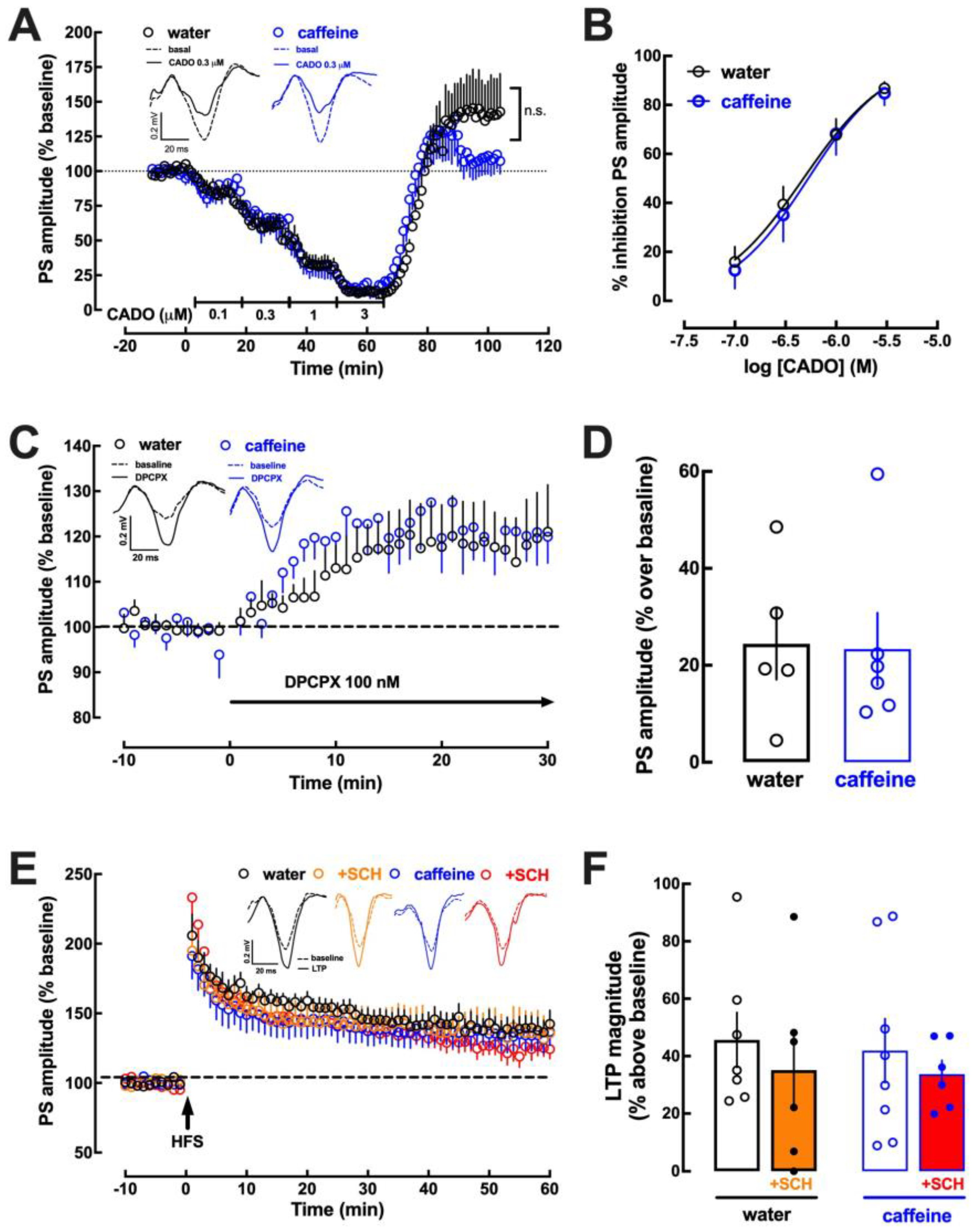
2.3. Impact of Caffeine Intake on Adenosine Modulation of Synaptic Transmission and Plasticity in the Hippocampus
2.4. Impact of Caffeine Intake on Adenosine Modulation of Synaptic Transmission and Plasticity in the Prefrontal Cortex
2.5. Impact of Caffeine Intake on Adenosine Modulation of Synaptic Transmission and Plasticity in the Amygdala
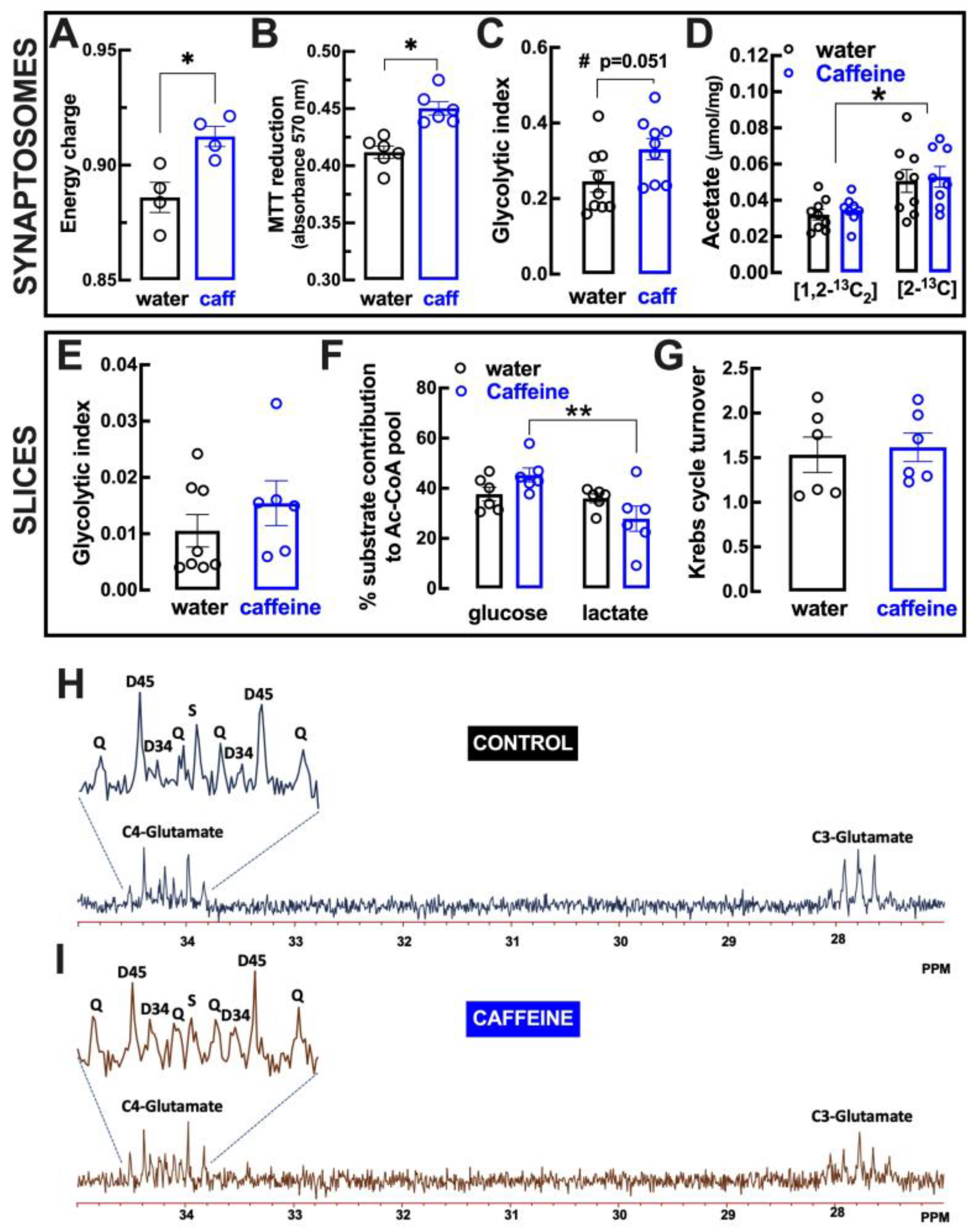
2.6. Impact of Caffeine Intake on Metabolic Features of Cortical Synapses
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs and Treatments
4.3. Behavioral Analysis
4.4. Caffeine Quantification
4.5. Slice Electrophysiology
4.6. Energy Charge, Redox Potential and Primary Metabolism in Synaptosomes
4.7. Superfusion of Cortical Slices
4.8. Nuclear Magnetic Resonance (NMR) Spectroscopy
4.9. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A1R | adenosine A1 receptors |
| A2AR | adenosine A2A receptors |
| ACSF | artificial cerebrospinal fluid |
| CADO | 2-chloroadenosine |
| DPCPX | 1,3-dipropyl-8-cyclopentylxanthine |
| EPSP | excitatory post-synaptic potential |
| LTP | long-term potentiation |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NMR | nuclear magnetic resonance |
| PCA | perchloric acid |
| PS | population spike |
| RT | room temperature |
| SCH58261 | 2-(2-furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine |
References
- Freedman, N.D.; Park, Y.; Abnet, C.C.; Hollenbeck, A.R.; Sinha, R. Association of coffee drinking with total and cause-specific mortality. N. Engl. J. Med. 2012, 366, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, R.M.; Hu, F.B.; Willett, W.C. Coffee, caffeine, and health. N. Engl. J. Med. 2020, 383, 369–378. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between caffeine intake and all-cause and cause-specific mortality: A population-based prospective cohort study. Mayo Clin. Proc. 2017, 92, 1190–1202. [Google Scholar] [CrossRef]
- Di Maso, M.; Boffetta, P.; Negri, E.; La Vecchia, C.; Bravi, F. Caffeinated coffee consumption and health outcomes in the US population: A dose-response meta-analysis and estimation of disease cases and deaths avoided. Adv. Nutr. 2021, 12, 1160–1176. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.Y.D.; Dobrachinski, F.; Silva, H.B.; Lopes, J.P.; Gonçalves, F.Q.; Soares, F.A.A.; Porciúncula, L.O.; Andrade, G.M.; Cunha, R.A.; Tomé, A.R. Neuromodulation and neuroprotective effects of chlorogenic acids in excitatory synapses of mouse hippocampal slices. Sci. Rep. 2021, 11, 10488. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Bättig, K.; Holmén, J.; Nehlig, A.; Zvartau, E.E. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 1999, 51, 83–133. [Google Scholar]
- Lopes, J.P.; Pliássova, A.; Cunha, R.A. The physiological effects of caffeine on synaptic transmission and plasticity in the mouse hippocampus selectively depend on adenosine A1 and A2A receptors. Biochem. Pharmacol. 2019, 166, 313–321. [Google Scholar] [CrossRef]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A2a receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Xu, K.; Oztas, E.; Petzer, J.P.; Castagnoli, K.; Castagnoli, N., Jr.; Chen, J.F. Neuroprotection by caffeine and more specific A2A receptor antagonists in animal models of Parkinson’s disease. Neurology 2003, 61, S55–S61. [Google Scholar] [CrossRef]
- Rudolphi, K.A.; Keil, M.; Fastbom, J.; Fredholm, B.B. Ischaemic damage in gerbil hippocampus is reduced following upregulation of adenosine A1 receptors by caffeine treatment. Neurosci Lett. 1989, 103, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R.; Peeling, J.; Lesiuk, H.J.; Brownstone, R.M.; Rydzy, M.; Saunders, J.K.; Geiger, J.D. The effects of caffeine on ischemic neuronal injury as determined by magnetic resonance imaging and histopathology. Neuroscience 1991, 42, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, V.; Johansson, B.; Fredholm, B.B. Long-term caffeine treatment leads to a decreased susceptibility to NMDA-induced clonic seizures in mice without changes in adenosine A1 receptor number. Brain Res. 1993, 612, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Rigoulot, M.A.; Leroy, C.; Koning, E.; Ferrandon, A.; Nehlig, A. Prolonged low-dose caffeine exposure protects against hippocampal damage but not against the occurrence of epilepsy in the lithium-pilocarpine model in the rat. Epilepsia 2003, 44, 529–535. [Google Scholar] [CrossRef]
- Cognato, G.P.; Agostinho, P.M.; Hockemeyer, J.; Müller, C.E.; Souza, D.O.; Cunha, R.A. Caffeine and an adenosine A2A receptor antagonist prevent memory impairment and synaptotoxicity in adult rats triggered by a convulsive episode in early life. J. Neurochem. 2010, 112, 453–462. [Google Scholar] [CrossRef]
- Li, W.; Dai, S.; An, J.; Li, P.; Chen, X.; Xiong, R.; Liu, P.; Wang, H.; Zhao, Y.; Zhu, M.; et al. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience 2008, 151, 1198–1207. [Google Scholar] [CrossRef]
- Lusardi, T.A.; Lytle, N.K.; Szybala, C.; Boison, D. Caffeine prevents acute mortality after TBI in rats without increased morbidity. Exp. Neurol. 2012, 234, 161–168. [Google Scholar] [CrossRef]
- Duarte, J.M.; Carvalho, R.A.; Cunha, R.A.; Gruetter, R. Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J. Neurochem. 2009, 111, 368–379. [Google Scholar] [CrossRef]
- Duarte, J.M.; Agostinho, P.M.; Carvalho, R.A.; Cunha, R.A. Caffeine consumption prevents diabetes-induced memory impairment and synaptotoxicity in the hippocampus of NONcZNO10/LTJ mice. PLoS One 2012, 7, e21899. [Google Scholar] [CrossRef]
- Gonçalves, N.; Simões, A.T.; Cunha, R.A.; de Almeida, L.P. Caffeine and adenosine A2A receptor inactivation decrease striatal neuropathology in a lentiviral-based model of Machado-Joseph disease. Ann. Neurol. 2013, 73, 655–666. [Google Scholar] [CrossRef]
- Gonçalves, N.; Simões, A.T.; Prediger, R.D.; Hirai, H.; Cunha, R.A.; de Almeida, L.P. Caffeine alleviates progressive motor deficits in a transgenic mouse model of spinocerebellar ataxia. Ann. Neurol. 2017, 81, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Chen, Y.Y.; Wang, X.S.; Wu, S.Z.; Yang, H.M.; Xu, H.Q.; He, J.C.; Wang, X.T.; Chen, J.F.; Zheng, R.Y. Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in Wistar rats. Brain Res. 2010, 1309, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xi, N.N.; Chen, Y.; Shang, X.F.; Hu, Q.; Chen, J.F.; Zheng, R.Y. Chronic caffeine treatment protects against experimental autoimmune encephalomyelitis in mice: Therapeutic window and receptor subtype mechanism. Neuropharmacology 2014, 86, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Pires, V.A.; Pamplona, F.A.; Pandolfo, P.; Prediger, R.D.; Takahashi, R.N. Chronic caffeine treatment during prepubertal period confers long-term cognitive benefits in adult spontaneously hypertensive rats (SHR), an animal model of attention deficit hyperactivity disorder (ADHD). Behav. Brain Res. 2010, 215, 39–44. [Google Scholar] [CrossRef]
- Pandolfo, P.; Machado, N.J.; Köfalvi, A.; Takahashi, R.N.; Cunha, R.A. Caffeine regulates frontocorticostriatal dopamine transporter density and improves attention and cognitive deficits in an animal model of attention deficit hyperactivity disorder. Eur. Neuropsychopharmacol. 2013, 23, 317–328. [Google Scholar] [CrossRef]
- Pechlivanova, D.M.; Tchekalarova, J.D.; Alova, L.H.; Petkov, V.V.; Nikolov, R.P.; Yakimova, K.S. Effect of long-term caffeine administration on depressive-like behavior in rats exposed to chronic unpredictable stress. Behav. Pharmacol. 2012, 23, 339–347. [Google Scholar] [CrossRef]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.; Porciúncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef]
- Machado, D.G.; Lara, M.V.S.; Dobler, P.B.; Almeida, R.F.; Porciúncula, L.O. Caffeine prevents neurodegeneration and behavioral alterations in a mice model of agitated depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 98, 109776. [Google Scholar] [CrossRef]
- Cunha, R.A.; Agostinho, P.M. Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J. Alzheimers Dis. 2010, 20, S95–S116. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Dzhala, V.; Desfreres, L.; Melyan, Z.; Ben-Ari, Y.; Khazipov, R. Epileptogenic action of caffeine during anoxia in the neonatal rat hippocampus. Ann. Neurol. 1999, 46, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Esmaili, Z.; Heydari, A. Effect of acute caffeine administration on PTZ-induced seizure threshold in mice: Involvement of adenosine receptors and NO-cGMP signaling pathway. Epilepsy Res. 2019, 149, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W. The effects of selective A1 and A2a adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res. 1995, 705, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Al Moutaery, K.; Al Deeb, S.; Ahmad Khan, H.; Tariq, M. Caffeine impairs short-term neurological outcome after concussive head injury in rats. Neurosurgery 2003, 53, 704–711. [Google Scholar] [CrossRef]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.C.; Lien, S.H.; Chang, J.H.; Yang, S.C. Caffeine alters resting-state functional connectivity measured by blood oxygenation level-dependent MRI. NMR Biomed. 2014, 27, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, S.H.; Kim, S.H.; Kim, S.H.; Hwang, J.; Kim, J.G.; Han, K.; Kim, J.B. Drinking coffee enhances neurocognitive function by reorganizing brain functional connectivity. Sci. Rep. 2021, 11, 14381. [Google Scholar] [CrossRef]
- Magalhães, R.; Picó-Pérez, M.; Esteves, M.; Vieira, R.; Castanho, T.C.; Amorim, L.; Sousa, M.; Coelho, A.; Fernandes, H.M.; Cabral, J.; et al. Habitual coffee drinkers display a distinct pattern of brain functional connectivity. Mol. Psychiatry 2021, 26, 6589–6598. [Google Scholar] [CrossRef]
- Nehlig, A.; Daval, J.L.; Boyet, S.; Vert, P. Comparative effects of acute and chronic administration of caffeine on local cerebral glucose utilization in the conscious rat. Eur. J. Pharmacol. 1986, 129, 93–103. [Google Scholar] [CrossRef]
- Dager, S.R.; Layton, M.E.; Strauss, W.; Richards, T.L.; Heide, A.; Friedman, S.D.; Artru, A.A.; Hayes, C.E.; Posse, S. Human brain metabolic response to caffeine and the effects of tolerance. Am. J. Psychiatry 1999, 156, 229–237. [Google Scholar] [CrossRef]
- Paiva, I.; Cellai, L.; Meriaux, C.; Poncelet, L.; Nebie, O.; Saliou, J.M.; Lacoste, A.S.; Papegaey, A.; Drobecq, H.; Le Gras, S.; et al. Caffeine intake exerts dual genome-wide effects on hippocampal metabolism and learning-dependent transcription. J. Clin. Investig. 2022, 132, e149371. [Google Scholar] [CrossRef] [PubMed]
- Conlay, L.A.; Conant, J.A.; deBros, F.; Wurtman, R. Caffeine alters plasma adenosine levels. Nature 1997, 389, 136. [Google Scholar] [CrossRef] [PubMed]
- Marangos, P.J.; Boulenger, J.P.; Patel, J. Effects of chronic caffeine on brain adenosine receptors: Regional and ontogenetic studies. Life Sci. 1984, 34, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Georgiev, V.; Lindström, K.; Fredholm, B.B. A1 and A2A adenosine receptors and A1 mRNA in mouse brain: Effect of long-term caffeine treatment. Brain Res. 1997, 762, 153–164. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K. Synaptic dysfunction in depression: Potential therapeutic targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Pickett, E.; Spires-Jones, T.L. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 2016, 28, 72–84. [Google Scholar] [CrossRef]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Adami, P.V.M.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef]
- Barthet, G.; Mulle, C. Presynaptic failure in Alzheimer’s disease. Prog. Neurobiol. 2020, 194, 101801. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and brain function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef]
- Rebola, N.; Pinheiro, P.C.; Oliveira, C.R.; Malva, J.O.; Cunha, R.A. Subcellular localization of adenosine A1 receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003, 987, 49–58. [Google Scholar] [CrossRef]
- Rebola, N.; Canas, P.M.; Oliveira, C.R.; Cunha, R.A. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 2005, 132, 893–903. [Google Scholar] [CrossRef]
- Serchov, T.; Clement, H.W.; Schwarz, M.K.; Iasevoli, F.; Tosh, D.K.; Idzko, M.; Jacobson, K.A.; de Bartolomeis, A.; Normann, C.; Biber, K.; et al. Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of homer1a. Neuron 2015, 87, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, D.A.; Harrison, E.; Stewart, M.G. Synapses in hippocampus occupy only 1–2% of cell membranes and are spaced less than half-micron apart: A quantitative ultrastructural analysis with discussion of physiological implications. Neuropharmacology 1998, 37, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Nagai, J.; Yu, X.; Papouin, T.; Cheong, E.; Freeman, M.R.; Monk, K.R.; Hastings, M.H.; Haydon, P.G.; Rowitch, D.; Shaham, S.; et al. Behaviorally consequential astrocytic regulation of neural circuits. Neuron 2021, 109, 576–596. [Google Scholar] [CrossRef]
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.; Jamison, S.; Robin, L.M.; Zhao, Z.; Martin, E.D.; Aguilar, J.; Benneyworth, M.A.; Marsicano, G.; Araque, A. Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci. 2017, 20, 1540–1548. [Google Scholar] [CrossRef]
- Kerkhofs, A.; Canas, P.M.; Timmerman, A.J.; Heistek, T.S.; Real, J.I.; Xavier, C.; Cunha, R.A.; Mansvelder, H.D.; Ferreira, S.G. Adenosine A2A receptors control glutamatergic synaptic plasticity in fast spiking interneurons of the prefrontal cortex. Front. Pharmacol. 2018, 9, 133. [Google Scholar] [CrossRef]
- Coelho, J.E.; Rebola, N.; Fragata, I.; Ribeiro, J.A.; de Mendonça, A.; Cunha, R.A. Hypoxia-induced desensitization and internalization of adenosine A1 receptors in the rat hippocampus. Neuroscience 2006, 138, 1195–1203. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Cunha, R.A.; de Mendonça, A.; Ribeiro, J.A. Modification of adenosine modulation of synaptic transmission in the hippocampus of aged rats. Br. J. Pharmacol. 2000, 131, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef]
- Goosens, K.A.; Maren, S. Long-term potentiation as a substrate for memory: Evidence from studies of amygdaloid plasticity and Pavlovian fear conditioning. Hippocampus 2002, 12, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.J.; Andres-Beck, L.G.; Nett, K.E.; Oliveros, A.; Choi, S.; Veldic, M.; Choi, D.S. Chronic caffeine exposure in adolescence promotes diurnal, biphasic mood-cycling and enhanced motivation for reward in adult mice. Behav. Brain Res. 2019, 370, 111943. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.F.; Deboer, T.; Landolt, H.P. Adenosine, caffeine, and sleep-wake regulation: State of the science and perspectives. J. Sleep Res. 2022, 31, e13597. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S. Mechanisms of the psychostimulant effects of caffeine: Implications for substance use disorders. Psychopharmacology 2016, 233, 1963–1979. [Google Scholar] [CrossRef]
- Wu, L.; Meng, J.; Shen, Q.; Zhang, Y.; Pan, S.; Chen, Z.; Zhu, L.Q.; Lu, Y.; Huang, Y.; Zhang, G. Caffeine inhibits hypothalamic A1R to excite oxytocin neuron and ameliorate dietary obesity in mice. Nat. Commun. 2017, 8, 15904. [Google Scholar] [CrossRef]
- Pelligrino, D.A.; Xu, H.L.; Vetri, F. Caffeine and the control of cerebral hemodynamics. J. Alzheimers Dis. 2010, 20, S51–S62. [Google Scholar] [CrossRef]
- Chen, X.; Ghribi, O.; Geiger, J.D. Caffeine protects against disruptions of the blood-brain barrier in animal models of Alzheimer’s and Parkinson’s diseases. J. Alzheimers Dis. 2010, 20, S127–S141. [Google Scholar] [CrossRef]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A receptors in the brain: Current research and their role in neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [PubMed]
- Daval, J.L.; Deckert, J.; Weiss, S.R.; Post, R.M.; Marangos, P.J. Upregulation of adenosine A1 receptors and forskolin binding sites following chronic treatment with caffeine or carbamazepine: A quantitative autoradiographic study. Epilepsia 1989, 30, 26–33. [Google Scholar] [CrossRef]
- Fukata, Y.; Fukata, M. Epilepsy and synaptic proteins. Curr. Opin. Neurobiol. 2017, 45, 1–8. [Google Scholar] [CrossRef]
- Van Koert, R.R.; Bauer, P.R.; Schuitema, I.; Sander, J.W.; Visser, G.H. Caffeine and seizures: A systematic review and quantitative analysis. Epilepsy Behav. 2018, 80, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, P.; Schirinzi, T.; Meringolo, M.; Mercuri, N.B.; Pisani, A. Centrality of early synaptopathy in Parkinson’s disease. Front. Neurol. 2018, 9, 103. [Google Scholar] [CrossRef]
- Ren, X.; Chen, J.F. Caffeine and Parkinson’s disease: Multiple benefits and emerging mechanisms. Front. Neurosci. 2020, 14, 602697. [Google Scholar] [CrossRef]
- Nortley, R.; Attwell, D. Control of brain energy supply by astrocytes. Curr. Opin. Neurobiol. 2017, 47, 80–85. [Google Scholar] [CrossRef]
- Deliktaş, M.; Ergin, H.; Demiray, A.; Akça, H.; Özdemir, Ö.M.A.; Özdemir, M.B. Caffeine prevents bilirubin-induced cytotoxicity in cultured newborn rat astrocytes. J. Matern. Fetal Neonatal. Med. 2019, 32, 1813–1819. [Google Scholar] [CrossRef]
- Marret, S.; Delpech, B.; Girard, N.; Leroy, A.; Maingonnat, C.; Menard, J.F.; Fessard, C. Caffeine decreases glial cell number and increases hyaluronan secretion in newborn rat brain cultures. Pediatr. Res. 1993, 34, 716–719. [Google Scholar] [CrossRef][Green Version]
- Zipp, F.; Aktas, O. The brain as a target of inflammation: Common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006, 29, 518–527. [Google Scholar] [CrossRef]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Brothers, H.M.; Marchalant, Y.; Wenk, G.L. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neurosci. Lett. 2010, 480, 97–100. [Google Scholar] [CrossRef]
- Ardais, A.P.; Rocha, A.S.; Borges, M.F.; Fioreze, G.T.; Sallaberry, C.; Mioranzza, S.; Nunes, F.; Pagnussat, N.; Botton, P.H.; Cunha, R.A.; et al. Caffeine exposure during rat brain development causes memory impairment in a sex selective manner that is offset by caffeine consumption throughout life. Behav. Brain Res. 2016, 303, 76–84. [Google Scholar] [CrossRef]
- Jee, H.J.; Lee, S.G.; Bormate, K.J.; Jung, Y.S. Effect of caffeine consumption on the risk for neurological and psychiatric disorders: Sex differences in human. Nutrients 2020, 12, 3080. [Google Scholar] [CrossRef]
- Von Borstel, R.W. Biological effects of caffeine: Metabolism. Food Technol. 1983, 37, 39–42. [Google Scholar]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef]
- Dias, L.; Lopes, C.R.; Gonçalves, F.Q.; Nunes, A.; Pochmann, D.; Machado, N.J.; Tomé, A.R.; Agostinho, P.; Cunha, R.A. Crosstalk between ATP-P2X7 and adenosine A2A receptors controlling neuroinflammation in rats subject to repeated restraint stress. Front. Cell Neurosci. 2021, 15, 639322. [Google Scholar] [CrossRef]
- Dellu, F.; Fauchey, V.; Le Moal, M.; Simon, H. Extension of a new two-trial memory task in the rat: Influence of environmental context on recognition processes. Neurobiol. Learn Mem. 1997, 67, 112–120. [Google Scholar] [CrossRef]
- Kovacevic, N.; Henderson, J.T.; Chan, E.; Lifshitz, N.; Bishop, J.; Evans, A.C.; Henkelman, R.M.; Chen, X.J. A Three-dimensional MRI atlas of the mouse brain with estimates of the average and variability. Cereb. Cortex 2005, 15, 639–645. [Google Scholar] [CrossRef]
- Costenla, A.R.; Diógenes, M.J.; Canas, P.M.; Rodrigues, R.J.; Nogueira, C.; Maroco, J.; Agostinho, P.M.; Ribeiro, J.A.; Cunha, R.A.; de Mendonça, A. Enhanced role of adenosine A2A receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur. J. Neurosci. 2011, 34, 12–21. [Google Scholar] [CrossRef]
- Simões, A.P.; Machado, N.J.; Gonçalves, N.; Kaster, M.P.; Simões, A.T.; Nunes, A.; de Almeida, L.P.; Goosens, K.A.; Rial, D.; Cunha, R.A. Adenosine A2A receptors in the amygdala control synaptic plasticity and contextual fear memory. Neuropsychopharmacology 2016, 41, 2862–2871. [Google Scholar] [CrossRef]
- Real, J.I.; Simões, A.P.; Cunha, R.A.; Ferreira, S.G.; Rial, D. Adenosine A2A receptors modulate the dopamine D2 receptor-mediated inhibition of synaptic transmission in the mouse prefrontal cortex. Eur. J. Neurosci. 2018, 47, 1127–1134. [Google Scholar] [CrossRef]
- Anderson, W.W.; Collingridge, G.L. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J. Neurosci. Methods 2007, 162, 346–356. [Google Scholar] [CrossRef]
- Cunha, R.A.; Sebastião, A.M.; Ribeiro, J.A. Ecto-5′-nucleotidase is associated with cholinergic nerve terminals in the hippocampus but not in the cerebral cortex of the rat. J. Neurochem. 1992, 59, 657–666. [Google Scholar] [CrossRef]
- Cunha, R.A.; Almeida, T.; Ribeiro, J.A. Parallel modification of adenosine extracellular metabolism and modulatory action in the hippocampus of aged rats. J. Neurochem. 2001, 76, 372–382. [Google Scholar] [CrossRef]
- Cunha, R.A.; Sebastião, A.M.; Ribeiro, J.A. Separation of adenosine triphosphate and its degradation products in innervated muscle of the frog by reversed phase high-performance liquid chromatography. Chromatographia 1989, 28, 610–612. [Google Scholar] [CrossRef]
- Silva, C.G.; Porciúncula, L.O.; Canas, P.M.; Oliveira, C.R.; Cunha, R.A. Blockade of adenosine A2A receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol. Dis. 2007, 27, 182–189. [Google Scholar] [CrossRef]
- Duarte, J.M.; Cunha, R.A.; Carvalho, R.A. Different metabolism of glutamatergic and GABAergic compartments in superfused hippocampal slices characterized by nuclear magnetic resonance spectroscopy. Neuroscience 2007, 144, 1305–1313. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, C.R.; Oliveira, A.; Gaspar, I.; Rodrigues, M.S.; Santos, J.; Szabó, E.; Silva, H.B.; Tomé, Â.R.; Canas, P.M.; Agostinho, P.; et al. Effects of Chronic Caffeine Consumption on Synaptic Function, Metabolism and Adenosine Modulation in Different Brain Areas. Biomolecules 2023, 13, 106. https://doi.org/10.3390/biom13010106
Lopes CR, Oliveira A, Gaspar I, Rodrigues MS, Santos J, Szabó E, Silva HB, Tomé ÂR, Canas PM, Agostinho P, et al. Effects of Chronic Caffeine Consumption on Synaptic Function, Metabolism and Adenosine Modulation in Different Brain Areas. Biomolecules. 2023; 13(1):106. https://doi.org/10.3390/biom13010106
Chicago/Turabian StyleLopes, Cátia R., Andreia Oliveira, Ingride Gaspar, Matilde S. Rodrigues, Joana Santos, Eszter Szabó, Henrique B. Silva, Ângelo R. Tomé, Paula M. Canas, Paula Agostinho, and et al. 2023. "Effects of Chronic Caffeine Consumption on Synaptic Function, Metabolism and Adenosine Modulation in Different Brain Areas" Biomolecules 13, no. 1: 106. https://doi.org/10.3390/biom13010106
APA StyleLopes, C. R., Oliveira, A., Gaspar, I., Rodrigues, M. S., Santos, J., Szabó, E., Silva, H. B., Tomé, Â. R., Canas, P. M., Agostinho, P., Carvalho, R. A., Cunha, R. A., Simões, A. P., Lopes, J. P., & Ferreira, S. G. (2023). Effects of Chronic Caffeine Consumption on Synaptic Function, Metabolism and Adenosine Modulation in Different Brain Areas. Biomolecules, 13(1), 106. https://doi.org/10.3390/biom13010106