
H2S in Critical Illness—A New Horizon for Sodium Thiosulfate?

 , , , ,
, , , ,
Abstract
1. Introduction
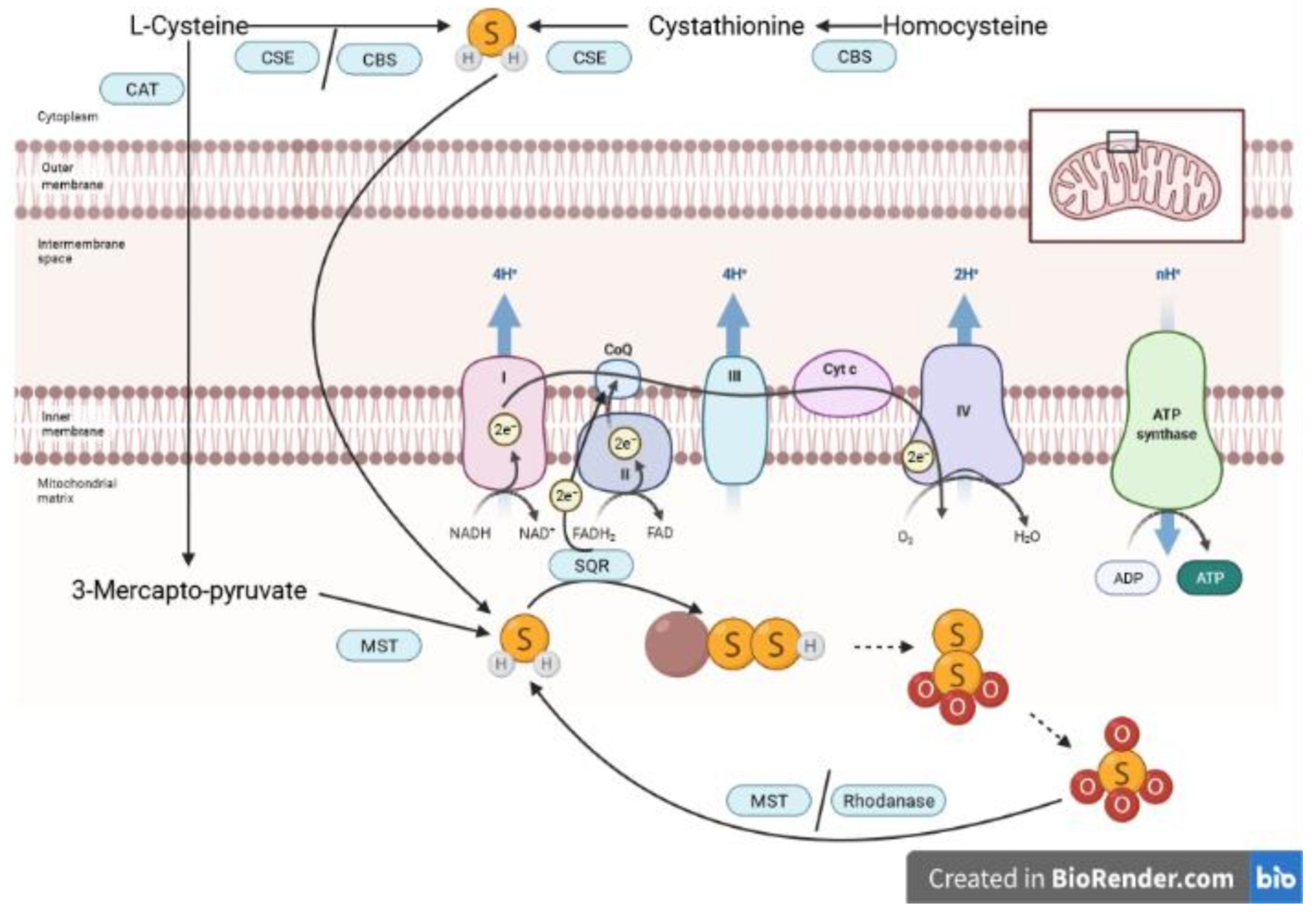
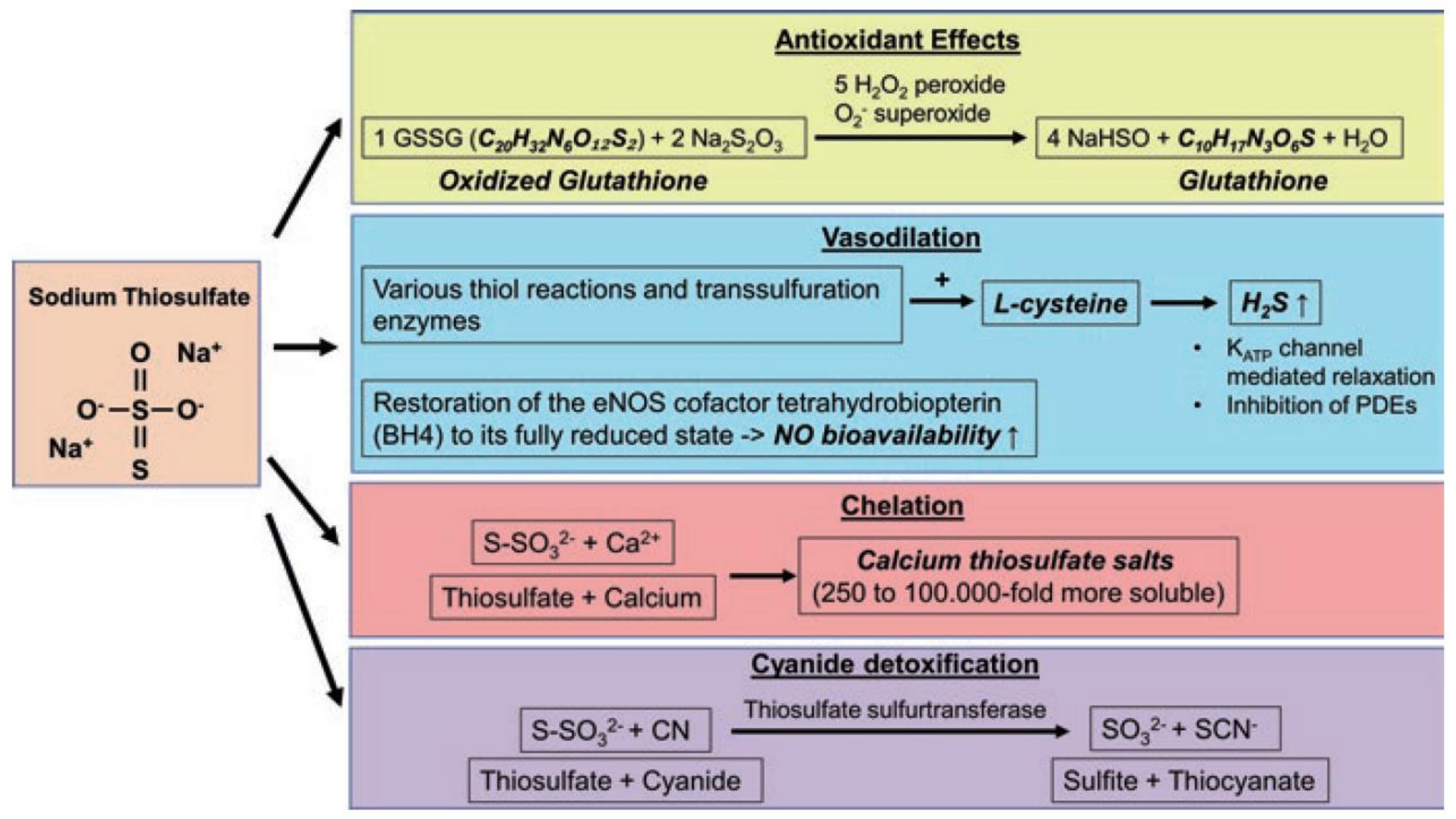
2. Biology of H2S and S2O32−
3. Experimental Evidence for Beneficial Effects of Na2S2O3 in Animal Models of Critical Illness
3.1. Langendorff Heart Model and In Vivo I/R Experiments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author | Animal Model | Condition | Na2S2O3 Administration | Effects of Na2S2O3 (Compared to Vehicle) |
|---|---|---|---|---|
| Mouse models | ||||
| Tokuda 2012 [48] | Male C57BL/6J mice, aged 8–10 weeks | Endotoxemia: LPS (10 mg/kg i.p.) with 1 mL of saline (i.p.) at 0, 6 and 24 h after LPS | 1 or 2 g/kg (i.p.) immediately after LPS | improved survival |
| Shirozu 2014 [49] | Male C57BL/6J mice, aged 8–12 weeks | Acute liver failure and endotoxemia: D-Galactosamine (300 mg/kg) and 1 mg/kg LPS (i.p.) with 1 mL of saline (i.p.) at 0, 6 and 24 h after LPS | 2 g/kg (i.p.) at 30 min before and 3 h after LPS | liver injury ↓, antioxidant and anti-apoptotic effects, preserved mitochondrial membrane potential |
| Sakaguchi 2014 [50] | Male C57BL6J mice, aged 8–10 weeks | Endotoxemia: intratracheal LPS (2 mg/kg) Polymicrobial septic shock: CLP with fluid resuscitation | 2 g/kg (i.p.) at 0 and 12 h after LPS 0.5 g/kg (i.v.) at 10 min after CLP | cell accumulation and MPO in BALF ↓, lung edema ↓, lung IL-6, TNFα, NOS2, MMP9 ↓, lung NFκB signaling ↓ |
| Marutani 2015 [47] | Male C57BL/6J mice, aged 8–9 weeks | Global cerebral I/R: bilateral common carotid artery occlusion, reperfusion after 40 min | 10 mg/kg (i.p.) in 1 mL of 5% dextrose-enriched lactated Ringer’s solution at 10 min after reperfusion and daily for 1 week | improved 20-day survival, neuronal function score ↑ at 24 h after reperfusion |
| Acero 2017 [51] | Female C57BL/6J mice, aged 2 months | Endotoxemia: LPS (3 mg/kg i.p.) | 100, 350, 500, and 750 mg/kg (i.p.) immediately after LPS and at 8, 24, and 32 h | brain IL1-β, COX-2, Iba-1, TSPO ↓ (at 500 mg/kg Na2S2O3) neuroinflammation ↓ |
| Renieris 2021 [52] | Male and female mice, WT and CSE−/− (C57BL/6) aged 7–8 weeks | Sepsis: P. aeruginosa (i.p.) infection | 2 g/kg (s.c.) daily for four days after infection | survival ↑ in WT mice |
| Gröger 2022a [53] | Male CSE−/− mice (C57BL/6J.129SvEv), aged 20–25 weeks | Polytrauma: blast wave-induced blunt chest trauma + hemorrhagic shock (1 h), retransfusion and intensive care management (6 h) | i.v. bolus (0.45 mg/g) at start of resuscitation | norepinephrine requirements, lactate ↓, Horowitz index and urine output ↑, lung IL-6 and MCP1 ↓, lung GR and NOS2 ↑, kidney IκBα and HO-1 ↑ |
| Rat models | ||||
| Ravindran 2017 [43] | Male Wistar rats Langendorff heart model | 30 min ischemia with 60 min reperfusion | 1 mM (15 min) post-conditioning | apoptosis ↓, anti-oxidant defense ↑, preserved mitochondrial enzyme activity |
| Ravindran 2017 b [39] | Langendorff heart model | 30 min ischemia with 60 min reperfusion | 0.1 mM and 1 mM (10 min) pre-conditioning | myocardial injury, inflammatory cell infiltration, and interstitial oedema ↓ |
| Ravindran 2017 b [39] | Rats | Left Anterior Descending Artery Ligation (30 min ischemia with 2h reperfusion) | 1 mM (i.v., 15 min prior to ischemia) | oxidative stress ↓, mitochondrial protection |
| Ravindran 2018 [54] | Male Wistar rats (aged 6 weeks) with adenine-induced vascular calcification, Langendorff heart model | 30 min ischemia with 60 min reperfusion | 400 mg/kg orally for 28 days | calcification ↓, I/R-induced cardiac injury ↓ in non-calcified hearts, oxidative stress ↓ in non-calcified hearts, no effect in calcified hearts after I/R |
| Ravindran 2018 b [44] | Langendorff heart model | 30 min ischemia with 60 min reperfusion | 1 mM at start of reperfusion | contractility ↑, myocardial injury ↓, mitochondrial enzyme activity ↑, oxidative stress ↓ |
| Ravindran 2019 [40] | Male Wistar rats, 200–250 g Langendorff heart model | 30 min ischemia with 60 min reperfusion | 1 mM 15 min before ischemia | LV contractility ↑, cardiac injury ↓, recovered ATP production, PGC1α and mitochondrial copy number ↑, mitochondrial proteins ↑, better mitochondrial ultrastructure |
| Kannan 2019 [41] | Male Wistar rats Langendorff heart model | 30 min ischemia with 60 min reperfusion | 1 mM at 15 min before I/R 1 mM at reperfusion with(out) additional PAG (CSE/CBS inhibitor) | Infarct size ↓, less pronounced with PAG co-treatment |
| Ravindran 2020 [45] | Male Wistar rats (250–300 g) | Isoproterenol-induced MI | 100 mg/kg (i.p.), 1 h before isoproterenol | cardiac injury ↓, ROS and caspase-3 and 9 ↓ (heart and brain), mitochondrial function ↑ (heart and brain) |
| Boovarahan 2021 [42] | Male Wistar rats (8–12 weeks, 250–300 g) Langendorff heart model | 30 min ischemia with reperfusion | 1 mM before ischemia or at reperfusion | Preconditioning protective through ↓oxidative stress, mitochondrial protection Postconditioning protective (different mechanism) |
| Schulz 2021 [55] | Male Wistar rats (320–380 g) | Polymicrobial sepsis: CASP | 1 g/kg (i.p.) immediately after sepsis induction and at 24 h | 24 h after sepsis induction: colonic and hepatic microcirculation ↑, mitochondria =, |
| large animal models | ||||
| Oksman 1982 [46] | Male and female dogs (15–25 kg) | Tourniquet shock experimental MI (descending coronary artery ligation) | 500 µg/kg (i.v., 30 min after removal) 500 mg/kg (i.v., 15 min after ligation) | heart function ↑, blood pressure ↑ heart function ↑, minimal effects on blood pressure |
| Broner 1989 [56] | New Zealand white rabbits (approx. 3 kg) | E. coli septicemia with fluid resuscitation | 660 mg/kg (i.v. bolus in combination with other antioxidants, then continuous infusion of 190 mg/kg/h) | well tolerated, no benefit |
| Datzmann 2020 [13] | Male and female Familial Hypercholesterolemia Bretoncelles Meishan pigs (reduced CSE protein levels), median age 24 months | Hemorrhagic shock (3 h), retransfusion and intensive care management | continuous i.v. infusion at start of retransfusion for 24 h (0.1 g/(kg∙h)) | PEEP and Horowitz-Index ↑ (48 h after shock), lung GR expression ↑, pH and BE ↓ |
3.2. Chronic Conditions
3.3. Endotoxemia and Sepsis
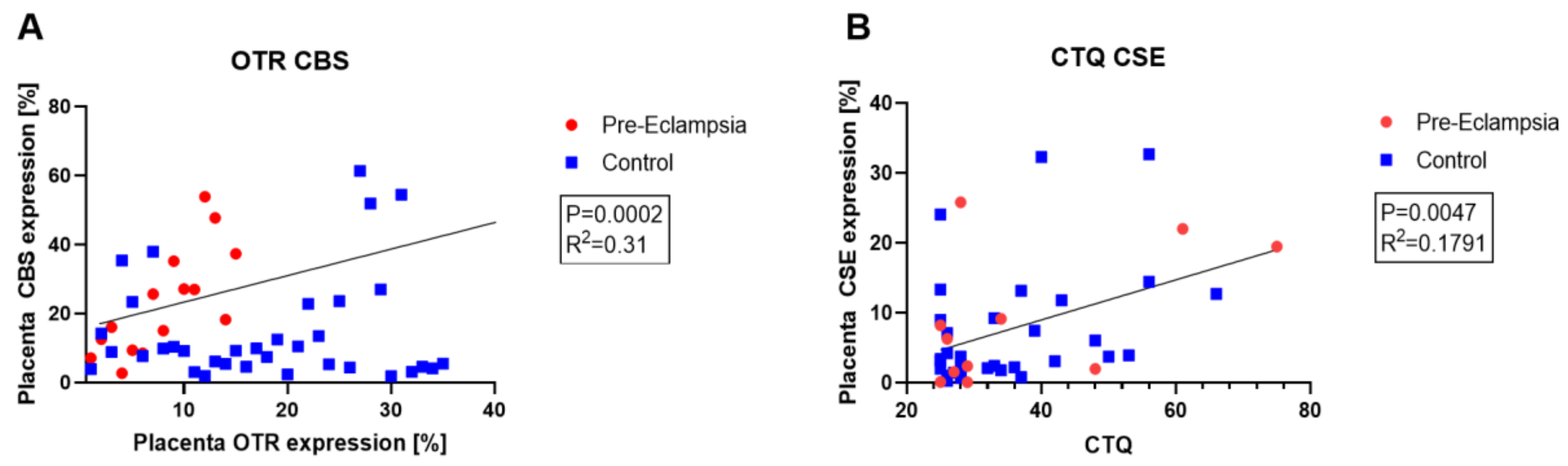
3.4. Role of Endogenous H2S Enzymes
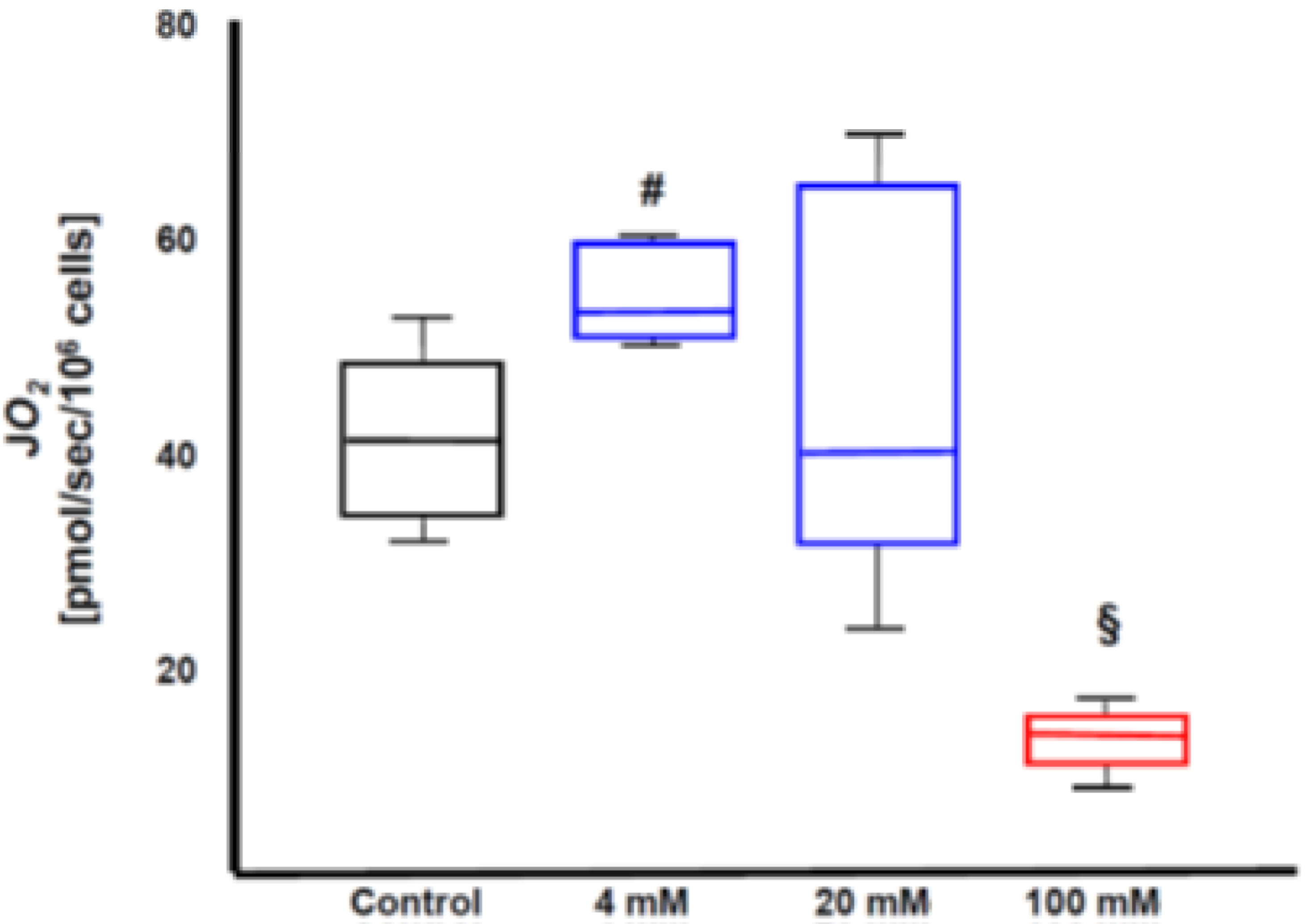
3.5. Role of the Interplay of H2S and Thiosulfate
4. Clinical Perspective
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Francis, R.C.; Vaporidi, K.; Bloch, K.D.; Ichinose, F.; Zapol, W.M. Protective and Detrimental Effects of Sodium Sulfide and Hydrogen Sulfide in Murine Ventilator-Induced Lung Injury. Anesthesiology 2011, 115, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Giudici, R.; Duy, C.N.; Schelzig, H.; Öter, S.; Gröger, M.; Wachter, U.; Vogt, J.; Speit, G.; Szabó, C.; et al. Hemodynamic and Metabolic Effects of Hydrogen Sulfide during Porcine Ischemia/Reperfusion Injury. Shock 2008, 30, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Derwall, M.; Westerkamp, M.; Löwer, C.; Deike-Glindemann, J.; Schnorrenberger, N.K.; Coburn, M.; Nolte, K.W.; Gaisa, N.; Weis, J.; Siepmann, K.; et al. Hydrogen Sulfide Does Not Increase Resuscitability in a Porcine Model of Prolonged Cardiac Arrest. Shock 2010, 34, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Asfar, P.; Calzia, E.; Radermacher, P. Is Pharmacological, H2S-Induced “suspended Animation” Feasible in the ICU? Crit. Care 2014, 18, 215. [Google Scholar] [CrossRef]
- Merz, T.; Denoix, N.; Wepler, M.; Gäßler, H.; Messerer, D.A.C.; Hartmann, C.; Datzmann, T.; Radermacher, P.; McCook, O. H2S in Acute Lung Injury: A Therapeutic Dead End(?). Intensive Care Med. Exp. 2020, 8, 33. [Google Scholar] [CrossRef]
- Bracht, H.; Scheuerle, A.; Gröger, M.; Hauser, B.; Matallo, J.; McCook, O.; Seifritz, A.; Wachter, U.; Vogt, J.A.; Asfar, P.; et al. Effects of Intravenous Sulfide during Resuscitated Porcine Hemorrhagic Shock. Crit. Care Med. 2012, 40, 2157–2167. [Google Scholar] [CrossRef]
- Wepler, M.; Merz, T.; Wachter, U.; Vogt, J.; Calzia, E.; Scheuerle, A.; Möller, P.; Gröger, M.; Kress, S.; Fink, M.; et al. The Mitochondria-Targeted H2S-Donor AP39 in a Murine Model of Combined Hemorrhagic Shock and Blunt Chest Trauma. Shock 2019, 52, 230–239. [Google Scholar] [CrossRef]
- Nußbaum, B.L.; Vogt, J.; Wachter, U.; McCook, O.; Wepler, M.; Matallo, J.; Calzia, E.; Gröger, M.; Georgieff, M.; Wood, M.E.; et al. Metabolic, Cardiac, and Renal Effects of the Slow Hydrogen Sulfide-Releasing Molecule GYY4137 during Resuscitated Septic Shock in Swine with Pre-Existing Coronary Artery Disease. Shock 2017, 48, 175–184. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Thadhani, R.; Brandenburg, V.M. Calciphylaxis. N. Engl. J. Med. 2018, 378, 1704–1717. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin Overdose: Toxicities and Management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef]
- Hunt, G.M.; Ryder, H.F. Metabolic Acidosis after Sodium Thiosulfate Infusion and the Role of Hydrogen Sulfide. Clin. Case Rep. 2018, 6, 1595–1599. [Google Scholar] [CrossRef]
- McGeer, P. Medical Uses of Sodium Thiosulfate. J. Neurol. Neuromedicine 2016, 1, 28–30. [Google Scholar] [CrossRef]
- Datzmann, T.; Hoffmann, A.; McCook, O.; Merz, T.; Wachter, U.; Preuss, J.; Vettorazzi, S.; Calzia, E.; Gröger, M.; Kohn, F.; et al. Effects of Sodium Thiosulfate (Na2S2O3) during Resuscitation from Hemorrhagic Shock in Swine with Preexisting Atherosclerosis. Pharmacol. Res. 2020, 151, 104536. [Google Scholar] [CrossRef]
- Olson, K.R.; DeLeon, E.R.; Gao, Y.; Hurley, K.; Sadauskas, V.; Batz, C.; Stoy, G.F. Thiosulfate: A Readily Accessible Source of Hydrogen Sulfide in Oxygen Sensing. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 305, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, L.R.; Francom, D.; Dieken, F.P.; Taylor, J.D.; Warenycia, M.W.; Reiffenstein, R.J.; Dowling, G. Determination of Sulfide in Brain-Tissue by Gas Dialysis Ion Chromatography-Postmortem Studies and 2 Case-Reports. J. Anal. Toxicol. 1989, 13, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Ogasawara, Y.; Kimura, H. Thioredoxin and Dihydrolipoic Acid Are Required for 3-Mercaptopyruvate Sulfurtransferase to Produce Hydrogen Sulfide. Biochem. J. 2011, 439, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Abou-Hamdan, A.; Guedouari-Bounihi, H.; Lenoir, V.; Andriamihaja, M.; Blachier, F.; Bouillaud, F. Oxidation of H2S in Mammalian Cells and Mitochondria. Methods Enzymol. 2015, 554, 201–228. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F. Sulfide Oxidation Evidences the Immediate Cellular Response to a Decrease in the Mitochondrial ATP/O2 Ratio. Biomolecules 2022, 12, 361. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of Mitochondrial Bioenergetic Function by Hydrogen Sulfide. Part I. Biochemical and Physiological Mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the Human Mitochondrial Hydrogen Sulfide Oxidation Pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Bilska-Wilkosz, A.; Iciek, M.; Górny, M.; Kowalczyk-Pachel, D. The Role of Hemoproteins: Hemoglobin, Myoglobin and Neuroglobin in Endogenous Thiosulfate Production Processes. Int. J. Mol. Sci. 2017, 18, 1315. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen Sulfide as an Oxygen Sensor. Antioxid. Redox Signal. 2015, 22, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen Sulfide as an Oxygen Sensor. Clin. Chem. Lab. Med. 2013, 51, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.W. Hemorrhagic Shock. N. Engl. J. Med. 2018, 378, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and Reperfusion-from Mechanism to Translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- McCook, O.; Radermacher, P.; Volani, C.; Asfar, P.; Ignatius, A.; Kemmler, J.; Möller, P.; Szabó, C.; Whiteman, M.; Woodg, M.E.; et al. H2S during Circulatory Shock: Some Unresolved Questions. Nitric Oxide-Biol. Chem. 2014, 41, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Winyard, P.G. Hydrogen Sulfide and Inflammation: The Good, the Bad, the Ugly and the Promising. Expert Rev. Clin. Pharmacol. 2011, 4, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Matallo, J.; Vogt, J.; Mccook, O.; Wachter, U.; Tillmans, F.; Groeger, M.; Szabo, C.; Georgieff, M.; Radermacher, P.; Calzia, E. Sulfide-Inhibition of Mitochondrial Respiration at Very Low Oxygen Concentrations. Nitric Oxide-Biol. Chem. 2014, 41, 79–84. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Módis, K.; Bos, E.M.; Calzia, E.; van Goor, H.; Coletta, C.; Papapetropoulos, A.; Hellmich, M.R.; Radermacher, P.; Bouillaud, F.; Szabo, C. Regulation of Mitochondrial Bioenergetic Function by Hydrogen Sulfide. Part II Pathophysiological and Therapeutic Aspects. Br. J. Pharmacol. 2014, 171, 2123–2146. [Google Scholar] [CrossRef] [PubMed]
- Dyson, A.; Dal-Pizzol, F.; Sabbatini, G.; Lach, A.B.; Galfo, F.; dos Santos Cardoso, J.; Pescador Mendonça, B.; Hargreaves, I.; Bollen Pinto, B.; Bromage, D.I.; et al. Ammonium Tetrathiomolybdate Following Ischemia/Reperfusion Injury: Chemistry, Pharmacology, and Impact of a New Class of Sulfide Donor in Preclinical Injury Models. PLoS Med. 2017, 14, e1002310. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, B.P.; Cardoso, J.D.S.; Michels, M.; Vieira, A.C.; Wendhausen, D.; Manfredini, A.; Singer, M.; Dal-Pizzol, F.; Dyson, A. Neuroprotective Effects of Ammonium Tetrathiomolybdate, a Slow-Release Sulfide Donor, in a Rodent Model of Regional Stroke. Intensive Care Med. Exp. 2020, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, P.D.; Lunev, S.; Aguilar Lozano, S.P.; de Assis Batista, F.; Al-dahmani, Z.M.; Joles, J.A.; Dolga, A.M.; Groves, M.R.; van Goor, H. Unraveling the Role of Thiosulfate Sulfurtransferase in Metabolic Diseases. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165716. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, I.T.N.; Wiggenhauser, L.M.; Bulthuis, M.; Hillebrands, J.L.; Feelisch, M.; Verhaar, M.C.; van Goor, H.; Joles, J.A. Cardiac Protection by Oral Sodium Thiosulfate in a Rat Model of L-NNA-Induced Heart Disease. Front. Pharmacol. 2021, 12, 650968. [Google Scholar] [CrossRef]
- Araya, C.E.; Fennell, R.S.; Neiberger, R.E.; Dharnidharka, V.R. Sodium Thiosulfate Treatment for Calcific Uremic Arteriolopathy in Children and Young Adults. Clin. J. Am. Soc. Nephrol. 2006, 1, 1161–1166. [Google Scholar] [CrossRef]
- Chen, K.; Rose, C. Nitrite and Thiosulfate Therapy in Cyanide Poisoning. J. Am. Med. Assoc. 1952, 149, 113–119. [Google Scholar] [CrossRef]
- Bauer, M.; Radermacher, P.; Wepler, M. Sodium Thiosulfate: A New Player for Circulatory Shock and Ischemia/Reperfusion Injury? In Annual Update in Intensive Care 2019; Vincent, J.-L., Ed.; Springer Nature: Cham, Switzerland, 2019; pp. 183–198. ISBN 978-3-030-06067-1. [Google Scholar]
- Ravindran, S.; Boovarahan, S.R.; Shanmugam, K.; Vedarathinam, R.C.; Kurian, G.A. Sodium Thiosulfate Preconditioning Ameliorates Ischemia/Reperfusion Injury in Rat Hearts Via Reduction of Oxidative Stress and Apoptosis. Cardiovasc. Drugs Ther. 2017, 31, 511–524. [Google Scholar] [CrossRef]
- Ravindran, S.; Kurian, G.A. Preconditioning the Rat Heart with Sodium Thiosulfate Preserved the Mitochondria in Response to Ischemia-Reperfusion Injury. J. Bioenerg. Biomembr. 2019, 51, 189–201. [Google Scholar] [CrossRef]
- Kannan, S.; Boovarahan, S.R.; Rengaraju, J.; Prem, P.; Kurian, G.A. Attenuation of Cardiac Ischemia-Reperfusion Injury by Sodium Thiosulfate Is Partially Dependent on the Effect of Cystathione Beta Synthase in the Myocardium. Cell Biochem. Biophys. 2019, 77, 261–272. [Google Scholar] [CrossRef]
- Boovarahan, S.R.; Venkatasubramanian, H.; Sharma, N.; Venkatesh, S.; Prem, P.; Kurian, G.A. Inhibition of PI3K/MTOR/KATP Channel Blunts Sodium Thiosulphate Preconditioning Mediated Cardioprotection against Ischemia–Reperfusion Injury. Arch. Pharmacal Res. 2021, 44, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Jahir Hussain, S.; Boovarahan, S.R.; Kurian, G.A. Sodium Thiosulfate Post-Conditioning Protects Rat Hearts against Ischemia Reperfusion Injury via Reduction of Apoptosis and Oxidative Stress. Chem.-Biol. Interact. 2017, 274, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Kurian, G.A. Effect of Sodium Thiosulfate Postconditioning on Ischemia-Reperfusion Injury Induced Mitochondrial Dysfunction in Rat Heart. J. Cardiovasc. Transl. Res. 2018, 11, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Gopalakrishnan, S.; Kurian, G.A. Beneficial Effect of Sodium Thiosulfate Extends beyond Myocardial Tissue in Isoproterenol Model of Infarction: Implication for Nootropic Effects. J. Biochem. Mol. Toxicol. 2020, 34, e22606. [Google Scholar] [CrossRef]
- Oksman, T.M.; Levandovskii, I.V.; Epishin, N.Y.; Vrana, M.; Blažek, Z. Sodium Thiosulfate in the Treatment of Early Postischemic Disorders. Bull. Exp. Biol. Med. 1982, 92, 275–278. [Google Scholar] [CrossRef]
- Marutani, E.; Yamada, M.; Ida, T.; Tokuda, K.; Ikeda, K.; Kai, S.; Shirozu, K.; Hayashida, K.; Kosugi, S.; Hanaoka, K.; et al. Thiosulfate Mediates Cytoprotective Effects of Hydrogen Sulfide against Neuronal Ischemia. J. Am. Heart Assoc. 2015, 4, e002125. [Google Scholar] [CrossRef]
- Tokuda, K.; Kida, K.; Marutani, E.; Crimi, E.; Bougaki, M.; Khatri, A.; Kimura, H.; Ichinose, F. Inhaled Hydrogen Sulfide Prevents Endotoxin-Induced Systemic Inflammation and Improves Survival by Altering Sulfide Metabolism in Mice. Antioxid. Redox Signal. 2012, 17, 11–21. [Google Scholar] [CrossRef]
- Shirozu, K.; Tokuda, K.; Marutani, E.; Lefer, D.; Wang, R.; Ichinose, F. Cystathionine γ-Lyase Deficiency Protects Mice from Galactosamine/Lipopolysaccharide-Induced Acute Liver Failure. Antioxid. Redox Signal. 2014, 20, 204–216. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Marutani, E.; Shin, H.; Chen, W.; Hanaoka, K.; Xian, M.; Ichinose, F. Sodium Thiosulfate Attenuates Acute Lung Injury in Mice. Anesthesiology 2014, 121, 1248–1257. [Google Scholar] [CrossRef]
- Acero, G.; Nava Catorce, M.; González-Mendoza, R.; Meraz-Rodríguez, M.A.; Hernández-Zimbron, L.F.; González-Salinas, R.; Gevorkian, G. Sodium Thiosulphate Attenuates Brain Inflammation Induced by Systemic Lipopolysaccharide Administration in C57BL/6J Mice. Inflammopharmacology 2017, 25, 585–593. [Google Scholar] [CrossRef]
- Renieris, G.; Droggiti, D.E.; Katrini, K.; Koufargyris, P.; Gkavogianni, T.; Karakike, E.; Antonakos, N.; Damoraki, G.; Karageorgos, A.; Sabracos, L.; et al. Host Cystathionine-γ Lyase Derived Hydrogen Sulfide Protects against Pseudomonas Aeruginosa Sepsis. PLoS Pathog. 2021, 17, e1009473. [Google Scholar] [CrossRef] [PubMed]
- Gröger, M.; Hogg, M.; Abdelsalam, E.; Kress, S.; Hoffmann, A.; Stahl, B.; Saub, V.; Denoix, N.; McCook, O.; Calzia, E.; et al. Effects of Sodium Thiosulfate During Resuscitation From Trauma-and-Hemorrhage in Cystathionine Gamma Lyase (CSE) Knockout Mice. Shock 2022, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Ramachandran, K.; Kurian, G.A. Sodium Thiosulfate Mediated Cardioprotection against Myocardial Ischemia-Reperfusion Injury Is Defunct in Rat Heart with Co-Morbidity of Vascular Calcification. Biochimie 2018, 147, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Kramer, S.; Kanatli, Y.; Kuebart, A.; Bauer, I.; Picker, O.; Vollmer, C.; Truse, R.; Herminghaus, A. Sodium Thiosulfate Improves Intestinal and Hepatic Microcirculation Without Affecting Mitochondrial Function in Experimental Sepsis. Front. Immunol. 2021, 12, 2121. [Google Scholar] [CrossRef] [PubMed]
- Broner, C.W.; Shenep, J.L.; Stidham, G.L.; Stokes, D.C.; Fairclough, D.; Schonbaum, G.R.; Rehg, J.E.; Hildner, W.K. Effect of Antioxidants in Experimental Escherichia Coli Septicemia. Circ. Shock. 1989, 29, 77–92. [Google Scholar]
- Mohan, D.; Balasubramanian, E.D.; Ravindran, S.; Kurian, G.A. Renal Mitochondria Can Withstand Hypoxic/Ischemic Injury Secondary to Renal Failure in Uremic Rats Pretreated with Sodium Thiosulfate. Indian J. Pharmacol. 2017, 49, 317–321. [Google Scholar] [CrossRef]
- Snijder, P.M.; Frenay, A.R.; de Boer, R.A.; Pasch, A.; Hillebrands, J.L.; Leuvenink, H.G.D.; van Goor, H. Exogenous Administration of Thiosulfate, a Donor of Hydrogen Sulfide, Attenuates Angiotensin II-Induced Hypertensive Heart Disease in Rats. Br. J. Pharmacol. 2015, 172, 1494–1504. [Google Scholar] [CrossRef]
- Nguyen, I.T.N.; Klooster, A.; Minnion, M.; Feelisch, M.; Verhaar, M.C.; van Goor, H.; Joles, J.A. Sodium Thiosulfate Improves Renal Function and Oxygenation in L-NNA–Induced Hypertension in Rats. Kidney Int. 2020, 98, 366–377. [Google Scholar] [CrossRef]
- Rein, J.L.; Miyata, K.N.; Dadzie, K.A.; Gruber, S.J.; Sulica, R.; Winchester, J.F. Successfully Treated Calcific Uremic Arteriolopathy: Two Cases of a High Anion Gap Metabolic Acidosis with Intravenous Sodium Thiosulfate. Case Rep. Nephrol. 2014, 2014, 765134. [Google Scholar] [CrossRef]
- Sen, U.; Vacek, T.P.; Hughes, W.M.; Kumar, M.; Moshal, K.S.; Tyagi, N.; Metreveli, N.; Hayden, M.R.; Tyagi, S.C. Cardioprotective Role of Sodium Thiosulfate on Chronic Heart Failure by Modulating Endogenous H2S Generation. Pharmacology 2008, 82, 201–213. [Google Scholar] [CrossRef]
- Gröger, M.; Hogg, M.; Abdelsalam, E.; Kress, S.; Hoffmann, A.; Stahl, B.; Calzia, E.; Wachter, U.; Vogt, J.A.; Wang, R.; et al. Effects of Sodium Thiosulfate during Resuscitation from Trauma-and-Hemorrhage in Cystathionine-γ-Lyase Knockout Mice with Diabetes Type 1. Front. Med. 2022; under review. [Google Scholar]
- Merz, T.; Denoix, N.; Wigger, D.; Waller, C.; Wepler, M.; Vettorazzi, S.; Tuckermann, J.; Radermacher, P.; McCook, O. The Role of Glucocorticoid Receptor and Oxytocin Receptor in the Septic Heart in a Clinically Relevant, Resuscitated Porcine Model With Underlying Atherosclerosis. Front. Endocrinol. 2020, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Merz, T.; Wepler, M.; Nußbaum, B.; Vogt, J.; Calzia, E.; Wang, R.; Szabo, C.; Radermacher, P.; McCook, O. Cystathionine-γ-Lyase Expression Is Associated with Mitochondrial Respiration during Sepsis-Induced Acute Kidney Injury in Swine with Atherosclerosis. Intensive Care Med. Exp. 2018, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Merz, T.; Stenzel, T.; Nußbaum, B.; Wepler, M.; Szabo, C.; Wang, R.; Radermacher, P.; McCook, O. Cardiovascular Disease and Resuscitated Septic Shock Lead to the Downregulation of the H2S-Producing Enzyme Cystathionine-γ-Lyase in the Porcine Coronary Artery. Intensive Care Med. Exp. 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Messerer, D.A.C.; Gässler, H.; Hoffmann, A.; Gröger, M.; Benz, K.; Huhn, A.; Calzia, E.; Radermacher, P.; Datzmann, T. The H2S Donor Sodium Thiosulfate (Na2S2O3) Does Not Improve Inflammation and Organ Damage after Hemorrhagic Shock in Cardiovascular Healthy Swine. Front. Immunol. 2022; under review. [Google Scholar]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide Catabolism Ameliorates Hypoxic Brain Injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Ichinose, F. Emerging Pharmacological Tools to Control Hydrogen Sulfide Signaling in Critical Illness. Intensive Care Med. Exp. 2020, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Dugbartey, G.J.; Juriasingani, S.; Sener, A. Hydrogen Sulfide Metabolite, Sodium Thiosulfate: Clinical Applications and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6452. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Sulphane Sulphur in Biological Systems: A Possible Regulatory Role. Biochem. J. 1989, 264, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I.; Cooper, A.J.L. Thiosulfoxide (Sulfane) Sulfur: New Chemistry and New Regulatory Roles in Biology. Molecules 2014, 19, 12789–12813. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Alendy, M.J.; Ahmed, K.I.; Leeuwenburgh, C.; Julian, D. The Hydrogen Sulfide Signaling System: Changes during Aging and the Benefits of Caloric Restriction. Age 2010, 32, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Paris, E.I.; Pelisov, M.G. Use of Sodium Thiosulfate in Shock Due to Burns. Voen.-Meditsinskii Zhurnal 1966, 5, 38–40. [Google Scholar]
- De Koning, M.S.L.Y.; Assa, S.; Maagdenberg, C.G.; van Veldhuisen, D.J.; Pasch, A.; van Goor, H.; Lipsic, E.; van der Harst, P. Safety and Tolerability of Sodium Thiosulfate in Patients with an Acute Coronary Syndrome Undergoing Coronary Angiography: A Dose-Escalation Safety Pilot Study (SAFE-ACS). J. Interv. Cardiol. 2020, 2020, 6014915. [Google Scholar] [CrossRef] [PubMed]
- De Koning, M.S.L.; van Dorp, P.; Assa, S.; Hartman, M.H.; Voskuil, M.; Anthonio, R.L.; Veen, D.; Pundziute-Do Prado, G.; Leiner, T.; van Goor, H.; et al. Rationale and Design of the Groningen Intervention Study for the Preservation of Cardiac Function with Sodium Thiosulfate after St-Segment Elevation Myocardial Infarction (GIPS-IV) Trial. Am. Heart J. 2022, 243, 167–176. [Google Scholar] [CrossRef]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.F.; et al. Dysregulation of Hydrogen Sulfide Producing Enzyme Cystathionine γ-Lyase Contributes to Maternal Hypertension and Placental Abnormalities in Preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef]
- Holwerda, K.M.; Bos, E.M.; Rajakumar, A.; Ris-Stalpers, C.; van Pampus, M.G.; Timmer, A.; Erwich, J.J.H.M.; Faas, M.M.; van Goor, H.; Lely, A.T. Hydrogen Sulfide Producing Enzymes in Pregnancy and Preeclampsia. Placenta 2012, 33, 518–521. [Google Scholar] [CrossRef]
- Wigger, D.C.; Gröger, N.; Lesse, A.; Krause, S.; Merz, T.; Gündel, H.; Braun, K.; McCook, O.; Radermacher, P.; Bock, J.; et al. Maternal Separation Induces Long-Term Alterations in the Cardiac Oxytocin Receptor and Cystathionine γ-Lyase Expression in Mice. Oxidative Med. Cell. Longev. 2020, 2020, 4309605. [Google Scholar] [CrossRef]
- Denoix, N.; McCook, O.; Ecker, S.; Wang, R.; Waller, C.; Radermacher, P.; Merz, T. The Interaction of the Endogenous Hydrogen Sulfide and Oxytocin Systems in Fluid Regulation and the Cardiovascular System. Antioxidants 2020, 9, 748. [Google Scholar] [CrossRef]
- Merz, T.; Lukaschewski, B.; Wigger, D.; Rupprecht, A.; Wepler, M.; Gröger, M.; Hartmann, C.; Whiteman, M.; Szabo, C.; Wang, R.; et al. Interaction of the Hydrogen Sulfide System with the Oxytocin System in the Injured Mouse Heart. Intensive Care Med. Exp. 2018, 6, 41. [Google Scholar] [CrossRef]
- Renieris, G.; Katrini, K.; Damoulari, C.; Akinosoglou, K.; Psarrakis, C.; Kyriakopoulou, M.; Dimopoulos, G.; Lada, M.; Koufargyris, P.; Giamarellos-Bourboulis, E.J. Serum Hydrogen Sulfide and Outcome Association in Pneumonia by the SARS-CoV-2 Coronavirus. Shock 2020, 54, 633–637. [Google Scholar] [CrossRef]
- Datzmann, T.; Merz, T.; McCook, O.; Szabo, C.; Radermacher, P. H2S as a Therapeutic Adjuvant Against COVID-19: Why and How? Shock 2021, 56, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Evgen’ev, M.B.; Frenkel, A. Possible Application of H2S-Producing Compounds in Therapy of Coronavirus (COVID-19) Infection and Pneumonia. Cell Stress Chaperones 2020, 25, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Offringa, A.K.; van Eijk, L.E.; Abdulle, A.E.; Hillebrands, J.L.; van der Voort, P.H.J.; van Goor, H.; van Hezik, E.J. N-Acetylcysteine and Hydrogen Sulfide in Coronavirus Disease 2019. Antioxid. Redox Signal. 2021, 35, 1207–1225. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merz, T.; McCook, O.; Brucker, C.; Waller, C.; Calzia, E.; Radermacher, P.; Datzmann, T. H2S in Critical Illness—A New Horizon for Sodium Thiosulfate? Biomolecules 2022, 12, 543. https://doi.org/10.3390/biom12040543
Merz T, McCook O, Brucker C, Waller C, Calzia E, Radermacher P, Datzmann T. H2S in Critical Illness—A New Horizon for Sodium Thiosulfate? Biomolecules. 2022; 12(4):543. https://doi.org/10.3390/biom12040543
Chicago/Turabian StyleMerz, Tamara, Oscar McCook, Cosima Brucker, Christiane Waller, Enrico Calzia, Peter Radermacher, and Thomas Datzmann. 2022. "H2S in Critical Illness—A New Horizon for Sodium Thiosulfate?" Biomolecules 12, no. 4: 543. https://doi.org/10.3390/biom12040543
APA StyleMerz, T., McCook, O., Brucker, C., Waller, C., Calzia, E., Radermacher, P., & Datzmann, T. (2022). H2S in Critical Illness—A New Horizon for Sodium Thiosulfate? Biomolecules, 12(4), 543. https://doi.org/10.3390/biom12040543