1. Introduction
Rice (
Oryza sativa L.) is the foremost staple food feeding half of the world’s population and more than half of China’s population [
1]. By 2050, the global population will be around 9.7 billion, making the global food and energy demand more challenging than ever. The decrease in suitable agricultural land due to several factors, including climatic changes, leads to strategies to improve the rice grain yields without further expanding farmland and environmental damage [
2]. Rice grain yield is a complex and highly variable quantitative trait that depends on three fundamental components: the weight of grain, the number of effective panicles per unit area, and the grain number per panicle (GNPP), each controlled by multiple genes (QTL). The GNPP has been revealed as the primary contributor to total yield per unit area and is essential for its improvement [
3].
The physiological network and genes underlying panicle and grain formation have been thoroughly studied. The primary mechanism includes the initiation during which the shoot apical meristem (SAM) is transformed to inflorescence meristem (IM), the formation of rachis branches, and spikelet specialization successively. These steps are vital in the panicle architecture and GNPP [
4,
5]. Recent studies highlighted several hormones, including cytokinin (CK), gibberellin acid (GA), abscisic acid (ABA), and ethylene (ET), that interfere to regulate the transcriptional and post-transcriptional activities of genes in rice, acting in different pathways involved in these processes [
6,
7,
8,
9]. Numbers of genes have been identified from high-throughput QTL mapping, and within the past ten years, the number of the QTLs related to the GNPP doubled from 315 QTL [
10], 369 [
11] to 722 QTL (
http://www.gramene.org; accessed on 10 April 2022) distributed along the 12 chromosomes in the rice genome. The first QTL grain number 1a (
GN1a) was mapped and cloned on chromosome1 coding for a cytokinin oxidase (
OsCKX2) which repressed the GNPP by reducing CK content [
12], and with aberrant panicle organization1 (
APO1) controlling the proliferation of cells in the meristem [
4]. These works set the foundation for GNPP-related gene mapping. CK is essential in rachis branch formation, and the high expression of
OsCKX2 weakened CK signaling in IM leads to few rachis branches and GNPP [
13,
14]. Besides regulating GA homeostasis,
OsCYP71D8L negatively controls the length of the panicles, and the rachis branch numbers, and subsequently the rice GNPP [
15]. Major QTL which controls, directly or indirectly, the GNPP and related traits include: heading date 7 (
Ghd7), which increases the differentiation period [
16];
LAX regulating the rachis branches initiation [
17]; dense erect panicle1 (
DEP1) controlling the secondary branches of inflorescence and GNPP [
18]; grain number 4-1
(GN4-1) and grain number per panicle1 (
GNP1) regulate the number of rachis branches and GNPP [
10,
19]; LONELY GUY
(LOG) positively regulates GNPP by controlling the concentration and distribution of CK [
13]; and Pyrabactin Resistance-Like
(PYL) positively regulates ABA signaling and negatively regulates GNPP in rice [
7].
The development of new molecular technology allowed the transition from the conventional method, and the integration of omics tools facilitated the identification of new functional genes and pathways. During the last decade, most of the studies for mapping QTL relied on traditional methods, which involve the construction of mapping the population through the F
2 generation, recombinant inbred lines (RIL), and near-isogenic lines (NIL), followed by the drawing of a genetic linkage map, and cloning. The following is a strategy by which numerous genes have been mapped and cloned. The most relevant include
qGN1c mapped using a set of chromosomal segment substitution lines (CSSLs) [
20],
SPP1 identified through NILs controlling the number of spikelets [
21],
qTGW3.2 for grain weight in rice using RIL [
22],
GNP1 [
19], and
qgnp7(
t) [
23] governing GNPP revealed through map-based cloning, and two steps substitution mapping, respectively. However, traditional gene mapping is tedious and time-consuming; most QTLs are inconsistent and span large genomic regions, making identifying candidate genes for a trait more challenging. New strategies which combine traditional methods and emerging technologies such as high-throughput whole-genome sequencing (NGS), alternative sequencing analysis, mapping by sequencing, bulked segregants analysis (BSA), RNA-sequencing (RNA-seq), and genome-wide association study (GWAS), have accelerated the identification of candidate genes for complex characters, and several studies have been successfully conducted to identify a new gene [
2,
24,
25,
26,
27]. BSA-Seq and RNA profiling were coupled to reveal the pathway and genes associated with the heading type in Chinese cabbage [
28], chilling tolerance in rice [
24], and plant architecture in
Brassica napus [
29]. Likewise, significant gene candidates have been identified by combining sequencing and a metabolome analysis in albino jackfruit seedlings [
30] and anthocyanin in cucumber fruit skin [
31].
Although comprehensive studies on QTL mapping for the GNPP have been carried out, discovering new QTL remains an excellent value for molecular and practical breeding. Considering the complexity of grain number related-genes and the limitation of traditional QTL mapping technology, in this study, we successfully integrated the BSA-seq, RNA-Seq, and metabolome analysis to identify two new genes linked to the GNPP.
4. Discussion
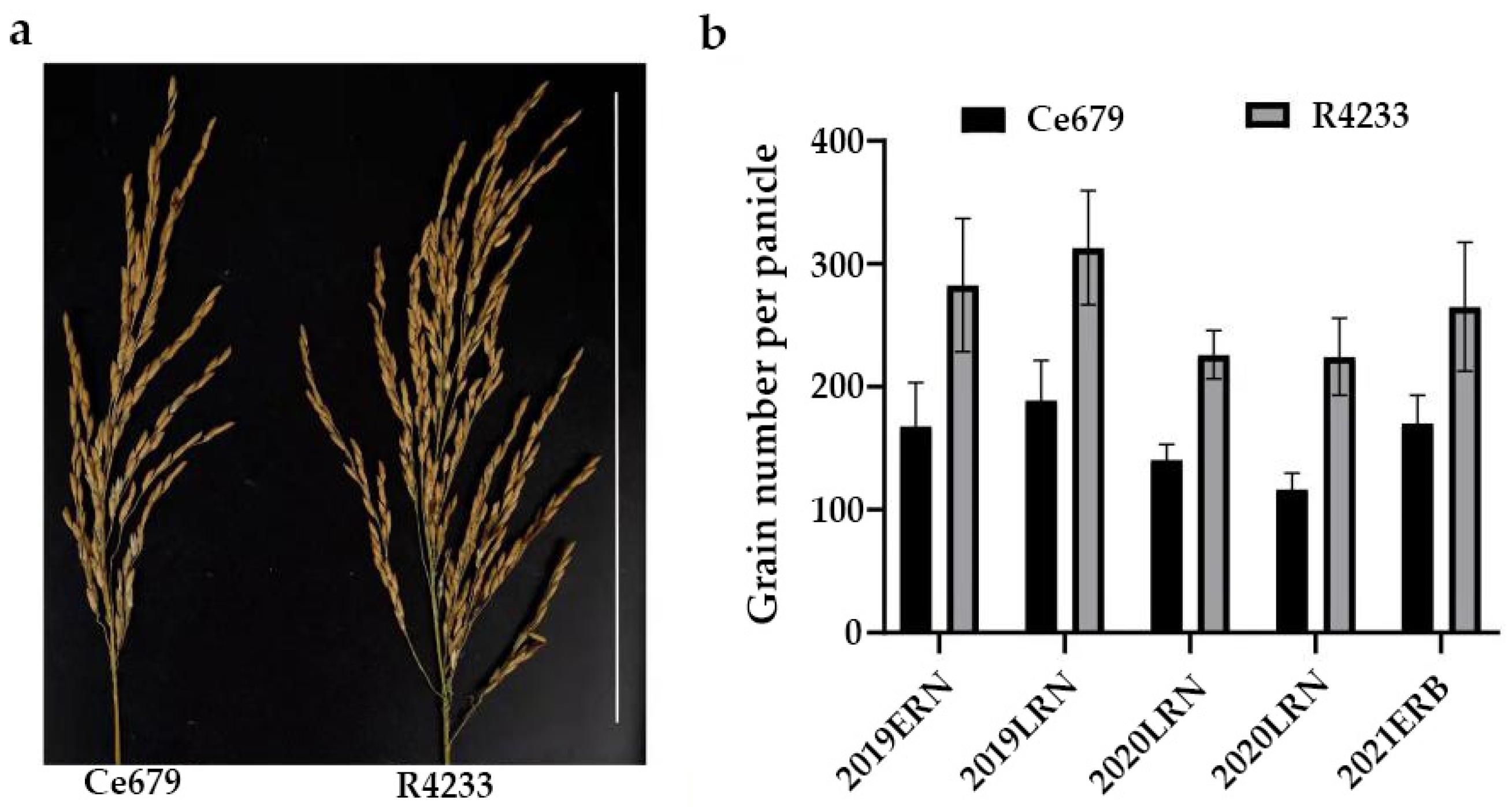
The GNPP is a primary agronomic trait that directly determines rice grain yield. Rice grain constituted one of the principal targets during artificial selection, and improving this trait has been integrated into the selection strategies by breeders and molecular biologists. It is profoundly affected by the panicle architecture-related components, such as the length of the central rachis and the number of primary and secondary rachis branches. The current study investigated two indica rice cultivars: Ce679 (low GNPP) and R4233 (high GNPP). R4233 is a restorer line developed from the successive backcrossing of Ce679. After evaluation, the results showed that it has gained some superior agronomic traits from its parents in addition to Pi9 for disease resistance. The combination of blast disease resistance and grain yield is essential in breeding. Although other traits such as flag leaf length and width that also showed a significant difference in this study are important for photosynthesis, the grain number remains the ultimate trait that can directly impact the food security. Thus, it is reasonable for breeders to seek a cultivar with a better yield.
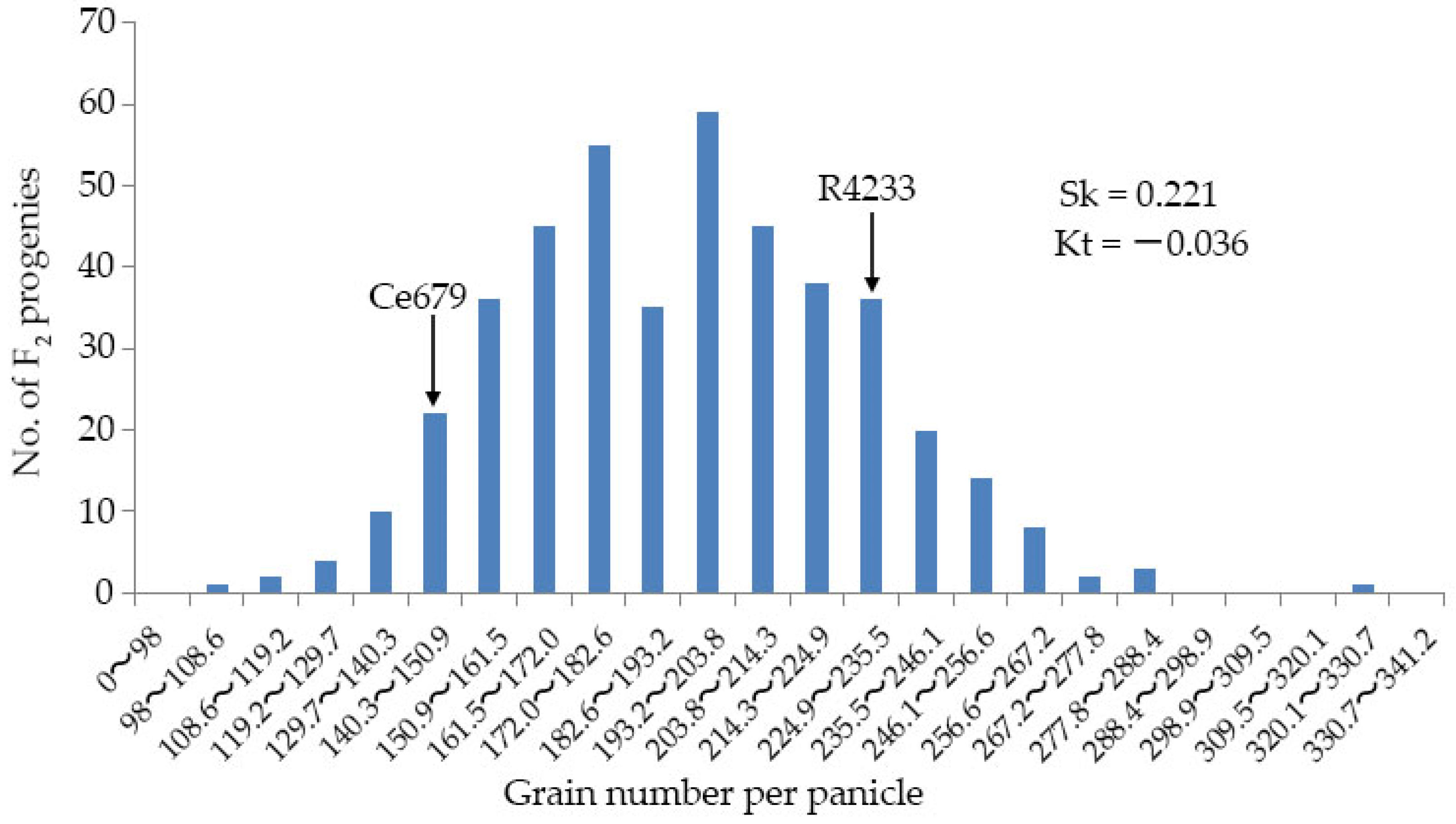
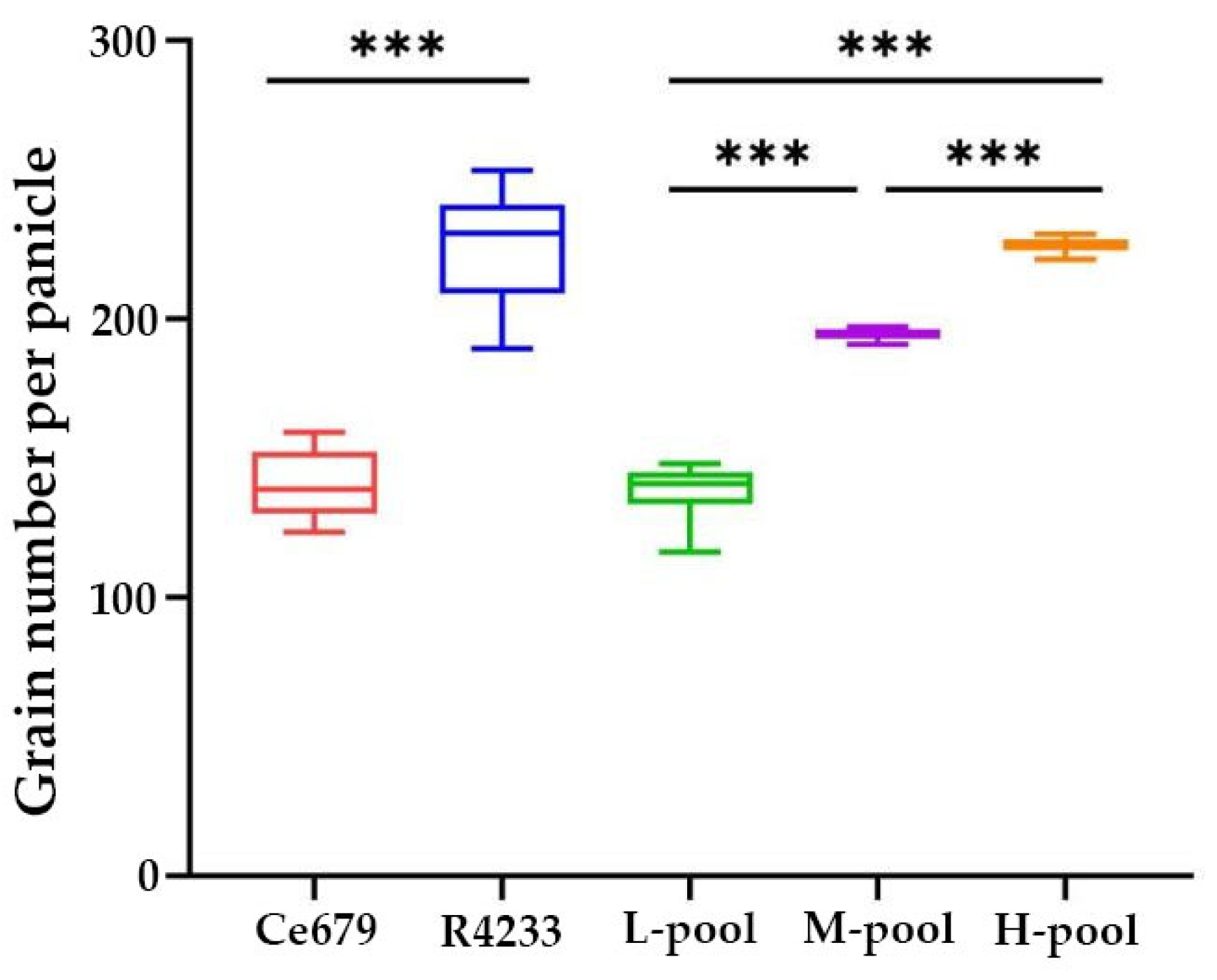
Since environmentally stable QTLs could be applied in a wide range of circumstances, the segregating population for QTL identification related to the GNPP was developed in the experimental field of Guangxi University and in Bobai Southern China during different growing periods. The quantification of different agronomic traits revealed remarkable differences in the panicle structure in R4233 compared with Ce679. We observed similar results for the GNPP in R4233 regardless of the location, which is important in breeding. The number of secondary branches per panicle was significantly higher in R4233 than the number of primary branches in Ce679 (
Table 2), suggesting that the secondary branches significantly contributed to the total branches and grain number. In our results, secondary branches contributed 82.3%, 86.6%, and 87.1% of the total branches in Ce679, R4233, and F1, respectively. The numbers of primary rachis branches and secondary rachis branches between Ce679 and R4233 were significantly different, but no difference was found concerning the length of the central rachis. Therefore, the GNPP in R4233 was significantly higher than Ce679 (
Figure 1). Conversely, the number of primary branches per panicle was not significantly different in F1 compared to Ce679. At the same time, this difference was significant in the number of secondary branches per panicle and GNPP. This result indicated that the development of the secondary branch per panicle might be the primary component that affects the GNPP in Ce679 and R4233, causing the grain number to double in R4233 compared with Ce679. In rice, panicle development is critical in grain production; mainly the transition to the reproductive phase, which involves the transformation of the shoot apical meristem (SAM) into the inflorescence meristem (IM). During this period, several lateral meristems (LM) initiate and grow as primary rachis branches (PRBs), which further produce next-order LM that grow as secondary rachis branches (SRBs). Later, the lateral spikelet will differentiate from the new LM, and the terminal spikelets are converted from rachis branch meristems. Several studies reported a positive regulation of the GNPP through the development of only the secondary rachis branch [
11,
19,
46,
47], or simultaneously primary and secondary rachis branch-related genes [
13,
18,
48,
49]. However, none of the previously identified genes have been found to directly relate to the number of grains in the present study, suggesting another mechanism contributing to the GNPP.
In a recent study, Guo et al. [
24] reported that the completion of mapping and cloning of the
Ctb1 gene related to the chilling resistance in rice took over 16 years with conventional breeding and intensive labor [
24]. This is because it involved several steps such as fine mapping, map-based cloning, and high-density linkage maps [
24]. This procedure has been simplified over the last decades with the development of high-throughput technologies, and the researcher can save much time, labor, and money. Lately, several “omics” techniques, which include, genomics, transcriptomics, proteomics, and metabolomics, have become effective technologies for plant functional genomic and breeding research [
50]. Our research took advantage of this technology to accelerate the identification of genes and pathways which take 4 years where more time is needed. Omics research is also undergoing a shift from a single-omics to a large-scale multi-omics approach. In most of the previous research, a single-omics approach was used [
51]. For instance, researchers employed BSA-Seq as an effective approach to identify the minor genes with various sequencing depths because of the precision and sensitivity of the sequencing. RNA-Seq was used to identify new genes and SNP loci, measure gene expression levels, and calculate fold changes in DEG. Proteomics was simply to detect gene products and metabolomics measured how proteins are expressed, and the pathway of metabolites, which influence how genes display the biochemical phenotype of the cell. The limitation of the single-omics approach is that it may not help to obtain a deeper understanding of the fundamental biological processes, a more accurate prediction of the response variable, and gain further insight into mechanistic aspects of the system [
52]. So, an integration of the different omics approaches is required to envisage overall comprehension of the gene, product, and phenotype under a set of conditions. This approach is gaining more interest and has been successfully used. Recently, in many crops, physiological activity, agronomic traits, responses to biotic and abiotic stress, and yield have been well documented via the use of integrative omic approaches. This robust approach has superseded conventional phenomics, resulting in a formidable tool for crop genetics and breeding sciences [
53]. Combining genetic data with prospective phenotyping technologies may offer information on complicated features to help improve crops [
54]. The combination of BSA-seq and RNA-seq allowed the identification of the candidate for the agronomic trait [
24,
28,
29,
55]. This approach enhanced the accurate identification of gene candidates for the grain number in rice [
26], where BSA-Seq alone would identify only the candidate interval. The combination of a transcriptome and metabolome analysis helped to predict molecular mechanisms of genes, and gene networks in crop science. For example, Wang et al. (2019) deciphered the complex response mechanisms involved in heat stress in pepper [
56]. The large number of specific response of genes and metabolites highlighted the complex regulatory mechanisms and metabolite networks related to various pathways associated with cold stress after combining a transcriptome and metabolome analysis in tobacco [
57] and wheat [
54]. Several gene and metabolite networks have been revealed as essential for melatonin-mediated salt tolerance in rice through transcriptome and metabolome investigations [
58]. However, the main inconvenience remains the huge datasets generated, which require bioinformatic tools for data mining and organizing [
59]. Furthermore, in some case it is necessary to carry out some additional molecular works for functional validation of the candidate gene such as RNAi and Crispr/cas9.
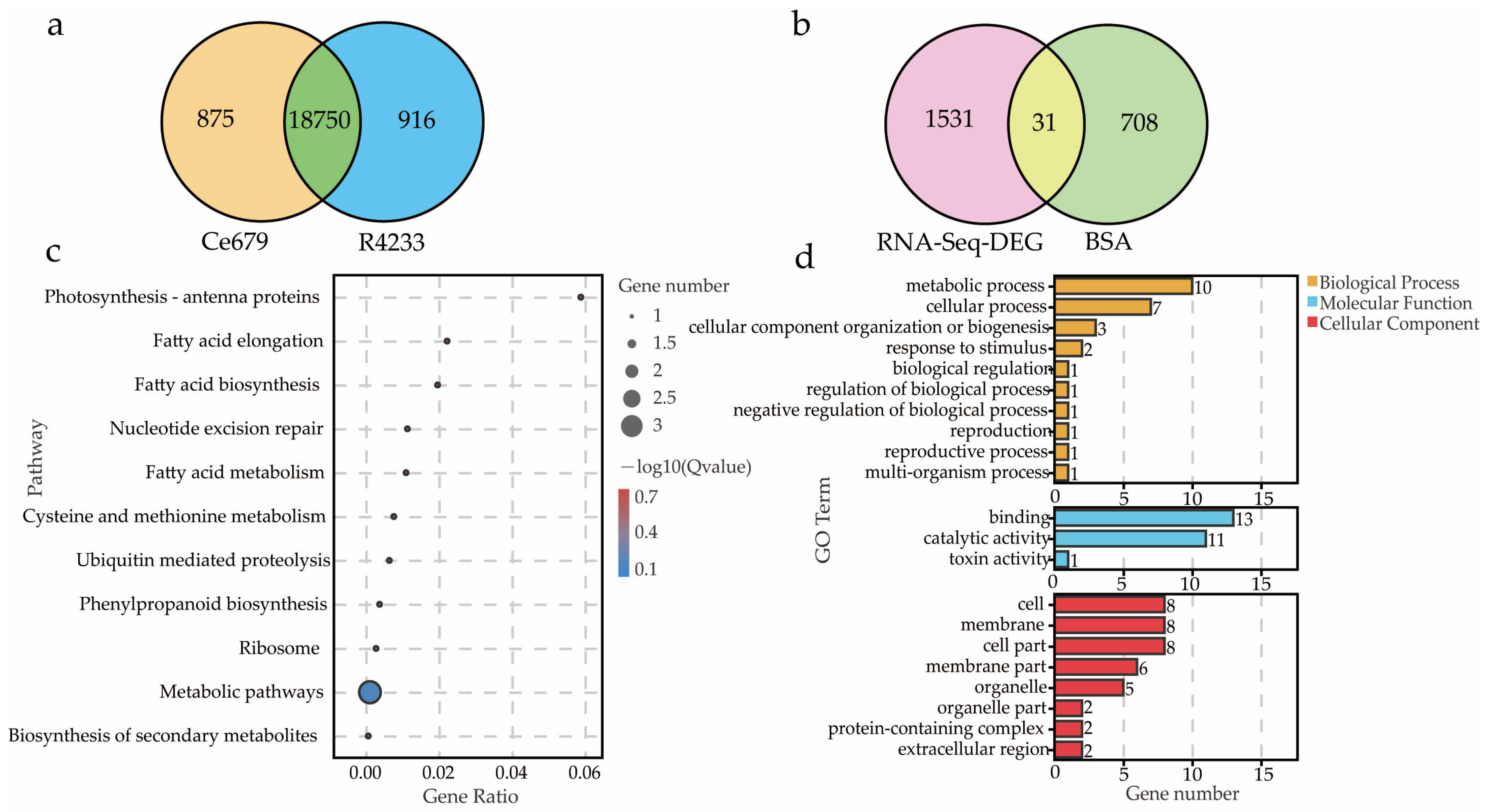
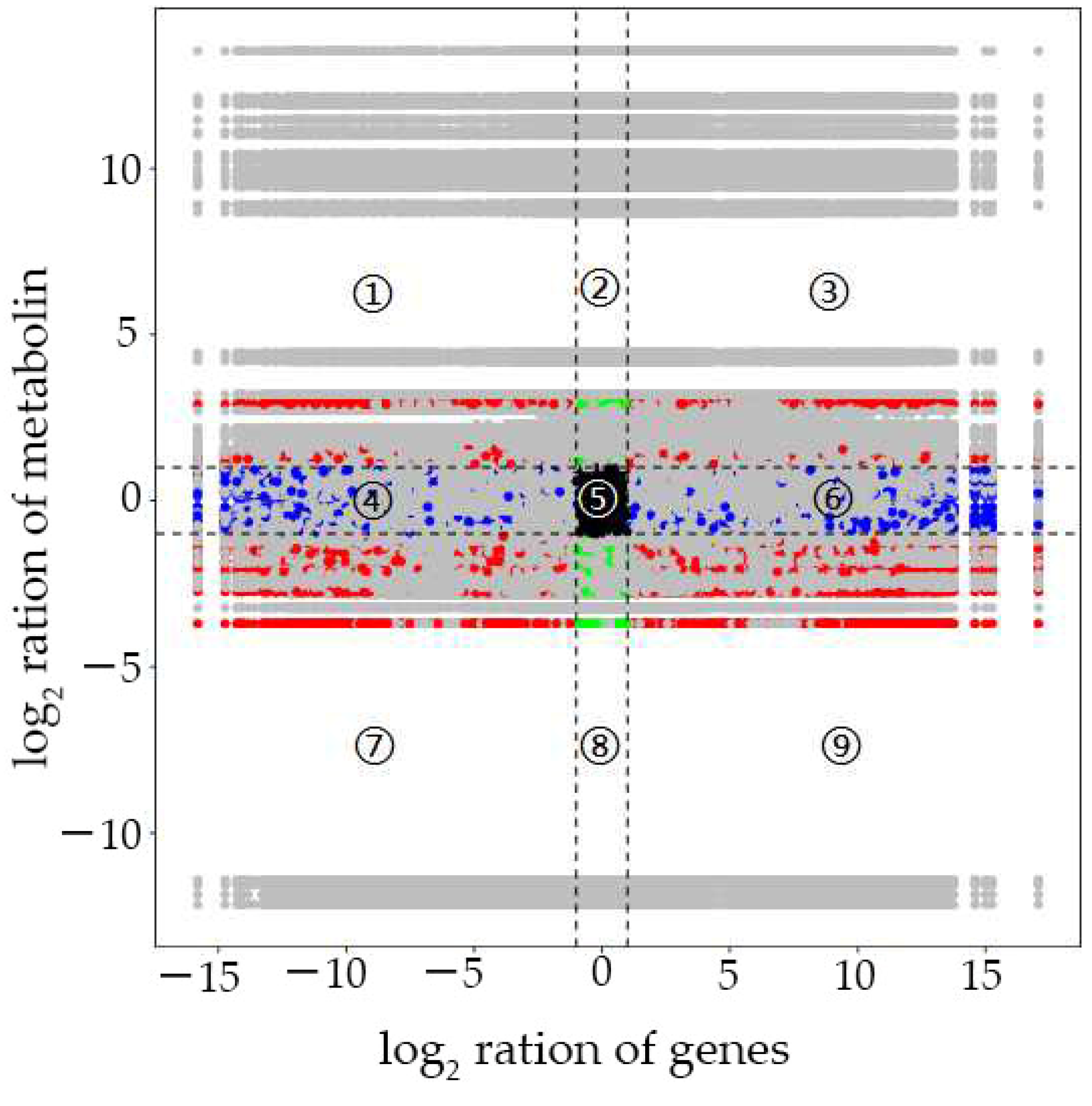
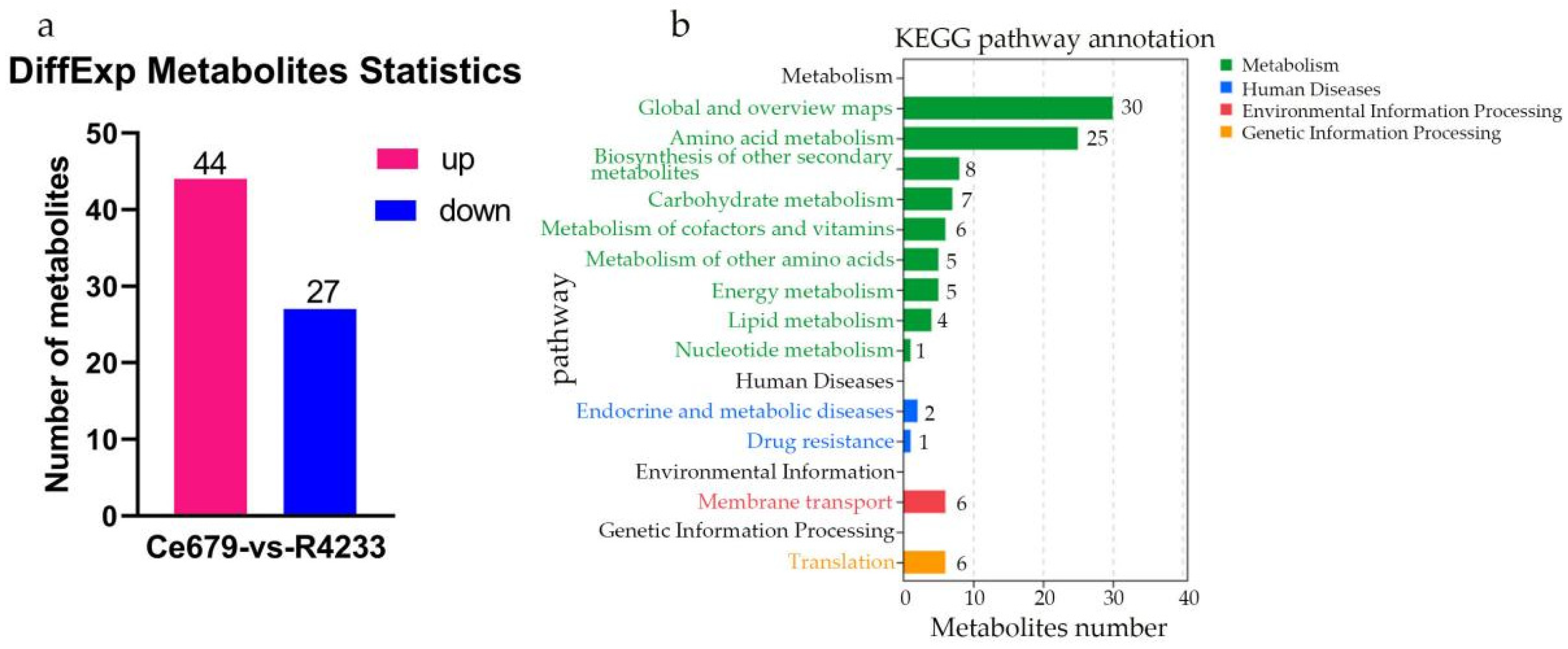
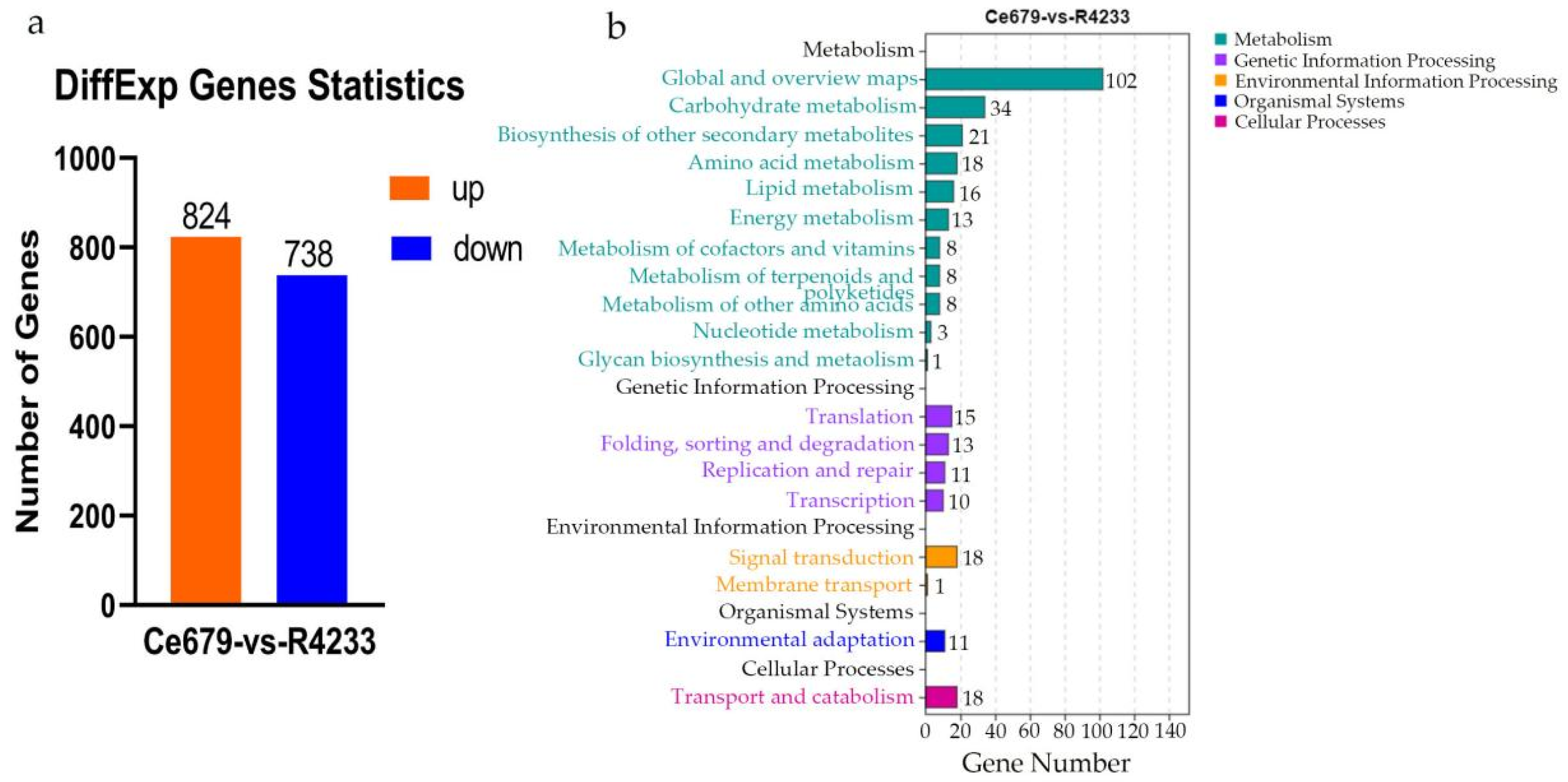
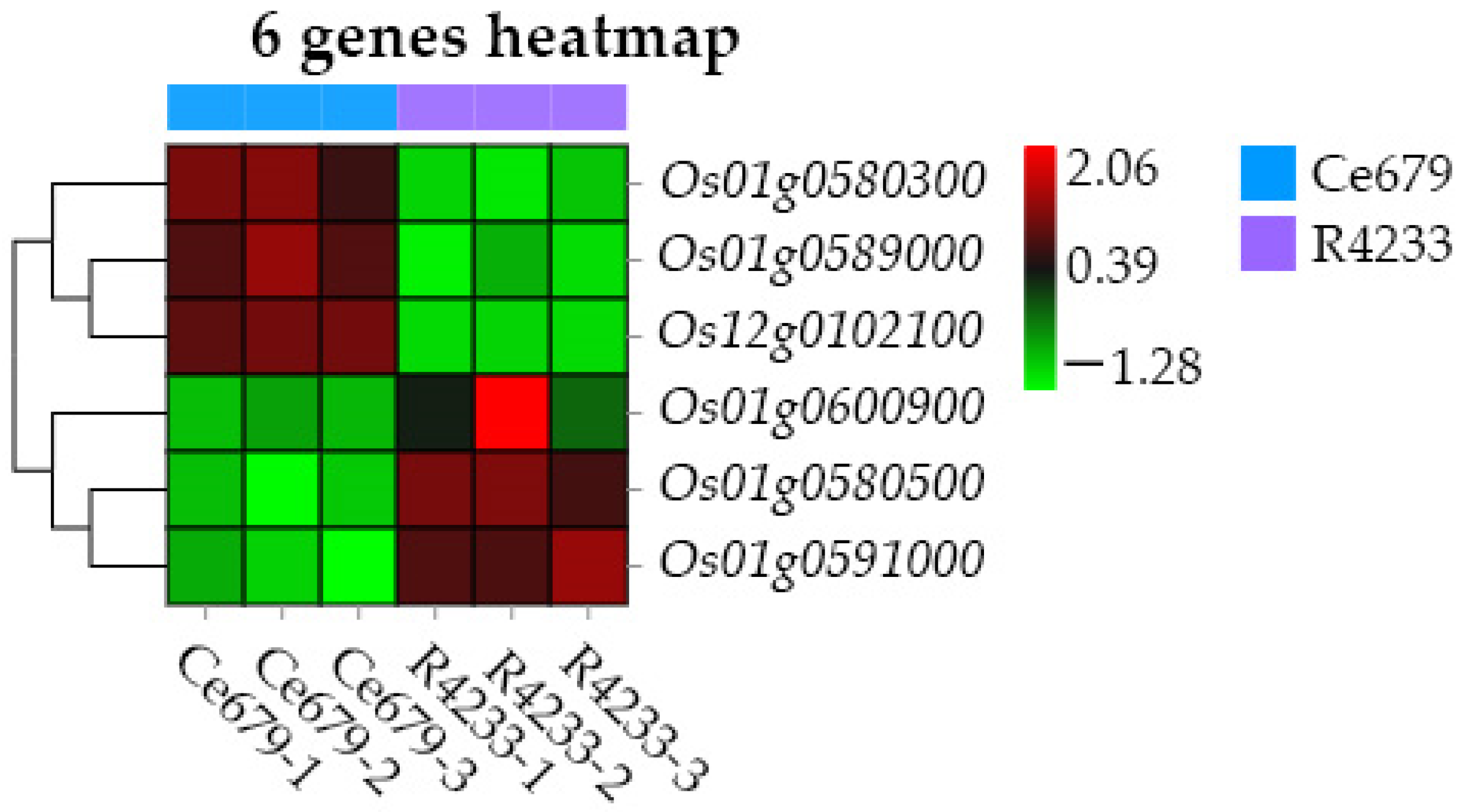
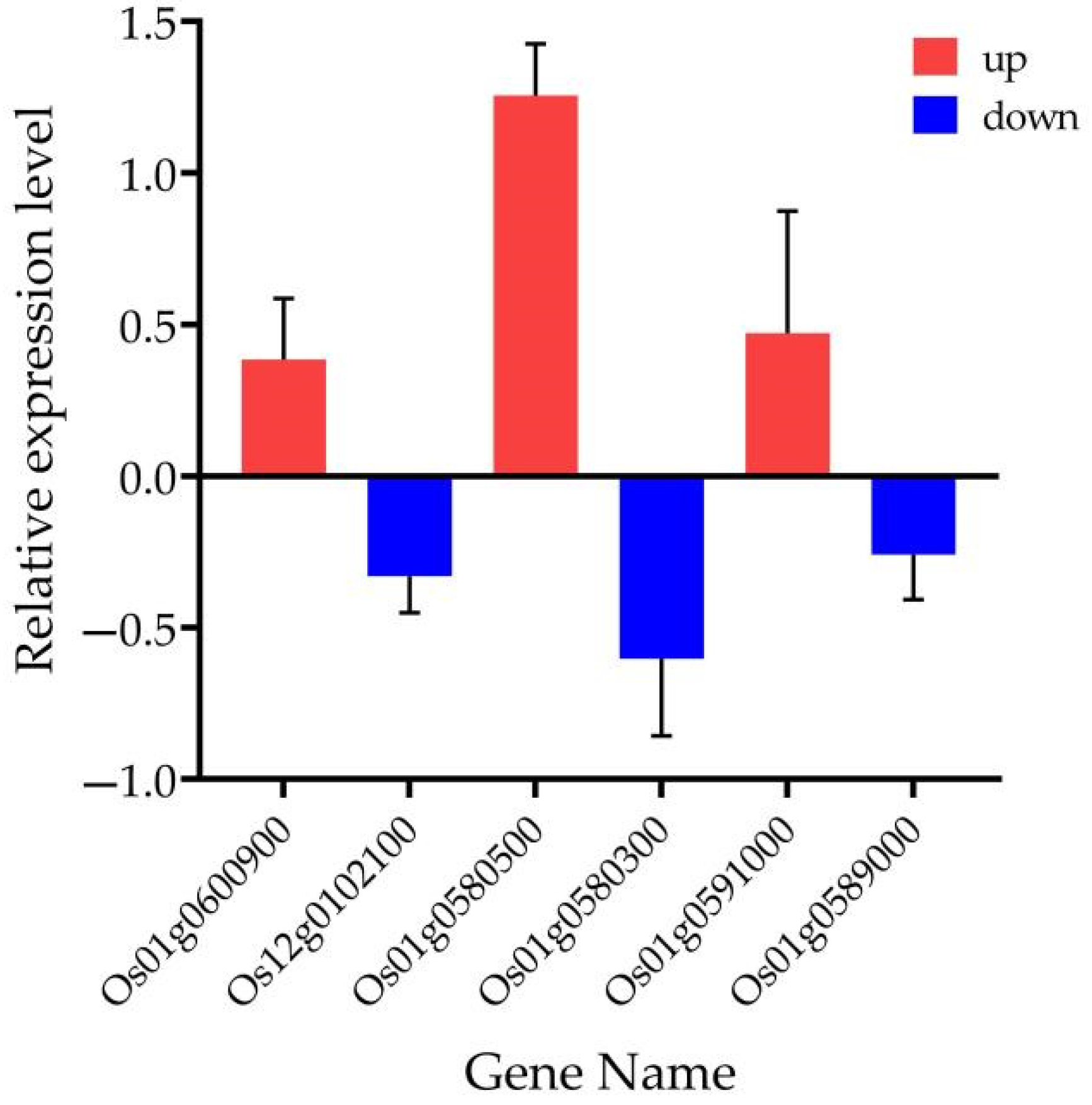
In the current study, we applied a multi-omics approach to investigate the GNPP. BSA-Seq was used to identify QTLs’ position. Further, integration of the BSA-Seq analysis and RNA-sequencing to mine QTL related to spikelet grain number at panicle initiation showed thirty-one DEGs, fourteen of which were located on chromosome 1, four on chromosome 5, eight on chromosome 10, and five on chromosome 12. The result reflected the Manhattan plot analysis from BSA-seq, indicating the success of this joint point approach. A KEGG enrichment analysis of these 31 DEGs and 71 differentially enriched metabolites was performed. Two genes,
Os12g0102100 and
Os01g0580500, and 12 metabolites were significantly enriched in 3 metabolic pathways.
Os12g0102100, the alcohol dehydrogenase superfamily zinc-containing protein, is a novel gene, and the contribution to the GNPP is not yet elucidated. This gene was downregulated in the cultivar 4233 and upregulated in Ce679, highlighting its negative effect on the GNPP. The
Os01g0580500 code for the enzyme 1-aminoclopropane-1-carboxylate oxidase (
OsACO7) is responsible for the final step of the ethylene biosynthesis pathway through the conversion of 1-aminocyclopropane-1-carboxylic acid (ACC) into ethylene in flowering plants under aerobic conditions. This gene was highly expressed in R4233 and downregulated in Ce679, suggesting that this gene positively regulated the GNPP through ethylene synthesis. Increasing evidence indicated that several hormones synthesized by plants, including cytokinin (CK), auxin, abscisic acid (ABA), and ethylene, played a crucial role in developing the panicle and indirectly the GNPP in rice by regulating transcriptional and post-transcriptional activities of GNPP-related genes [
6,
7,
60]. A previous study showed that
OsACO7 was linked to ethylene biosynthesis, enhancing the resistance of young rice plants to the infection of blast fungus [
61]. Ethylene is a group of plant growth regulators involved in coordinating numerous plant development processes such as germination, growth, ripening, senescence, and biotic and abiotic stress responses. A recent study showed that it also contributed to important agronomic traits in rice, including the regulation of panicle architecture, grain filling rate, and size [
60]. Yin and coworkers (2015), revealed that ethylene deficient mutant mhz5/crtiso had smaller panicles, fewer branches, and more excessive tillers than wild-type plants. In addition, several studies reported that the difference in filling rate between the higher and lower spikelets resulted from the level of ethylene in the two parts. A cultivar with a compact panicle had higher ethylene content than lax-panicle rice [
62,
63,
64]. These suggested that the level of ethylene at the panicle initiation stage may positively affect the development of the primary and secondary branches in R4233.
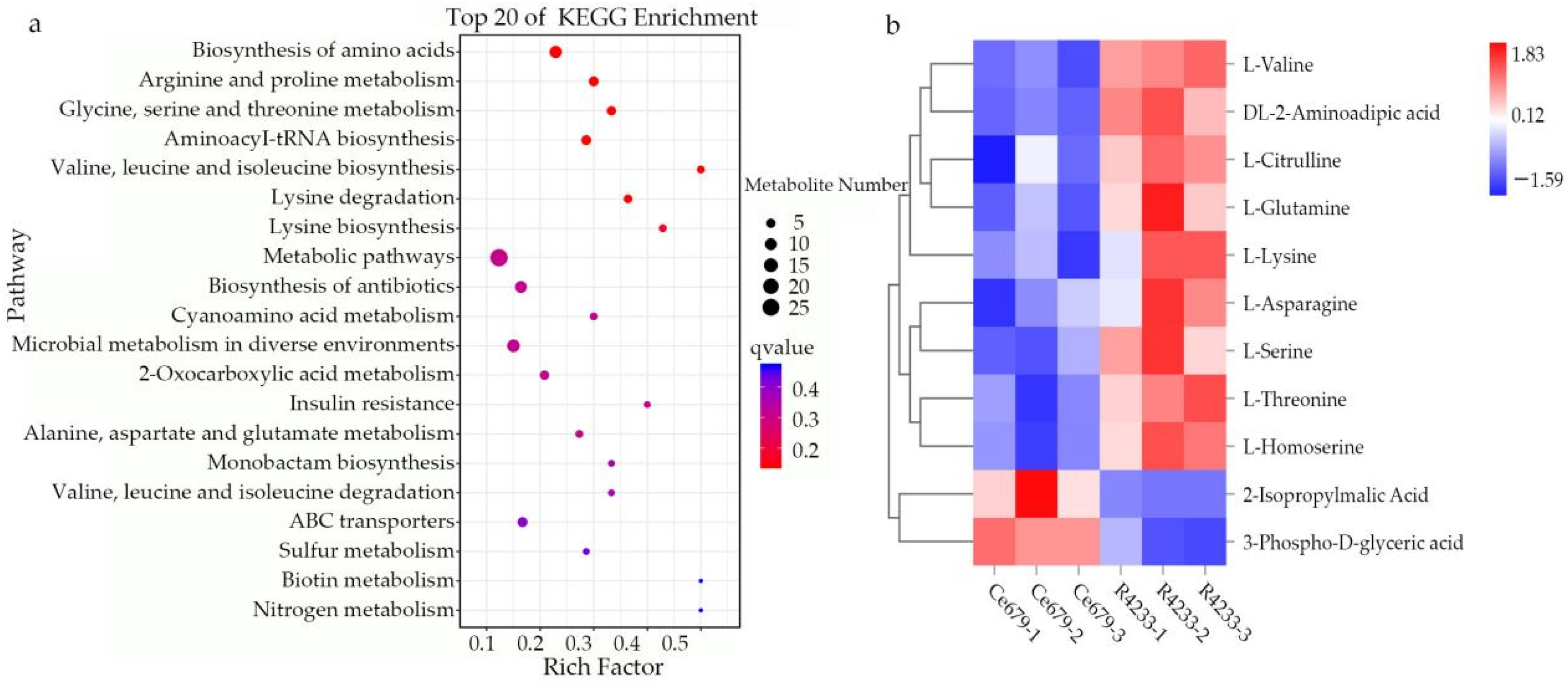
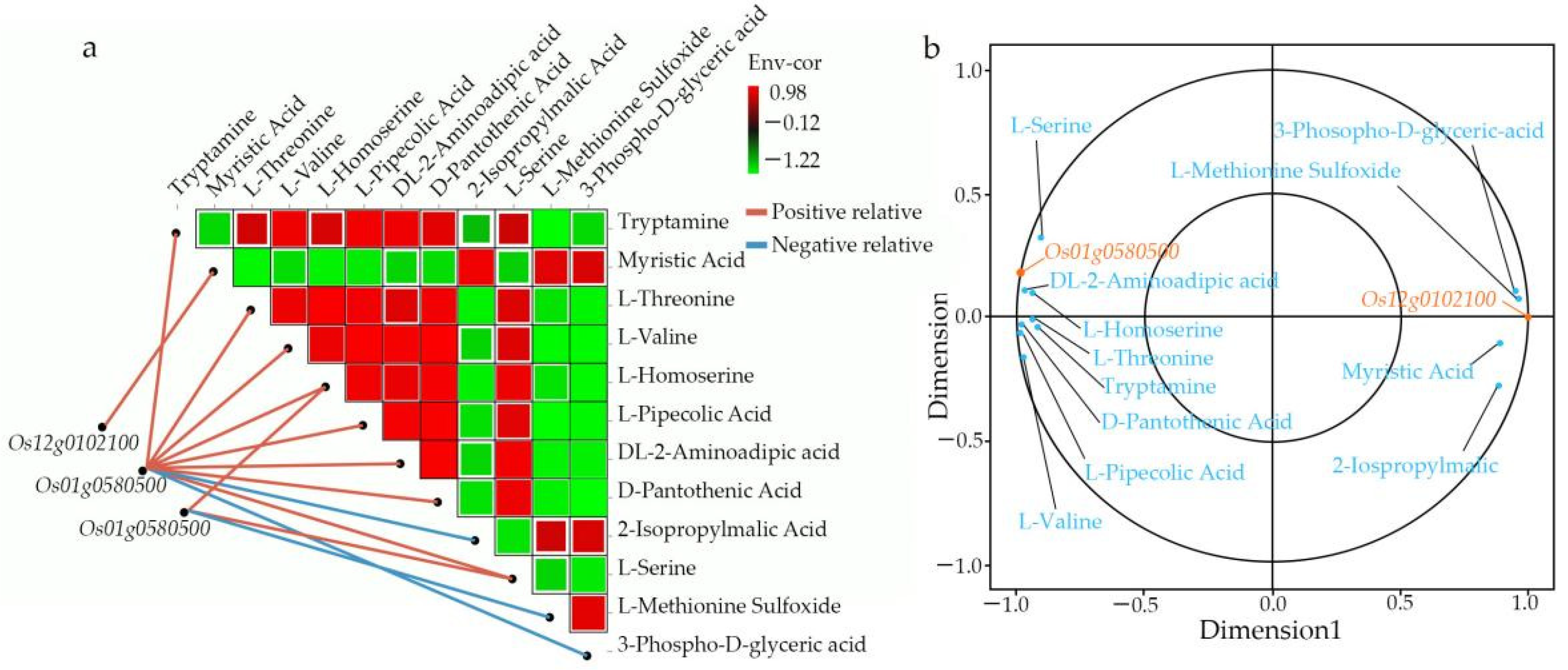
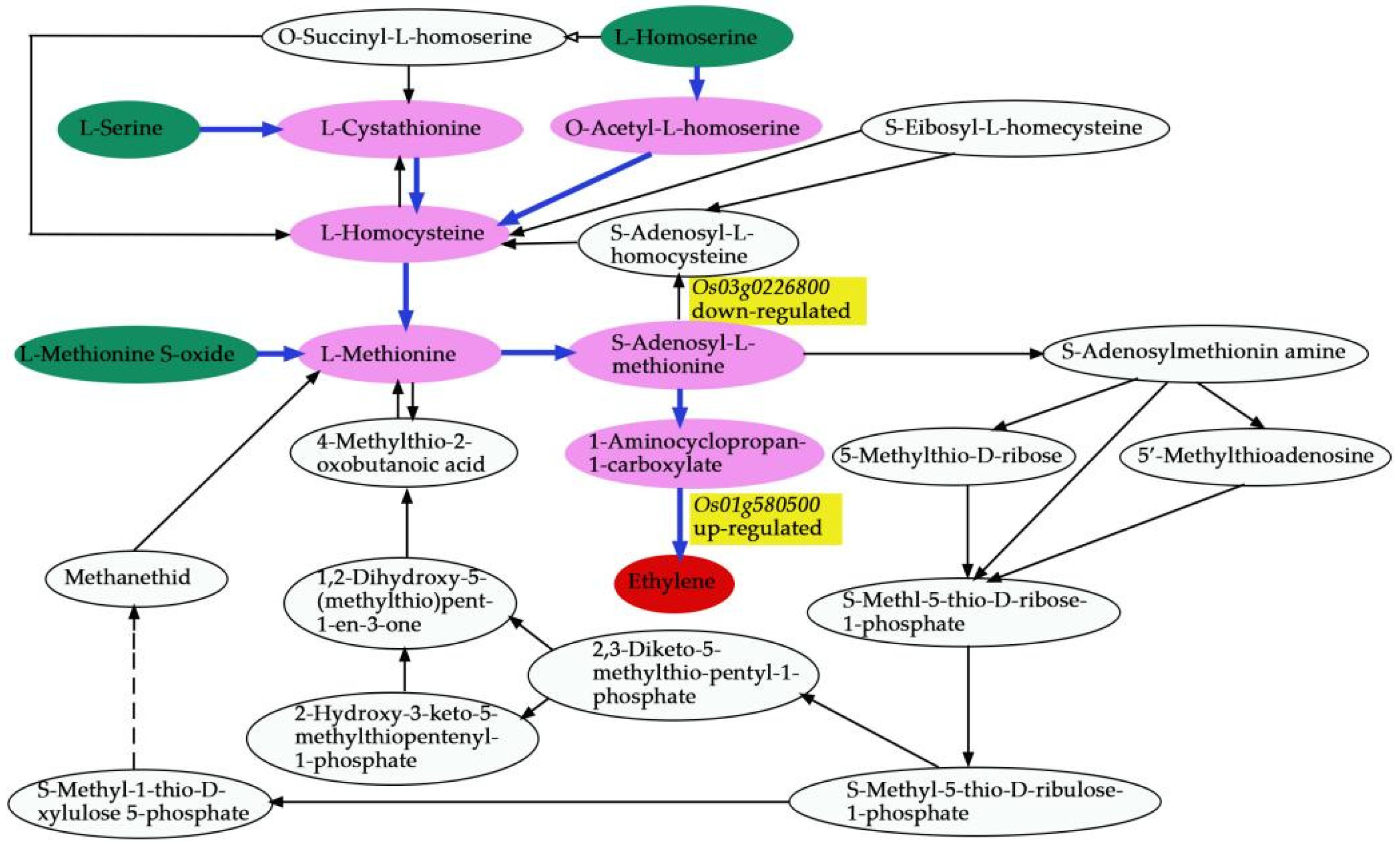
We comprehensively assessed the endogenous metabolites involved in these processes. Our results revealed that
OsACO7 interacted with 11 metabolites classified into four groups: amino acids and derivatives (the most prominent group), alkaloids, organic acids, and others sharing two essential pathways. Among the twelve metabolites, ten were found in the biosynthesis of secondary metabolites, suggesting that this pathway contributed substantially to the development of primary and secondary branches of the panicle. This result was in line with Ke et al. (2018), who found that the biosynthesis of secondary metabolites contributed to rice panicle development. Secondary metabolites are generally regarded as indispensable to maintaining normal metabolism and completing the normal life cycle in the plant. Cysteine and methionine metabolism pathways involved three of twelve metabolites: L-Serine, L-Homoserine, and L-Methionine Sulfoxide. Although only three metabolites were identified in this pathway, they seemed to contribute because they can be considered the primary substrate from which ethylene is synthesized (
Figure 15). L-Serine and L-Homoserine were also identified in the biosynthesis of the secondary metabolites’ pathway, suggesting that these metabolites were essential in ethylene biosynthesis and the GNPP. Juan and co-authors (2014) reported that ethylene was synthesized from S-adenosylmethionine (SAM) through 1-aminocyclopropane-1-carboxylic acid (ACC), and L-serine was the primary source of one-carbon units for methylation reactions that occurred with the generation of S-adenosylmethionine [
65]. Other studies also revealed that ethylene was produced from methionine [
66,
67].
Another essential pathway was fatty acid biosynthesis, in which the gene
Os12g0102100 coding for mitochondrial trans-2-enoyl-CoA reductase was implicated in the biosynthesis of myristic acid, also called tetradecanoic acid, which is a long-chain saturated fatty acid. Qin and colleagues (2007) showed that saturated, very-long-chain fatty acids promote the development of cotton fiber and the elongation of cells in arabidopsis through the activation of the biosynthesis of ethylene. A previous study reported that myristic acid is vital in cell regulation because it modifies the number of proteins through acylation and N-myristoylation in the signal transduction cascade [
68,
69]. The high expression of this gene increases the production of miristic acid, which may interfere with the ethylene production pathway in Ce679, leading to the repression of the development process of secondary branches. The differential regulatory mechanism may coincide with the specific ethylene responses to secondary and primary branches’ development, suggesting a possibility for a novel ethylene-GNPP regulatory mechanism in rice.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}